

Abstract
Basic architecture of ABC transporters includes two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs). Although the transport process takes place in the TMDs, which provide the substrate translocation pathway across the cell membrane and control its accessibility between the two sides of the membrane, the energy required for the process is provided by conformational changes induced in the NBDs by binding and hydrolysis of ATP. Nucleotide-dependent conformational changes in the NBDs, therefore, need to be coupled to structural changes in the TMDs. Using molecular dynamics simulations, we have investigated the structural elements involved in the conformational coupling between the NBDs and the TMDs in the Escherichia coli maltose transporter, an ABC importer for which an intact structure is available both in inward-facing and outward-facing conformations. The prevailing model of coupling is primarily based on a single structural motif, known as the coupling helices, as the main structural element for the NBD-TMD coupling. Surprisingly, we find that in the absence of the NBDs the coupling helices can be conformationally decoupled from the rest of the TMDs, despite their covalent connection. That is, the structural integrity of the coupling helices and their tight coupling to the core of the TMDs rely on the contacts provided by the NBDs. Based on the conformational and dynamical analysis of the simulation trajectories, we propose that the core coupling elements in the maltose transporter involve contributions from several structural motifs located at the NBD-TMD interface, namely, the EAA loops from the TMDs, and the Q-loop and the ENI motifs from the NBDs. These three structural motifs in small ABC importers show a high degree of correlation in motion and mediate the necessary conformational coupling between the core of TMDs and the helical subdomains of NBDs. A comprehensive analysis of the structurally known ABC transporters shows a high degree of conservation of the identified 3-motif coupling elements only in the subfamily of small ABC importers, suggesting a distinct mode of NBD-TMD coupling from the other two major ABC transporter folds, namely large ABC importers and ABC exporters.
Introduction
ATP-binding cassette (ABC) transporters constitute one of the largest families of membrane transporters (1). They use the energy of ATP binding and hydrolysis to drive the transport of various substrates across the cell membrane, often against the concentration gradient. In recent years, several different ABC transporters have been structurally resolved as full transporter complexes (2–13). These structures have characterized the fundamental architecture of all ABC transporters as a complex of two nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs, also called the permease or the transporter domains), assembled as homodimers or pseudodimers. The NBDs bind and hydrolyze ATP to provide energy for active transport, whereas the TMDs provide the physical pathway for substrate translocation. In addition to these four basic domains, prokaryotic ABC importers are associated with a periplasmic or extracellular substrate-binding protein (BP), whose function is believed to be substrate acquisition and its delivery to the TMDs. In addition to numerous crystal structures of isolated BPs (reviewed by Berntsson et al. (14)), several ABC transporters have been crystallized with their BPs bound at the periplasmic side, either with or without the substrate (6–8).
All ABC transporters share highly conserved NBDs. Numerous crystal structures of isolated NBDs from different ABC transporters have been reported (reviewed by Moussatova et al. (15)). In particular, several structures of the isolated NBDs of the maltose transporter (MalK) have been resolved in different dimeric conformations (16,17), demonstrating the ability of the NBDs to adopt different conformational states in response to different nucleotide binding conditions. Structurally, each NBD monomer is composed of two subdomains: the RecA-like subdomain and the helical subdomain, with the dimer formed through a head-to-tail assembly of the two monomers. The two symmetrically related nucleotide-binding sites reside mostly within the RecA-like subdomains in a dimer, but the binding sites also involve a strictly conserved (LSGGQ) motif provided by the helical subdomain of the opposing NBD monomers. Therefore, in a closed NBD dimer, each of the two bound ATPs is sandwiched between a RecA-like subdomain and a helical subdomain provided by different monomers.
Based on the crystal structures of isolated NBDs, along with their resolved conformations in intact ABC transporters, it has been established that the NBDs of all ABC transporters share a common mechanism, despite opposite transport directions in ABC importers and exporters. The two NBD monomers form a closed dimer when bound to ATP, whereas they separate after ATP hydrolysis (18,19), or in the absence of a bound nucleotide. The dynamics associated with dimer opening (separation of the two monomers) in the nucleotide-free or the ADP-bound forms has been characterized in molecular dynamics (MD) simulations of different ABC transporters (20–22). Earlier simulations from our laboratory have shown that the opening of the NBD dimer is a direct effect of ATP hydrolysis, rather than that of the dissociation of the hydrolysis product, and that ATP hydrolysis in either of the two active sites (single hydrolysis event) is sufficient to trigger the dimer opening (21).
In contrast to the NBDs, little conservation exists among the TMDs of ABC transporters. Based on the available crystal structures, the TMDs can be divided into at least three topologically different folds: 1), a small importer fold (19), also known as type I ABC importers (23,24), represented by the crystal structures of the maltose transporter (8,12), the molybdate/tungstate transporters (6,10), and the methionine transporter (11); 2), a large importer fold (19), or type II ABC importers (23,24), exemplified by the vitamin B12 transporter (2,7) and one of its homolog (4); and 3), an ABC exporter fold (19,23,24) captured in the crystal structures of the multidrug resistance protein P-glycoprotein (13) and several of its bacterial homologs (3,5,9). To simplify the discussion we will refer to these TMD folds as small ABC importers, large ABC importers, and ABC exporters, respectively.
For the transporter to function, the opening and closure of the dimeric NBDs are highly coupled to conformational changes in the TMDs. Once the NBDs separate in the posthydrolysis, or the nucleotide-free states, the TMDs open toward the cytoplasm, forming a state termed inward-facing. The ATP-dependent closure of the NBDs, on the other hand, facilitates the conformational transition of the TMDs to the outward-facing state. This NBD-dependent control of the conformational state of the TMDs, which is shared by all diverse TMD structural folds, seemed to be attributed to a universal coupling mechanism that conveys the conformational cues from the NBDs to the TMDs. Evidenced by all TMD structures, the only structural element in the TMDs that is shared by all three TMD folds appears to be a single helix lying approximately parallel to the membrane surface on the cytoplasmic side, right between the core of the TMDs and their flanking NBDs. Due to the universal presence and its location, this helix has been suggested to be responsible for the NBD-TMD coupling, and thus termed the coupling helix (18,25).
Here, we report the results of a series of MD simulations investigating the dynamics of the maltose transporter in different forms. Starting from the nucleotide-bound, outward-facing intermediate state (8), we model and simulate the transporter in various combinations of its components. We show that the coupling helices are conformationally decoupled from the rest of the TMDs in the absence of the NBDs, due to the essential role of the NBDs consolidating the structure of the coupling helices with the core of the TMDs. The tight coupling between the NBDs and the TMDs is achieved by the formation of rigid bodies that include the core of the TMDs and the helical subdomain of the NBDs, through the association of three conserved motifs provided by both the TMDs and the NBDs. The three conserved motifs include the EAA loop from the TMDs (where the coupling helix belongs to), and the Q-loop together with its flanking ENI motif from the NBDs. Examining several crystal structures of full ABC transporter complexes, we demonstrate that the three structural motifs are conserved among small ABC importers, and form a unique feature only shared by this subset of ABC transporters.
Methods
Detailed descriptions for the construction of the simulation systems, as well as for the methods used in data analysis are provided in the Supporting Material. In brief, MD simulations were carried out using the crystal structure of the nucleotide-bound, substrate-associated state of the Escherichia coli maltose transporter (MalEFGK, Protein Data Bank (PDB) entry: 2R6G (8)). The transporter was simulated in an explicit lipid-bilayer environment composed of 1-palmitoyl-2-oleoyl-phosphatidylethanolamine molecules. After initial equilibration of 10 ns, the simulations were branched into four different configurations: 1), control simulations of the intact transporter with MgATP-bound; 2), nucleotide-free, full transporter; 3), removing the periplasmic section from System B; and 4), further removing the two MalK monomers from System C, i.e., only the TMDs without the periplasmic loop of MalF. Two independent 70 ns equilibrium simulations were carried out for each system, with different initial random seeds. In the following, the eight simulations will be named as System A1, System A2, System B1, System B2, and so on.
To illustrate the conformational changes during the simulations, four sets of distances were measured at each time point in the trajectories: two center-of-mass distances between the EAA1 and EAA2 helices of MalF and MalG, and two distances measured between the centers of masses of the RecA-like subdomain of one NBD and the helical subdomain from its opposing NBD. The detailed definitions for these distances are provided in the Supporting Material. To measure the internal coupling in the EAA loop, and the cross-domain coupling between the NBDs and the TMDs, generalized correlations were calculated for the Cα atoms of these regions using the g_correlation algorithm provided by Lange and Grubmüller (26). The NBD-TMD coupling was further examined by structural aligning of the EAA helices, for all structures in the simulations trajectories of Systems A–C, as well as for crystal structures of small ABC importers, and calculating the structural deviations at the NBDs neighboring to the aligned EAA helices. Similar superpositions were done for other ABC transporters: the large ABC importer structures were superimposed using their EAA loops, whereas for ABC exporters the intracellular loops (ICLs) were used in structural superposition. Relative displacements of Cα atoms after such superpositions were compared to identify the coupling elements among different domains.
Results and Discussions
Conformational response of the NBDs to nucleotide removal
In small ABC importers, the coupling helices are part of a conserved, two-helix motif termed the EAA loop, a motif exhibiting highly homologous sequences among both small and large ABC importers (27). The EAA loop had been predicted to be composed of two amphipathic α-helices connected by a loop (27), a feature that was later verified by all crystal structures of ABC importers. Interestingly, the orientations of the two helices in the EAA loop are opposite in the small and large ABC importers. In small ABC importers, the coupling helix is the first helix (sequence-wise) of the EAA loop (green helices in Fig. 1, A–D), making direct contacts with the NBDs, whereas the second EAA helix (purple helices in Fig. 1, A–D) is located right above the coupling helix, in a closer proximity to the core of the TMDs. The two helices will be referred to as EAA1 (the coupling helix) and EAA2 (the second EAA helix) hereafter.
Figure 1.
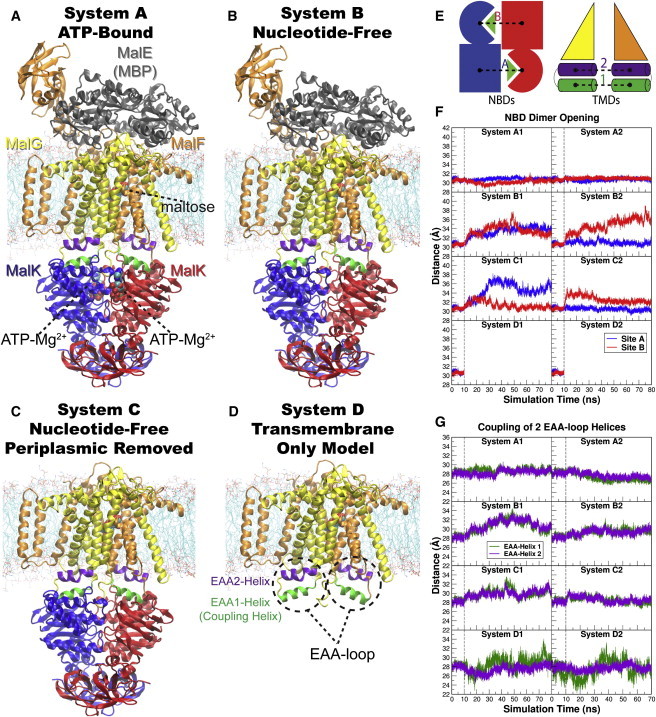
Overview of the equilibrium simulations. (A–D) Initial structure of the four simulation systems, showing proteins (in ribbons), bound substrates (including maltose and MgATP, shown as van der Waals spheres and labeled in panel A), and lipids (in line representations). The color scheme for this and all following figures is: MalK monomers in blue and red, MalF in orange, MalG in yellow, MalE in dark gray (labeled in panel A); the two EAA1 (the coupling helices) and EAA2 helices are highlighted in green and purple, respectively (labeled in panel D). (E) Schematic representation of the distances measured to quantify the NBD opening and the separation of the EAA helix pairs for panels F and G. Left: the NBD opening in each simulation system is measured as the center-of-mass distance between the helical subdomain of one MalK monomer (P88–E151, squares) and the RecA-like subdomain of the opposite MalK monomer (A2–Y87 and P152–G235, pac-man shaped); the bound nucleotides are shown as green triangles occupying the binding sites A and B. Right: the separation of the two sets of EAA helices are measured as the distances between their centers of masses (MalF:P396–G407 with MalG:D185–G196 as set 1, and MalF:F411–L422 with MalG:W200–S211 as set 2). (F) The conformational changes in the NBDs of each simulation system measured by the degree of NBD dimer opening. (G) The conformational changes of the EAA loops of the TMDs, measured as the separation of the two EAA helix pairs. The vertical dashed lines at 10 ns in panels F and G denote the time point at which all the simulations were branched out from their common parent equilibration simulation system.
Removal of the nucleotides from the NBDs in the nucleotide-free systems (Systems B and C) is expected to result in significant conformational changes. Two types of conformational changes are considered relevant to the transport mechanism: the change of the dimerization state of the NBDs, and the degree of conformational changes transmitted from the NBDs to the TMDs. To quantify the extent of these conformational changes, the former is measured as the degree of NBD opening at both active sites (Fig. 1 F), and the latter is measured as the separation of EAA1/EAA2 helices (Fig. 1 G) because the EAA loops are the structural elements in the TMDs that are in physical contact with the NBDs, and thus are expected to move in a correlated manner to the NBDs. These quantities are described in detail in the Supporting Material.
System A preserves all the components of the intact transporter, and thus serves as a control system. The transporter in two repeated simulations (Systems A1 and A2 in Fig. 1, F and G) maintains a stable conformation both in the NBDs and in the TMDs. Examining the degree of opening in binding sites A and B, it is clear that the NBD dimer maintains its fully closed conformation throughout the two 70 ns simulations of Systems A1 and A2 (Fig. 1 F), a behavior that was also observed during the simulation of the ATP-bound MalK dimer in its isolated form (21).
Removing the nucleotides (MgATP) from the NBDs (Systems B and C), result in large conformational changes in the NBDs. In both Systems B1 and B2, an immediate opening of at least one nucleotide-binding site is observed (Fig. 1 F). The NBD opening appears to be symmetric in System B1, with an average opening of 2.5–3.5 Å at both nucleotide-binding sites, reaching a conformational state similar to the crystal structure of the semiopen MalK (16). In System B2, on the other hand, significant NBD opening is only observed in one of the two nucleotide-binding sites. This asymmetric NBD opening is also observed in both Systems C1 and C2. The NBD opening does not occur at the same site in the three simulations with asymmetric NBD openings, that is, site A opens in System C1 and Site B opens in Systems B2 and C2. The degree of opening in these sites is mostly comparable to, if not larger than, the symmetric opening of binding sites in System B1, except for System C2 where the opening of site B is only ∼2 Å.
The degree of NBD opening in all the four simulations with removed nucleotide (Systems B1, B2, C1, and C2) are much smaller than what is observed in the crystal structure of nucleotide-free state, and three of the four simulations show the opening at only one of the two binding sites. Considering that the dimer opening due to nucleotide removal from an isolated, completely closed NBD can be captured with relatively short MD simulations (28), we attribute the small degree of NBD opening observed here to the structural constraints imposed by the presence of the TMDs preventing the NBDs from fully opening within the limited time scale of the simulations. Within the vacant yet closed nucleotide-binding sites, hydrogen bonds between the side chains of the Walker A and LSGGQ motifs from the opposing NBDs are frequently observed, especially between S38 and S135 of MalK. Furthermore, a number of van der Waals contacts and other hydrogen bonds can also be occasionally observed between the two NBD monomers at the Q-loops and the D-loops. It is not clear whether these transient contacts and interactions help to maintain the closed binding sites or are merely the structural consequences due to the trapped closed conformation. The fact that the NBD opening can occur at either site or both, suggests that the NBD opening is a stochastic event that can be delayed by the structural restraints from the TMDs, and possibly by transient interactions between the two NBD monomers.
It should be noted that even though the degree of NBD opening in none of the nucleotide-free simulations (Systems B1, B2, C1, and C2) is large enough to induce inversion of the TMDs to open toward the cytoplasm (discussed later), all of the simulations clearly show structural changes toward a conformational state resembling the resting state crystal structure (PDB: 3FH6 (12)). These structural changes therefore only represent early events along the transport cycle captured within the limited time scale of our simulations.
Effect of NBD opening on the EAA loops
Depending on the nature of the opening at the nucleotide-binding sites, the conformational changes in the NBDs result in various degrees of separation of the EAA helices in Systems B and C. In contrast, in both control simulations (Systems A1 and A2), the distances between the EAA1 helices and that of the EAA2 helices are relatively constant. The two sets of EAA helices in both Systems A1 and A2 exhibit only a slight distance variation from the original crystal structure (on the order of ∼1 Å, Fig. 1 G).
The largest separation of the EAA helices among all the simulations occurs in the middle of simulation System B1, reaching ∼4 Å larger separation than in the starting crystal structure. None of the other three nucleotide-free simulations (Systems B2, C1, and C2) yielded comparable separation of the EAA helices, a behavior that might be related to the observed asymmetric NBD opening in these simulations. The separation of the EAA helices is naturally affected by the number of binding sites that exhibit opening during the simulations. For example, with both nucleotides-binding sites open, the EAA helices in System B1 are able to separate almost twice the amount in System B2, even though the opening at site B in the latter is larger than that of either site in the former. One might speculate that the conformational conversion of the TMDs from the outward-facing state to the inward-facing state requires the opening of both nucleotide-binding sites.
Although the opening of both binding sites might be necessary for a successful conformational change of the TMDs, this does not necessarily entail that both bound nucleotides have to be hydrolyzed. Using MD simulations performed on isolated NBD, we have shown that a single hydrolysis event is able to trigger the opening of both nucleotide-binding sites due to the destabilization of the dimer interface (21). In the context of an intact transporter, the symmetric NBD opening induced by single hydrolysis may still hold true, however, its description would possibly require a much longer time scale than what has been simulated here. This notion can be supported by many examples of full functional ABC transporters containing one degenerate nucleotide-binding site (summarized by Procko et al. (29)).
NBD dependence of the coupling of the EAA helices
The most unexpected results are obtained when the two NBDs are completely removed from the simulation system (Systems D1 and D2). In Systems A–C, regardless of the degree of separation in EAA1 and EAA2 helices, the distances between EAA1 and EAA2 pairs are always closely related (Fig. 1 G). On the contrary, in the absence of the NBDs (Systems D1 and D2), the EAA1 helices exhibit very large fluctuations, resulting in EAA1 distances ranging from ∼5 Å longer and shorter than the crystallographic distance, whereas the EAA2 helices mostly maintain the same level of separation throughout both simulation runs (Fig. 1 G). The residue-wise correlations within the EAA loops also show that the internal coupling of the EAA loops is largely abolished in both Systems D1 and D2 (Fig. 1 A). These results clearly indicate that, in the absence of the NBDs, the coupling helices (EAA1 helices) are largely decoupled from the rest of the TMDs, whereas the EAA2 helices maintain their tight coupling to the core of the TMDs.
Examining the detailed structures of the EAA loops, especially at its interface with the NBDs, it is found that the decoupled behavior observed in the absence of the NBDs is likely due to the partial disruption of the tertiary structure of the EAA loop. Here, the NBDs provide essential contacts to stabilize the native structure, in a way analogous to a latch locking in between two gates. The participation of the NBDs in the folding of EAA loops seems to be a conserved feature among small ABC importers. Upon close inspection of the contacts between the EAA loops and the NBDs in all simulation trajectories of Systems A–C, as well as in all available crystal structures of small ABC importers, we found that the positions of NBD-TMD contacting residues are almost identical in all these structures. Specifically, besides the majority of the EAA1 helices, five residues in each of the EAA2 helices contact the NBDs. The TMD contacting residues in the NBDs are highly clustered in and around the Q-loop region, except for a few others that are positioned in the first loop after the Walker A motif, and the first helical loop in the helical subdomain. Notably, the latter regions belong to the structurally diverse region (SDR, a nonconserved region between the Q-loop and the LSGGQ motif of the NBDs (30)) of ABC transporters. The high degree of spatial resemblance of the NBD-TMD contacts, and the positional identity of the contact residues along the polypeptide sequence, despite their low degree of sequence conservation, suggest a common mechanical coupling and mechanism of molecular recognition between the NBDs and the EAA loops in small ABC importers.
Aside from the EAA loops, the core of the TMDs (TM5–TM7 of MalF and TM3–TM5 of MalG (12)) does not show any noticeable structural changes during the two simulations of System D. Although the absence of EAA2 structural changes in System D might well be related to the limited time scale of our simulations, capturing such changes within the same time scales in other simulation systems presented here (Systems B and C) might suggest otherwise. We might speculate that the zero separation of EAA2 helices in System D may be an indication of the presence of another low-energy conformational state (resting state) for the isolated TMDs, and that the resting state adopted by the TMDs is selected by the conformational state of the associated NBDs. The conformational energy landscape of the TMDs can be further characterized by MD simulations using the inward-facing crystal structure of the maltose transporter (12).
Identifying the NBD-TMD coupling elements
In the maltose transporter, the coupling helix (EAA1) of each TMD inserts into a complementary deep groove of the neighboring NBD, which is located right between the RecA-like and the helical subdomains. Mutagenesis and chemical cross-linking studies have suggested that the NBDs of the maltose transporter interact with the TMDs through the helical subdomains (31,32), while based on the crystal structures, the Q-loop of the NBDs (which connects the RecA-like and the helical subdomains) appears to be the binding partner of the coupling helices (18,25). Indeed, examining the simulation trajectories, the Q-loop residues (especially Y87–H89) show the highest contact frequencies with the EAA loop in all of Systems A–C. Our simulations provide a more detailed and dynamical view of the interdomain coupling mechanism. The correlation of the Cα fluctuations between each NBD (MalK) and its flanking TMD (MalF or MalG) was calculated for Systems A–C (Fig. 2 B), using the generalized correlation method (26). The highest interdomain correlation was found to be always between the EAA loops of the TMDs and a region connecting the two subdomains of the NBD, including the Q-loop and nearby residues. Furthermore, the correlation of the NBD residues to the TMD residues is generally higher in the helical subdomains than in the RecA-like subdomains.
Figure 2.
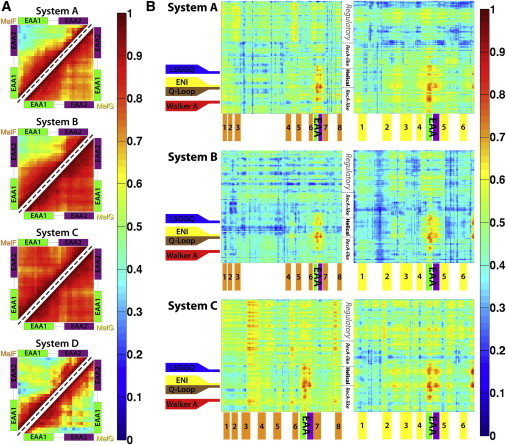
Generalized correlations between residues in different parts of the maltose transporter. The generalized correlation matrix of each simulation is calculated with the combination of the two repeated trajectories for each system, instead of averaged values for each trajectory. (A) Internal correlations for residues within the EAA-loops of (top left triangle) MalF and (bottom right triangle) MalG in all four simulation systems. (B) Cross correlations between the residues from the NBDs and the TMDs in Systems A–C, the left panels show the correlations between polypeptide chains B (MalK) and chain F (MalF), whereas the right panels are the correlations between chains A (MalK) and chain G (MalG). The locations of key structural motifs, transmembrane helices (in numbers), and major domains of the NBDs are labeled.
To further characterize the structural elements involved in the coupling of the NBDs and the TMDs, the trajectories were aligned using the EAA loops, which show the highest correlation to the motions of the NBD residues (Fig. 3, A and B). After such superpositions, NBD residues showing the lowest degrees of relative Cα displacements and fluctuations can be considered to have the strongest coupling to the EAA helices, and thus to the TMDs (Fig. 4, A–C). Analyzing the distribution of Cα displacements in Systems A–C, it is found that the residues in the helical subdomain indeed show both lower mean deviations and smaller fluctuations, when compared to residues in the RecA-like subdomain. The helical subdomain, therefore, exhibits closer coupling to the TMDs than the RecA-like subdomain. In particular, the region between G78–L102 (highlighted regions in Fig. 4, A–C, and cyan colored regions in Fig. 4 E) shows least structural deviations and fluctuations relative to the EAA helices, matching the region exhibiting highest correlations in Fig. 2 B.
Figure 3.
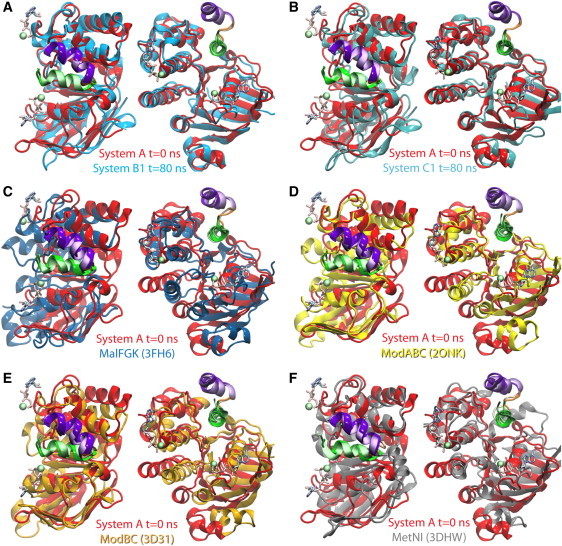
NBD-TMD coupling in small ABC importers. The starting structure of simulation System A (equivalent to the crystal structure 2R6G (8) with the coordinates of MgATP) is superimposed with end structures of simulation Systems B1 and C1, as well as with several other crystal structures of small ABC importers. The superposition is performed using the EAA loop (green and purple helices connected by an orange loop, colored as in Fig. 1), and the orientation of the flanking NBDs are compared. In the reference structure, labeled System A, the EAA loop is shown in a glossy representation and the NBD in red; in other structures the EAA loop is drawn using faded colors and the NBDs in various colors: (A) the end structure of System B1 in cyan; (B) the end structure of System C1 in teal; (C) the resting state crystal structure of the maltose transporter (PDB:3FH6 (12)) in dark blue; (D) the molybdate/tungstate transporter of Archaeoglobus fulgidus (PDB:2ONK (6)) in yellow; (E) the molybdate transporter of Methanosarcina acetivorans (PDB:3D31 (10)) in brown; (F) the methionine transporter of Escherichia coli (PDB:3DHW (11)) in gray. In each panel, the structures are shown both in top (extracellular) view (left), and in side view (right). MgATP is shown in System A as a point of reference to highlight the ATP-binding sites.
Figure 4.
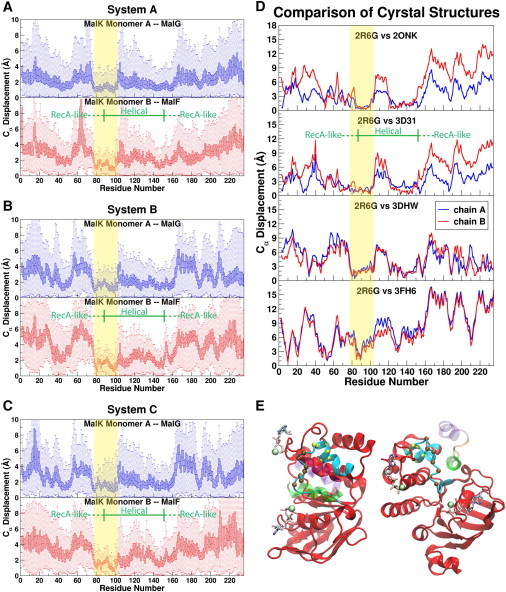
Identifying the NBD structural motifs responsible for the EAA loop coupling. (A–C) Box plots showing the distribution of the displacement of each Cα atom of the two NBD monomers relative to the flanking EAA loops in simulations of Systems A–C. The Cα displacements are measured after aligning the structures using the EAA loop (MalF:P396–L422 for chain B and MalG:D185–S211 for chain A), and with reference to the Cα positions in the crystal structure 2R6G (8). The boxed region covers 25–75 percentile of the distribution with a line in the middle for the mean displacement, whereas the full range (0 and 100 percentile) is indicated by the dotted lines. Regions of low displacement and small fluctuation are highlighted with yellow bands and are suggested to be responsible for the EAA loop coupling mechanism. The charts are plotted in blue and red for each MalK monomer, respectively, corresponding to their colors in Fig. 1. (D) Comparison of the NBD-TMD coupling among small ABC importers. The crystal structures in Fig. 3, C–F, are superimposed onto the maltose transporter structure 2R6G using the EAA loops and the Cα positions in each NBD are compared with the equivalent Cα positions of MalK in 2R6G. The low displacement region of MalK defined in panels A–C is also highlighted with yellow bands for reference. (E) The structural motif in the NBD responsible for the EAA loop coupling. Showing the NBD and EAA loop structures of the starting structure of System A; from the same view as in Fig. 3A. The EAA loop of MalF is shown in transparent. The region showing highly coupled motion in the simulations are colored in cyan, where the Cα atoms of the Q-loop and the ENI motif (following definitions by Jones and George (33)) are shown as brown and yellow spheres, respectively.
Given that the EAA loops are structurally highly conserved among small ABC importers, the above structural analysis can also be applied to other crystal structures in this family (Fig. 3, C–F, Fig. 4 D). The results indicate that the structural elements identified above for the maltose transporter are also present in other homologous ABC importers, which all show low relative Cα displacements at equivalent positions (corresponding to MalK:G78–L102, highlighted in Fig. 4 D). The results of the analysis indicate the presence of a structurally conserved region in the NBDs that is in charge of close coupling to the conserved counterpart in the TMDs, that is, the EAA loops. In the next section, we will provide a detailed description of the conserved sequence motifs involved in this region.
Note that the crystal structure serving as the reference here (PDB:2R6G) is the only one with nucleotide-bound, closed NBDs, whereas all other structures (the resting maltose transporter 3FH6 (12), two molybdate/tungstate transporters 2ONK (6) and 3D31 (10), and the methionine transporter 3DHW (11)) are inclusively crystallized in the absence of nucleotides, hence exhibiting various degrees of NBD opening. Because these structures cover a broad spectrum of crystallizing conditions, including the presence and the absence of nucleotides and/or the BP/substrates, as well as different inhibitory states of the NBD-associated regulatory domains, the NBD-TMD coupling elements characterized through our analysis appear not to be affected and consistently present in all the structures.
Structural characterization of the NBD-TMD coupling motifs
Structurally, the above TMD-coupling region of the NBDs (MalK:G78–L102) corresponds to the Q-loop, its N-terminal flanking β-strand, and the first α-helix of the helical subdomain at the C-terminus of the Q-loop (the cyan region in Fig. 4 E). Interestingly, this region includes two conserved motifs right at its center: one is the aforementioned Q-loop (brown spheres in Fig. 4 E), and the other is named the ENI motif (yellow spheres in Fig. 3 E), identified by Jones and George (33) after structural comparison of several monomeric NBDs (Fig. 5 A), but without assigning any functional or structural role to it. In the crystal structures of small ABC importers, the TMD contacts in the NBDs are primarily formed by nonspecific, hydrophobic interactions provided by the Q-loop and a few residues in the helical subdomain, which are immediately before and after the ENI motif. The ENI motif itself, however, is not involved in the NBD-TMD interface.
Figure 5.
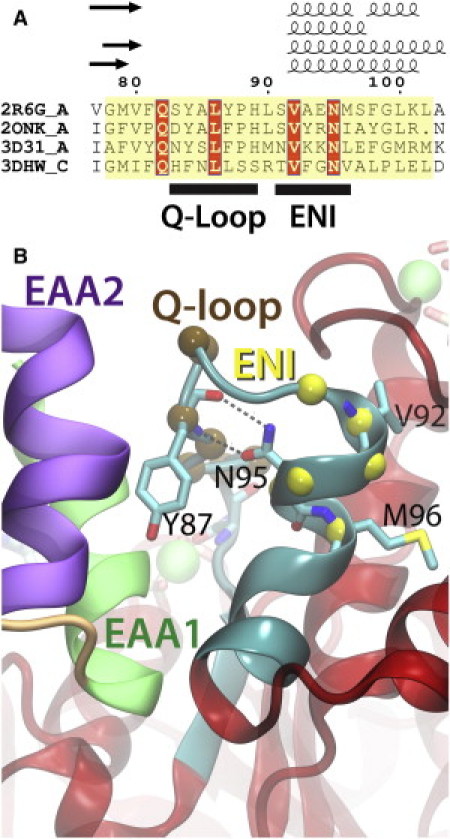
TMD-coupling motifs in MalK. (A) Structure-based sequence alignment of the NBDs in small ABC importers at the NBD-TMD coupling region. The NBD of each crystal structure is structurally aligned to reach the best fit up to the position of G235 of MalK, neglecting the attached regulatory domains or the associated dimerizing helices. The structure-based alignment is performed using Multiseq (40), manually optimized at the Walker A motifs (due to structural variations resulted from nucleotide binding), and formatted with ESPript (41). The full alignment is shown in Fig. S1. (B) A close-up of the Q-loop and ENI motifs in MalK. Structures are colored as in Fig. 4E, and key residues involving the functional role of the ENI motif (Y87, V92, N95, and M96) are shown in stick models. The hydrogen bonds connecting the side chain of N95 and the backbone of Y87 are highlighted.
A clue for the role of the ENI motif is provided by the observed stronger correlation of the entire helical subdomain with the EAA helices when compared to the RecA-like subdomain. Examining the conserved sequences and the local structure of the ENI motif, it is found that the conserved hydrophobic residues in the ENI motif form a significant part of the hydrophobic core of the helical subdomain. In the case of the maltose transporter, these include V92 and M96 of MalK (Fig. 3 B) (33). In addition, the strictly conserved asparagine in this motif makes two hydrogen bonds with the backbone of the Q-loop right at a position contacting the EAA helices (MalK:N95 and MalK:Y87, Fig. 5 B). This hydrogen bond stabilizes the ENI motif in a fixed orientation with respect to the Q-loop, thus providing close coupling to the EAA helices for the entire helical subdomain. One can expect that the mutation of this strictly conserved asparagine, even to a structurally similar aspartate or glutamine, might result in decoupling between the helical subdomain and the EAA loop, whose phenotype might be similar to a mutation at the contacting residues between the Q-loop and the EAA loop, such as an L86F mutant of MalK (34). In contrast to a mutation at the Q-loop or the EAA loop, a mutation at the ENI motif might not affect the NBD-TMD interface directly, but impair the transport function by structural decoupling of the helical subdomains.
Note that when superimposing different structures with the EAA loop, the area near the LSGGQ motif is generally the region showing the second lowest Cα displacements besides the ENI motif (Fig. 4 D), where the residues in between the two motifs generally show much weaker coupling (higher Cα deviations). This suggests that the structural connection between the ENI motif and the LSGGQ motif is likely through direct contacts in the hydrophobic core of the helical subdomain and not related to their sequence proximity along the peptide chain. In summary, the NBD-TMD coupling is achieved through a network of interactions extending from the core of the TMDs to the EAA loop, then to the Q-loop, and through the ENI motif to reach the helical subdomain of the NBDs, particularly the strictly conserved LSGGQ motif, which is an essential part of the nucleotide-binding site. These structural components can be viewed together as an internally tightly coupled (rigid) body during the conformational switching of the transporter.
The rotation of the RecA-like subdomain in the NBD
As discussed previously, the helical subdomains of the NBDs display a higher degree of TMD-coupling than the RecA-like subdomains. During the transport cycle, therefore, the helical subdomains are expected to maintain their relative orientation to the TMDs through their close association to the EAA loops, while the RecA-like subdomains enjoy more freedom to change their relative orientation in response to nucleotide binding and hydrolysis. This internal conformational change within the NBDs is identical to the traditional notion of the rotation of the helical subdomain (35,36), only viewed from a different perspective. However, in the context of the full transporter, attributing the rotation to the RecA-like subdomain seems to provide a more accurate description of the dynamical phenomenon, since the helical subdomains are highly coupled to the TMDs.
For example, in comparison of the crystal structures of the resting and the intermediate states of the maltose transporter, the conformational changes at the MalK/TMD interface was described as a 30° rotation of the coupling helix, the core of the TMDs, and the helical subdomains, against the RecA-like subdomain (12). The same description can be substantially reduced into a rotation of only the RecA-like subdomain, against the TMDs and the helical subdomain. Such relative motions are even better demonstrated when superimposing the intermediate state structure of the maltose transporter to other nucleotide-free crystal structures of small ABC importers, using their EAA loops (Fig. 3, D–F, Fig. 4 D), Therefore, we propose that due to its tighter coupling to TMDs, the helical subdomain offers a better reference point for structural and dynamical comparison of the NBDs, at least among small ABC importers.
NBD-TMD coupling in other ABC transporter folds
Through the analysis presented previously, we have characterized several motifs responsible for the NBD-TMD coupling in small ABC importers. Equivalent analysis of the other two major ABC transporter folds (large ABC importers and ABC exporters), however, does not yield the same conclusions, implying that the NBD-TMD coupling mechanism may differ among different types of ABC transporters.
Despite the high sequence homology of the EAA loop among different ABC importers (27), its structure is completely different between the small and the large ABC importers. As demonstrated by the crystal structures of the vitamin B12 transporter BtuCDF (7) and its homolog HI1470/1 (4), not only are the orientation of the EAA helices in large ABC importers opposite to that in the maltose transporter, but the relative positions of the two EAA helices are also reversed. That is, the EAA1 helices in large ABC importers are closer to the core of the TMDs and away from the NBDs, and it is the EAA2 helices that are flanked by the NBDs. The loop connecting the two EAA helices in large ABC importers is also oppositely oriented to its counterpart in small ABC importers: this loop faces the interior of the transporter in BtuCDF and the periphery of the maltose transporter. In addition, most of the TMD-contacts from the NBDs are provided by the SDR in large ABC importers. It appears that the coupling mechanisms of the two types of ABC importers might be different, even though the seemingly homologous EAA loop is part of the coupling mechanism in both families.
Indeed, superimposing the crystal structures of BtuCDF and HI1470/1 at their EAA loops suggests that the Q-loop region is unlikely to participate in the NBD-TMD coupling mechanism in large ABC importers, which is in sharp contrast to its role in the small ABC importers (Fig. S2). Instead, in large ABC transporters, the region of highest TMD-coupling is closer to the periphery of the NBDs and far from the Q-loops. Apparently, the NBD-TMD coupling mechanism in large ABC importers is likely mediated by direct contacts between the TMDs and the helical subdomain, especially between the TMDs and the SDR of the helical subdomains, which are located at the periphery of the NBDs. This might explain the lower homology in the Q-loop sequences, as well as the absence of the ENI motif in large ABC importers.
Among ABC exporters, the ENI motif is strictly conserved in the NBDs (Fig. S3) and the EAA loop is absent from the TMDs. Instead, each of the two TMDs in an ABC exporter contacts both NBDs through two separate ICLs; often supplemented with the peptide linker directly connecting the TMDs and the NBDs. To investigate the NBD-TMD coupling, several crystal structures of ABC exporters in different conformational states were superimposed using their ICLs, and the Cα deviations in the NBDs were compared. However, the results do not reveal strong coupling between the NBDs and the ICLs in ABC exporters (Fig. S4).
The structural analysis of both large ABC importers and ABC exporters shows limited NBD-TMD coupling, although most NBD-TMD interfaces among small ABC importers are highly coupled. This might explain the tightly regulated ATPase activity reported for small ABC importers. For example, the NBDs of the maltose transporter are known to dimerize only when both the substrate-bound BP and MgATP are present (37), while BtuCDF shows significant substrate-independent ATPase activity in vitro (38). Moreover, it is known that the basal ATPase activity composes an important part of the mechanism of the multidrug exporter P-glycoprotein (39). Through the structural comparison presented in this study, we suggest that the NBDs and the TMDs are exceptionally tightly coupled in small ABC importers but only loosely coupled in the other two types of ABC transporters. Members of small ABC importers likely use the interaction network among the EAA loop, the Q-loop, the ENI motif, and the hydrophobic core of the helical subdomain of the NBDs, to conformationally couple the core of the TMDs to the nucleotide binding site and achieve the highly coherent motions among these domains in a nucleotide-dependent manner during the transport cycle.
Acknowledgments
Simulations in this study have been performed at the Ranger cluster of Texas Advanced Computing Center, the Bigred cluster of Indiana University, and the Abe cluster at National Center for Supercomputing Applications (TeraGrid grant MCA06N060).
This study is supported by National Institutes of Health grants R01-GM086749, R01-GM067887, P41-RR05969, and U54-GM087519.
Supporting Material
References
- 1.Davidson A.L., Dassa E., Chen J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 2008;72:317–364. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Locher K.P., Lee A.T., Rees D.C. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 3.Dawson R.J., Locher K.P. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 4.Pinkett H.W., Lee A.T., Rees D.C. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 5.Dawson R.J., Locher K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 6.Hollenstein K., Frei D.C., Locher K.P. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 7.Hvorup R.N., Goetz B.A., Locher K.P. Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF. Science. 2007;317:1387–1390. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- 8.Oldham M.L., Khare D., Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 9.Ward A., Reyes C.L., Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl. Acad. Sci. USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerber S., Comellas-Bigler M., Locher K.P. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science. 2008;321:246–250. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]
- 11.Kadaba N.S., Kaiser J.T., Rees D.C. The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science. 2008;321:250–253. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khare D., Oldham M.L., Chen J. Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell. 2009;33:528–536. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aller S.G., Yu J., Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berntsson R.P.-A., Smits S.H., Poolman B. A structural classification of substrate-binding proteins. FEBS Lett. 2010;584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 15.Moussatova A., Kandt C., Tieleman D.P. ATP-binding cassette transporters in Escherichia coli. Biochim. Biophys. Acta. 2008;1778:1757–1771. doi: 10.1016/j.bbamem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Chen J., Lu G., Quiocho F.A. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 2003;12:651–661. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Lu G., Westbrooks J.M., Chen J. ATP hydrolysis is required to reset the ATP-binding cassette dimer into the resting-state conformation. Proc. Natl. Acad. Sci. USA. 2005;102:17969–17974. doi: 10.1073/pnas.0506039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollenstein K., Dawson R.J., Locher K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Oldham M.L., Davidson A.L., Chen J. Structural insights into ABC transporter mechanism. Curr. Opin. Struct. Biol. 2008;18:726–733. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oloo E.O., Fung E.Y., Tieleman D.P. The dynamics of the MgATP-driven closure of MalK, the energy-transducing subunit of the maltose ABC transporter. J. Biol. Chem. 2006;281:28397–28407. doi: 10.1074/jbc.M513614200. [DOI] [PubMed] [Google Scholar]
- 21.Wen P.-C., Tajkhorshid E. Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys. J. 2008;95:5100–5110. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones M.K., Catte A., Segrest J.P. Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics. Biophys. J. 2009;96:354–371. doi: 10.1016/j.bpj.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Locher K.P. Review. Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:239–245. doi: 10.1098/rstb.2008.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rees D.C., Johnson E., Lewinson O. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson R.J., Hollenstein K., Locher K.P. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol. Microbiol. 2007;65:250–257. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 26.Lange O.F., Grubmüller H. Generalized correlation for biomolecular dynamics. Proteins. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 27.Saurin W., Köster W., Dassa E. Bacterial binding protein-dependent permeases: characterization of distinctive signatures for functionally related integral cytoplasmic membrane proteins. Mol. Microbiol. 1994;12:993–1004. doi: 10.1111/j.1365-2958.1994.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 28.Newstead S., Fowler P.W., Iwata S. Insights into how nucleotide-binding domains power ABC transport. Structure. 2009;17:1213–1222. doi: 10.1016/j.str.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Procko E., O'Mara M.L., Gaudet R. The mechanism of ABC transporters: general lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 2009;23:1287–1302. doi: 10.1096/fj.08-121855. [DOI] [PubMed] [Google Scholar]
- 30.Schmitt L., Benabdelhak H., Stubbs M.T. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J. Mol. Biol. 2003;330:333–342. doi: 10.1016/s0022-2836(03)00592-8. [DOI] [PubMed] [Google Scholar]
- 31.Wilken S., Schmees G., Schneider E. A putative helical domain in the MalK subunit of the ATP-binding-cassette transport system for maltose of Salmonella typhimurium (MalFGK2) is crucial for interaction with MalF and MalG. A study using the LacK protein of Agrobacterium radiobacter as a tool. Mol. Microbiol. 1996;22:655–666. doi: 10.1046/j.1365-2958.1996.d01-1724.x. [DOI] [PubMed] [Google Scholar]
- 32.Hunke S., Mourez M., Schneider E. ATP modulates subunit-subunit interactions in an ATP-binding cassette transporter (MalFGK2) determined by site-directed chemical cross-linking. J. Biol. Chem. 2000;275:15526–15534. doi: 10.1074/jbc.275.20.15526. [DOI] [PubMed] [Google Scholar]
- 33.Jones P.M., George A.M. Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc. Natl. Acad. Sci. USA. 2002;99:12639–12644. doi: 10.1073/pnas.152439599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunke S., Landmesser H., Schneider E. Novel missense mutations that affect the transport function of MalK, the ATP-binding-cassette subunit of the Salmonella enterica serovar typhimurium maltose transport system. J. Bacteriol. 2000;182:1432–1436. doi: 10.1128/jb.182.5.1432-1436.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith P.C., Karpowich N., Hunt J.F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davidson A.L., Chen J. ATP-binding cassette transporters in bacteria. Annu. Rev. Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 37.Orelle C., Ayvaz T., Davidson A.L. Both maltose-binding protein and ATP are required for nucleotide-binding domain closure in the intact maltose ABC transporter. Proc. Natl. Acad. Sci. USA. 2008;105:12837–12842. doi: 10.1073/pnas.0803799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borths E.L., Poolman B., Rees D.C. In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for vitamin B12 uptake. Biochemistry. 2005;44:16301–16309. doi: 10.1021/bi0513103. [DOI] [PubMed] [Google Scholar]
- 39.Al-Shawi M.K., Omote H. The remarkable transport mechanism of P-glycoprotein: a multidrug transporter. J. Bioenerg. Biomembr. 2005;37:489–496. doi: 10.1007/s10863-005-9497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts E., Eargle J., Luthey-Schulten Z. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics. 2006;7:382. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gouet P., Courcelle E., Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.