

Abstract
Aminochrome, the precursor of neuromelanin, has been proposed to be involved in the neurodegeneration neuromelanin-containing dopaminergic neurons in Parkinson’s disease. We aimed to study the mechanism of aminochrome-dependent cell death in a cell line derived from rat substantia nigra. We found that aminochrome (50μM), in the presence of NAD(P)H-quinone oxidoreductase, EC 1.6.99.2 (DT)-diaphorase inhibitor dicoumarol (DIC) (100μM), induces significant cell death (62 ± 3%; p < 0.01), increase in caspase-3 activation (p < 0.001), release of cytochrome C, disruption of mitochondrial membrane potential (p < 0.01), damage of mitochondrial DNA, damage of mitochondria determined with transmission electron microscopy, a dramatic morphological change characterized as cell shrinkage, and significant increase in number of autophagic vacuoles. To determine the role of autophagy on aminochrome-induced cell death, we incubated the cells in the presence of vinblastine and rapamycin. Interestingly, 10μM vinblastine induces a 5.9-fold (p < 0.001) and twofold (p < 0.01) significant increase in cell death when the cells were incubated with 30μM aminochrome in the absence and presence of DIC, respectively, whereas 10μM rapamycin preincubated 24 h before addition of 50μM aminochrome in the absence and the presence of 100μM DIC induces a significant decrease (p < 0.001) in cell death. In conclusion, autophagy seems to be an important protective mechanism against two different aminochrome-induced cell deaths that initially showed apoptotic features. The cell death induced by aminochrome when DT-diaphorase is inhibited requires activation of mitochondrial pathway, whereas the cell death induced by aminochrome alone requires inhibition of autophagy-dependent degrading of damaged organelles and recycling through lysosomes.
Keywords: dopamine, aminochrome, autophagy, apoptosis, neurons, DT-diaphorase
Mutated alpha-synuclein has been found to be associated to Familial Parkinson’s disease by generating neurotoxic protofibrils (Conway et al., 2000), but this mutation is very rare and cannot explain a possible role in sporadic Parkinson’s disease. However, the discovery that the formation of neurotoxic protofibrils is enhanced and stabilized by aminochrome (Norris et al., 2005) suggests a possible role of alpha-synuclein in sporadic Parkinson’s disease because aminochrome is the product of dopamine oxidation and the precursor of neuromelanin. The fact that the grafts transplanted into Parkinson’s patients developed subcellular aggregates of alpha-synuclein and ubiquitin that resembles Lewy bodies support this idea (Braak and Del Tredici, 2008).
At physiological pH, dopamine oxidizes to dopamine o-quinone that automatically cyclizes in several steps to aminochrome. Aminochrome is the precursor of neuromelanin that accumulates with age in human substantia nigra (Zecca et al., 2002, 2008). Neuromelanin is the pigment that accumulates in dopaminergic neurons in substantia nigra from healthy individuals, but in Parkinson’s disease, these neurons containing neuromelanin are lost. A possible explanation why dopaminergic neurons containing neuromelanin are lost in Parkinson’s disease is that aminochrome is able to participate in two neurotoxic pathways such as (1) aminochrome forms adducts with alpha-synuclein inhibiting alpha-synuclein fibrillization and enhancing and stabilizing neurotoxic protofibrils (Conway et al., 2001; Norris et al., 2005) and (2) aminochrome can be one electron reduced by flavoenzymes to leukoaminochrome o-semiquinone radical, which is extremely reactive with oxygen and neurotoxic when NAD(P)H-quinone oxidoreductase, EC 1.6.99.2 (DT)-diaphorase is inhibited with dicoumarol (DIC) (Arriagada et al., 2004; Fuentes et al., 2007; Paris et al., 2001, 2005a,b, 2009a,b, 2010; Segura-Aguilar et al., 1989).
In previous studies, we demonstrated the effect of aminochrome in the catecholaminergic cell line RCSN-3 cells by using different oxidizing agents to oxidize dopamine such as manganese (III), copper (II), iron (III) or in the presence of reserpine an inhibitor of vesicular monoamine transporter-2 (VMAT-2) in order to promote dopamine oxidation by increasing intracellular concentration of cytosolic dopamine (Arriagada et al., 2004, Fuentes et al., 2007; Paris et al., 2001, 2005a, b, 2009a, 2010) or in experiments in vivo (Diaz-Veliz et al., 2008; Segura-Aguilar et al., 2002). However, in all these experiments, the oxidizing agents or inhibitor such as reserpine were present, and therefore, we cannot discard an additive effect of the oxidizing agent because an extensive cell death was observed at 2 h when we used manganese (III) as an oxidizing agent (Arriagada et al., 2004). Therefore, the aim of this work was to study the cell death induced by purified aminochrome on RCSN-3 cells.
MATERIALS AND METHODS
Chemicals.
Dopamine, DIC, menadione, Dulbecco's Modified Eagle's Medium/Nutrient Mixture F-12 Ham (DME/HAM-F12) nutrient mixture (1:1), and Hank’s solution were purchased from Sigma Chemical Co. (St Louis, MO). Vector green fluorescent-protein light chain 3 (GFP-LC3) was gift from Zsolt Tallocky Ph D, Columbia University Medical Center. LIVE/DEAD Viability/Cytotoxicity Kit, Molecular Probes (Eugene, OR). Aminochrome was prepared by oxidizing dopamine with tyrosinase and purified according Paris et al. (2010).
Cell culture.
The RCSN-3 cell line grows in monolayer, with a doubling time of 52 h, a plating efficiency of 21%, and a saturation density of 56,000 cells/cm2 in normal growth media composed of DME/HAM-F12 (1:1), 10% bovine serum, 2.5% fetal bovine serum, and 40 mg/l gentamicine sulphate. The cultures were kept in an incubator at 37°C with 100% humidity, and the cells grew well in atmospheres of both 5% CO2. RCSN-3 cells are a cloned cell line derived from the substantia nigra of an adult rat. The cell line grows in monolayer and does not require differentiation to express catecholaminergic traits, such as tyrosine hydroxylase, dopamine release, dopamine and norepinephrine transport, formation of neuromelanin, VMAT-2 and monoamine oxidase (MAO)-A expression but not MAO-B. This cell line also expresses serotonin transporters, divalent metal transporter 1 (DMT1), dopamine receptor 1 messenger RNA (mRNA) under proliferating conditions, and dopamine receptor 5 mRNA after incubation with dopamine or DIC an inhibitor of DT-diaphorase (Paris et al., 2008).
Cell death.
For cell death experiments, cells were incubated with cell culture medium but in the absence of bovine serum and phenol red. However, in experiment 6, 10 % bovine serum was included that may decrease aminochrome concentration as a consequence of aminochrome ability to form adduct with proteins. Cell death was measured by counting live and dead cells after staining with 0.5μM calcein AM and 5μM ethidium homodimer-1 for 45 min at room temperature in the dark (LIVE/DEAD Viability/Cytotoxicity Kit, Molecular Probes). Calcein AM is a marker for live cells, and ethidium homodimer-1 intercalates into the DNA of dead cells. Cells were counted with a phase contrast microscope equipped with fluorescence using the following filters: Calcein AM 450–490 nm (excitation) and 515–565 nm (emission) and ethidium homodimer-1, 510–560 nm (excitation) and LP-590 nm (emission). The cell death was measured by incubating the cells during 24 or 48 h with cell culture medium, 100μM DIC, 50μM aminochrome, 50μM aminochrome, and 100μM DIC. The effect of vinblastine on aminochrome-induced cell death was measured by incubating the cells with cell culture medium, 100μM DIC, 10μM vinblastine, 30μM aminochrome, 30μM aminochrome, and 10μM vinblastine; 30μM aminochrome and 100μM DIC; or 30μM aminochrome, 100μM DIC, and 10μM vinblastine. The effect of rapamycin on aminochrome-induced cell death was measured by preincubating the cells during 24 h with 10μM rapamycin before the addition of 50μM aminochrome or 50μM aminochrome and 100μM DIC. Control cells were incubated with cell culture medium, 100μM DIC, or 10μM rapamycin (R).
Caspase-3 activation.
The RCSN-3 cells were incubated with aminochrome as described above for 24 h and were washed and incubated with a solution of 0.01% trypsin for 15 min. The cells are subsequently collected by centrifugation at 2000 rpm and fixed at −20°C with 1 ml methanol (100%) for 30 min. After washing with PBS and incubation with 1.5% milk for 40 min at room temperature, the cells are incubated with 500 μl antibodies against activated caspase-3 diluted 1:250 in PBS containing 1% bovine serum albumin (BSA) and 0.02% Na-azide overnight at 4°C. After washing, the samples were incubated with biotinylated IgG anti-rabbit, diluted 1:500 for 90 min at room temperature under dark conditions. The samples were washed and incubated with Cy-3-conjugated streptavidin at 1.5 μg/ml diluted in PBS for 1 h.
Cytochrome C release.
Cytochrome C release from mitochondria was measured by isolating RCSN-3 cell mitochondria by homogenizing the cells and centrifuge at 2000 × g during 10 min. The supernatant was centrifuged again at 12,000 × g during 10 min, and the pellet was washed and resuspended in 0.25 M sucrose, Tris-HCl pH 7.5. A nitrocellulose membrane was placed on Dot-Blot device (BioRad) and 100 μl Tris Buffered Saline (TBS) buffer (20mM Tris-HCl pH 7.6, 137mM NaCl) was added to each well prior addition of 25 μg mitochondria protein. Apply vacuum and when the nitrocellulose membrane is dry, incubate the membrane in TBS containing 0.2% Tween-20 and 5% low fat milk during 3 h at room temperature. After incubation, wash three times during 5 min with TBS buffer containing 0.1% Tween-20 and incubate overnight the membrane with TBS buffer containing 0.1% Tween-20, 5% BSA protein, and antibody against cytochrome C (Cell Signaling) diluted 1:1000. The membrane was washed and incubated with goat anti-rabbit IgG-horseradish peroxidase-linked antibodies and detected by using luminol and a Kodak BioMax light film.
Determination of mitochondrial membrane potential.
RCSN-3 cells were incubated as described above for 24 h at 37° C with 50μM aminochrome in the absence and presence of 100μM DIC, 100μM DIC alone, or cell culture medium for control cells. The cells were incubated in the dark for 40 min at room temperature with freshly prepared 1.5 μg/ml JC-1 (5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethylbenzimidazolylcarbocyanine iodide) in PBS and then washed three times with PBS. The cells were observed immediately after labeling using a confocal microscope to quantify the intensity of fluorescence simultaneously for the green monomeric form at 488 nm (excitation) and 510–525 nm (emission) and for red fluorescent aggregates-JC-1 at 543 nm (excitation) and 570 nm (emission). Background images were obtained from a cell-free section of the cover slip. A ratio image generated by dividing the fluorescence intensity at 590 nm by the fluorescence intensity at 527 nm is reported as a relative mitochondrial membrane electric potential value (Reers et al., 1995). The fluorescence quantification was performed by using Scion Image software (the National Institutes of Health).
Transmission electron microscopy.
Cells incubated as described above were pelleted and fixed with 3% glutaraldehyde in 0.1 M cacodylate buffer pH 7.4 for 120 min, washed three times, and postfixed in osmium tetroxide 1% for 60 min. The cells were dehydrated in an ascending ethanol battery ranging from 20 to 100% and were later placed in 3:1, 2:1, 1:1, 1:2, and 1:3 ratios of propylene oxide or epom-812 resin for 1 h at room temperature, respectively. Ultrathin sections of 70 nm were made and impregnated with 2% uranyl acetate and Reynold’s lead citrate. The sections were visualized in a Zeiss EM-900 transmission electron microscope at 50 kV and photographed. The negatives were scanned at 600 × 600 dpi resolution, and the images obtained were analyzed later with a computer with a windows operative system using customized software.
GFP-LC3 plasmid transfection.
Cells were grown in culture medium in 24-well plates for 48 h after being transfected with GFP-LC3 plasmid (gift from Zsolt Tallocky PhD, Columbia University Medical Center). To form the transfection complex, we used 2 μl Fugene HD transfection reagent (Roche Diagnostics) and 0.5 μg GFP-LC3 plasmid DNA in 25 μl total volume of medium for each plate, kept in the dark for 15 min. Thereafter, the transfection complex in 150 μl culture medium was added to each plate and incubated for 1 h. Then, 350 μl culture medium was added to each plate and cultured for 2 days.
Confocal microscopy.
Cover slips were mounted on to slides with fluorescent mounting medium (Dako, Carpinteria, CA) and kept in the dark at 4° C. Confocal microscopy (model LSM-410 Axiovert-100; Zeiss, Göttingen, Germany) was used to study the cells. Sample illumination was carried out via a He–Ne laser with 543-nm excitation filter and emission filter over 560 nm.
DNA isolation and quantification.
Cells for DNA isolation were initially trypsinized and then centrifuged at 1300 × g for 3 min. The supernatant was removed and then resuspended in isolation buffer I (sucrose 80mM, EDTA 1mM, 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES)-K-salt 10mM, pH 7.4) and then incubated on ice for 10 min. Equal volume of isolation buffer II (sucrose 300mM, EDTA 1mM, HEPES-K-salt 10mM, pH 7.4) was then added and mechanically homogenized using 50 strokes. To obtain both the nuclear and the mitochondrial fraction, cells were centrifuged at 10,000 × g for 20 min at 4°C. The pellet was stored at −20°C until used. To obtain high-molecular weight DNA, DNA isolation was performed using the DNA purification kit according to the manufacturer’s instructions (QIAGEN Genomic tip). Quantification of the DNA was accomplished using the Picogreen dsDNA quantitation assay (Molecular Probes). DNA samples were aliquoted and stored at −20°C.
Quantitative PCR of a large mitochondrial DNA and nuclear DNA fragment.
The quantitative PCR (QPCR) method to detect DNA damage was performed as previously described (Ayala-Torres et al., 2000; Santos et al., 2006). To verify that the QPCR was in the linear range, the optimal number of cycles for each DNA template was found where a 50% decrease in the amount of DNA resulted in roughly a 50% reduction in PCR amplification. QPCR products that demonstrated a 40–60% amplification of the target sequence when using 50% of the original template were considered acceptable. The PCR amplification profile for the 13.4-kb rat mitochondrial fragment was as follows: hot start for 10 min at 75°C when the DNA polymerase was added, an initial denaturation step for 1 min at 94°C, followed by 22 cycles of denaturation for 15 s at 94°C and then annealing/extension at 66°C for 14 min. To complete the profile, a final extension for 10 min at 72°C was performed. Primers 10633 and 13559 were used for the amplification of the rat mitochondrial fragment, and the primer nucleotide sequences were described previously (2). The PCR amplification for the 12.5-kb rat fragment from the nuclear TRPM-2 gene was the same as for the mitochondrial fragments except that the annealing/extension time was 13 min and the cycles were 27. Primers 5781 and 18312 were used for the amplification of the TRPM-2 fragment and the primer nucleotide sequences were described previously (Ayala-Torres et al., 2000).
Calculation of DNA lesion frequencies.
Assuming a random distribution, the Poisson equation was used to calculate the number of lesions (Ayala-Torres et al., 2000). Based on this equation, the amplification is directly proportional to the fraction of undamaged DNA templates. As such, the average lesion frequency per strand is calculated as −lnAD/AO, where AD is the amplification of the damaged or experimental template, whereas AO is the amplification of the undamaged or control template. Therefore, the results are shown as the number of lesions per strand normalized to 10kb.
Data analysis.
All data are expressed as mean ± SE values. Statistical significance was assessed using ANOVA for multiple comparisons and Student’s t-test for comparison between two given groups.
RESULTS
Incubation of RCSN-3 cells with purified aminochrome (50μM) during 24 h induces a moderate cell death in RCSN-3 cells in the absence (8.0 ± 1.6 %) and the presence of 100μM DIC (16 ± 0.4 %), whereas the cell death in control cells incubated with cell culture medium or in the presence of 100μM DIC was 1.8 ± 1.3% and 1.1 ± 0.7%, respectively. However, incubation of RCSN-3 cells with 50μM aminochrome at 48 h induces a 24 ± 2% (p < 0.01) cell death that significantly increase to 62 ± 3% cell death (p < 0.01) in the presence of 100μM DIC, whereas the cell death in control cells incubated with cell culture medium or in the presence of 100μM DIC was 1.6 ± 0.4% and 3.3 ± 0.5%, respectively (Fig. 1). No cell death was observed when RCSN-3 cells was incubated with 100μM aminochrome in the presence and absence of 100μM DIC at 2, 6, and 12 h (not shown).
FIG. 1.

The effect of aminochrome on cell death in RCSN-3 cells. The cell death of RCSN-3 cells incubated during 24 or 48 h was measured as described under “Materials and Methods” section. A significant cell death induced 50μM aminochrome (AM) in the absence and presence of 100μM DIC (AM + DIC) after 48-h incubation. No significant cell death was observed in cells treated with 100μM DIC or incubated with cell culture medium. The values are the mean ± SD (n = 3), and the statistical significance was assessed using ANOVA for multiple comparisons and Student’s t-test (**p < 0.01).
The slow cell death observed with purified aminochrome suggests the possible involvement of an apoptotic cell death, and therefore, we observed caspase-3 activation using immunohistochemical technique when the cells were incubated with 50μM aminochrome in the absence and presence of 100μM DIC (Figs. 2C and D, respectively). Quantification of numbers of caspase-3 positive cells we observed a significant caspase-3 activation in cells treated with 50μM aminochrome (24 ± 0.2 cells, p < 0.001) and 50μM aminochrome and 100μM DIC (26 ± 0.6 cells, p < 0.001). Cells incubated with cell culture medium or 100μM DIC alone presented 4.6 ± 1 and 3.8 ± 0.1 positive cells, respectively (Fig. 2E). We measured cytochrome C release from mitochondria by using dot-blot technique and a decrease in cytochrome C immunostaining in the mitochondrial fraction was observed in RCSN-3 cells treated with 50μM aminochrome and 100μM DIC (41 % of control, Figs. 2F and G).
FIG. 2.

Determination of caspase-3 activation, release of cytochrome C, and DNA fragmentation. Caspase-3 activation was determined in control cells (A), cells treated during 24 h with 100μM DIC (B), 50μM aminochrome (C), or 50μM aminochrome and 100μM DIC (D) by using immunohistochemistry as described under “Materials and Methods” section. Quantification of immunopositive casapase-3 was determined by confocal microscopy (E) and revealed caspase-3 activation when RCSN-3 cells were treated with 50μM aminochrome in the absence (AM) or presence of 100μM DIC (AM + DIC). The cells were also incubated with DIC alone (DIC) or in the presence of cell culture medium (C). The release of cytochrome C was measured with dot blot technique (F) as described under “Materials and Methods” section. The cells were treated with cell culture medium (1), 100μM DIC (2), 50μM aminochrome (3), or 50μM aminochrome and 100μM DIC (4) during 24 h. The quantification of dot blot was done by measuring pixel number (G). The statistical significance was assessed by using ANOVA for multiple comparisons and Student’s t-test (***p < 0.001).
In experiments reported with aminochrome in the presence of oxidizing agents, mitochondrial damage was observed (Arriagada et al., 2004; Fuentes et al., 2007), and therefore, we analyzed mitochondria membrane potential by using JC-1 marker after 24 h incubation. Incubation of RCSN-3 cells with 50μM aminochrome in the presence of 100μM DIC indicates a significant change in mitochondrial membrane potential (0.37 ± 0.03 relative fluorescence [RF]; p < 0.01). No significant changes in mitochondrial membrane potential were observed in cells treated with 50μM aminochrome (0.55 ± 0.12 RF) or 100μM DIC (0.86 ± 0.05 RF) in comparison with control cells incubated with cell culture medium (0.79 ± 0.07 RF; Fig. 3). Ultrastructure analysis of RCSN-3 cells was performed by using transmission electron microscopy in RCSN-3 cells incubated during 48 h. Normal morphology of mitochondria and regular chromatin aspect within the nucleus could be observed under control conditions (Fig. 4A). Cells treated with 100μM DIC showed normal mitochondrial aspects and nucleus chromatin appears normal (Fig. 4B). Aminochrome (50μM)-treated cells display normal mitochondria (white arrow) (Fig. 4C), whereas we can observe a damaged mitochondria (white arrow) (Fig. 4D) in cells treated with 50μM aminochrome and 100μM DIC.
FIG. 3.

Changes in mitochondrial membrane potential were measured using JC-1 staining in RCSN-3 cells by using confocal microscopy. The picture of cells with JC-1 staining in cells incubated with (A) cell culture medium (C), (B) 100μM DIC (DIC), (C) 50μM aminochrome (AM), or (D) 50μM aminochrome and 100μM DIC (AM + DIC) for 24 h. The values are the ratio between red/green fluorescence of JC-1 obtained from the confocal are plotted in (E). The statistical significance was assessed using ANOVA for multiple comparisons and Student’s t-test (**p < 0.01).
FIG. 4.

Ultrastructural analysis of RCSN-3 cells treated with aminochrome. The cell ultrastructure was studied by using transmission electron microscopy in control conditions (A), cells treated for 48 h with 100μM DIC (B), 50μM aminochrome (C), and 50μM aminochrome and 100μM DIC (D). We observed mitochondria (white arrows), organelles like mitochondria inside of double membrane autophagic vacuoles (white arrow head), double membrane autophagic vacuoles (black arrows). Bars = 0.5μm; nucleus, N.
We analyzed the formation autophagosome vacuoles by transfecting RCSN-3 cells with a plasmid coding for LC3-GFP that produce green fluorescence. Incubation of RCSN-3 cells with 50μM aminochrome (14 ± 2 positive cells; p < 0.05) or 50μM aminochrome and 100μM DIC (16 ± 0.3 positive cells (p < 0.001) during 48 h induced the formation of autophagic vacuoles. No significant number of positive cells with autophagic vacuoles was induced by 100μM DIC (3.5 ± 0.2 number of positive cells) in comparison with control cells incubated with cell culture medium (1.1 ± 0.4 positive cells, Fig. 5E). Ultra structure analysis of RCSN-3 cells by using transmission electron microscopy showed that RCSN-3 cells incubated with 50μM aminochrome for 48 h accumulate autophagic vacuoles (black arrow) containing membranous components like degraded mitochondria within a double membrane compartment (Fig. 4C). In RCSN-3 cells treated with 50μM aminochrome and 100μM DIC appears with highly vacuolated membranous components and autophagic vacuoles (black arrows) with membranous remanent inside (Fig. 4D). Cells treated with 100μM DIC or incubated with cell culture medium do not show autophagic vacuoles (Figs. 4B and A, respectively). To study the role of autophagic vacuoles on aminochrome-induced cell death, we used vinblastine that is an inhibitor of autophagosome fusion with lysosomes (Seglen and Brinchmann, 2010). A significant increase in cell death was observed when we compared RCSN-3 cells incubated during 48 h with 30μM aminochrome and 10μM vinblastine or 30μM aminochrome, 10μM vinblastine, and 100μM DIC in comparison with cells treated with 30μM aminochrome in the absence or presence of 100μM DIC (Fig. 6 A). The possible protective role of autophagy on aminochrome-induced cell death was studied by using rapamycin induce an upregulation of macroautophagy as a consequence of mTor pathway inactivation (Schmelzle and Hall, 2000). A significant decrease in cell death was observed when RCSN-3 cells were preincubated during 24 h with 10μM rapamycin before the addition of 50μM aminochrome or 50μM aminochrome and 100μM DIC during 48 h in comparison with cells incubated in the absence of rapamycin with 50μM aminochrome or 50μM aminochrome and 100μM DIC (Fig. 6B).
FIG. 5.
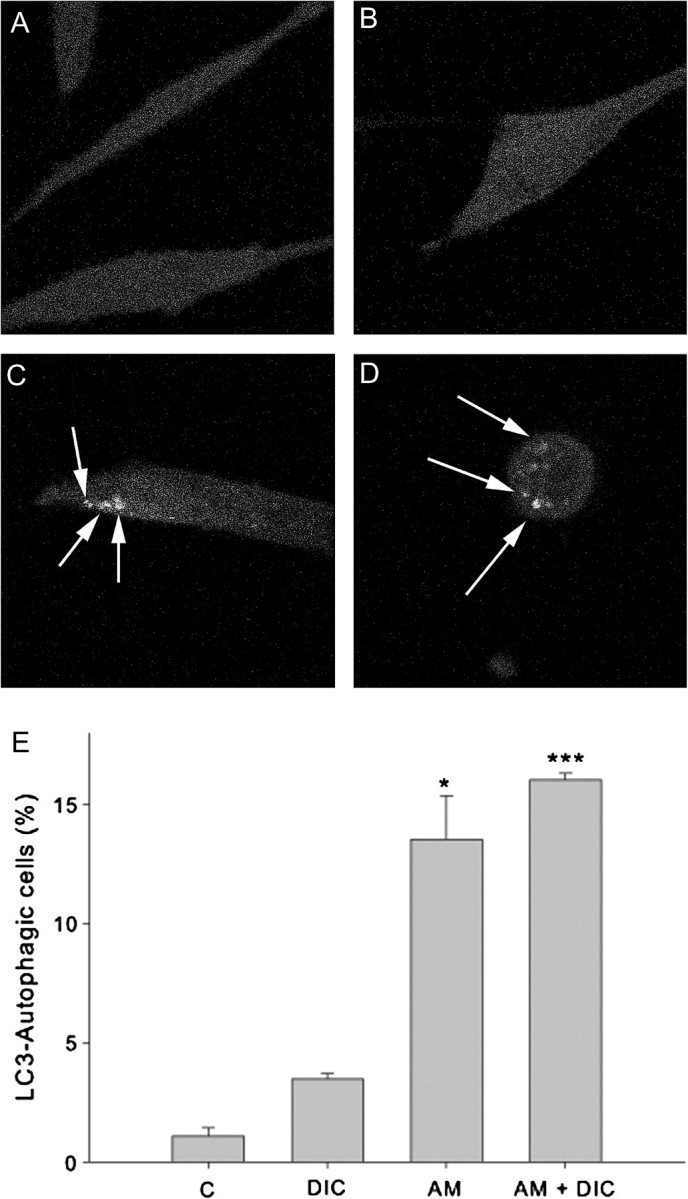
Determination of autophagosome vesicles in RCSN-3 cells transfected with GFP-LC3. (A) GFP-LC3 transfected RCSN-3 cells were treated with cell culture medium (A), 100μM DIC (B), 50μM aminochrome (C), and 50μM aminochrome and 100μM DIC during 48 h (D). The quantification of number of cells with autophagic vacuoles was plotted (n = 3 ± SD), and the statistical significance was assessed using ANOVA for multiple comparisons and Student’s t-test (*p < 0.05; ***p < 0.001).
FIG. 6.

The effect of autophagy on aminochrome-induced cell death. The effect of autophagy on aminochrome-induced cell death was measured by using vinblastine (A) that is an inhibitor of the fusion between autophagic vacuoles and lysosomes and rapamycin (B) that is an inducer of autophagy. (A) A significant increase in cell death was observed when RCSN-3 cells were incubated during 48 h with 30μM aminochrome and 10μM vinblastine (AM + V) in comparison with cells incubated with 30μM aminochrome alone (AM). A significant increase in cell death was also observed in cells incubated 30μM aminochrome, 100μM DIC, and 10μM vinblastine (AM + DIC + V) in comparison with cells incubated with 30μM aminochrome and 100μM DIC (AM + DIC). As control, we incubated the cells with cell culture medium (C), 100μM DIC (DIC), or 10μM vinblastine (V). (B) A significant decrease in cell death was observed when RCSN-3 cells were preincubated during 24 h with 10μM rapamycin (R) before the addition of 50μM aminochrome (AM + R) or 50μM aminochrome and 100μM DIC (AM + DIC + R) in comparison with cells incubated in the absence of rapamycin with 50μM aminochrome (AM) or 50μM aminochrome and 100μM DIC (AM + DIC). The control cells (C) were incubated with cell culture medium, 100μM DIC (DIC), or 10μM rapamycin (R). The statistical significance was assessed using ANOVA for multiple comparisons and Student’s t-test (***p < 0.001; **p < 0.01).
A dramatic morphological change characterized as an apparent volume reduction and loss of cell elongated shape to spherical shape determined with confocal fluorescence microscopy was observed when the cells RCSN-3 were incubated with 50μM aminochrome and 100μM DIC (Fig. 5D). Interestingly, RCSN-3 cells treated with 50μM aminochrome alone (Fig. 5C) or 100μM DIC (Fig. 5B) presented the typical elongated shape without significant volume reduction as cells incubated with cell culture medium (Fig. 5A).
Having observed phenotypic changes in mitochondria following aminochrome and DIC treatment, we decided to measure DNA damage in both the genomic and the mitochondrial DNA (mtDNA). To do this, we utilized a gene-specific QPCR-based assay (Ayala-Torres et al., 2000; Santos et al., 2006). Nuclear DNA (nDNA) and mtDNA were extracted and QPCR performed using previously tested primers to amplify the rat (nuclear) clusterin (TRPM-2) gene (12.5 kb) and a large mtDNA fragment (13.4 kb) (Ayala-Torres et al., 2000). Exposure of RCSN-3 cells to DIC (100μM), aminochrome (50μM), or a combination of the two for 48 h resulted in increased nDNA lesions (0.35 lesions/10 kb, 1.2 lesions/10 kb, and 0.39 lesions/10 kb, respectively; Fig. 7A) as compared with control cells incubated with cell culture medium. In addition, cells treated with DIC (100μM) or aminochrome (50μM) alone showed minimal increase in mtDNA lesions (0.06 lesions/10 kb, 0.08 lesions/10 kb, respectively, Fig. 7B) relative to cells incubated with cell culture medium. However, treatment of RCSN-3 cells with both DIC and aminochrome together resulted in increased mtDNA damage (0.2 lesions/10 kb; Fig. 7B).
FIG. 7.

nDNA and mtDNA damage in RCSN-3 cells. These data represent technical replicates of the number of excess lesions found per 10 kb from both nuclear and mitochondrial genomes in RCSN-3 cells treated with aminochrome, DIC, or both as compared with controls treated with cell culture medium alone. (A) An increase in the number of nDNA and (B) mtDNA lesions was observed in each condition. Error bars represent the standard error of the mean.
DISCUSSION
The fact that aminochrome is the precursor of neuromelanin and neuromelanin-containing neurons are lost during the neurodegenerative process underlying Parkinson’s disease and the discovery that aminochrome induces and stabilizes neurotoxic alpha-synuclein protofibrils (Conway et al., 2001; Norris et al., 2005), suggests that aminochrome may play an important role in the neurodegeneration of dopaminergic neurons observed in Parkinson’s disease. Aminochrome oxidation and its polymerization to neuromelanin (Zecca et al., 2002, 2008) seem to be a normal event because dopaminergic neurons contain neuromelanin in individuals without the disease. However, aminochrome is able to participate in two neurotoxic reactions: (1) the formation of adducts with proteins such as complex I and III of the electron transport chain in mitochondria (Van Laar et al., 2009), alpha-synuclein, inhibiting alpha-synuclein fibrillization and enhancing and stabilizing the formation of neurotoxic protofibrils (Conway et al., 2001; Norris et al., 2005), tyrosine hydroxylase (Xu et al., 1998), dopamine transporter (Whitehead et al., 2001) and parkin (LaVoie et al., 2005) and (2) aminochrome can also be reduced by flavoenzymes that catalyze one electron of quinones to form the neurotoxic leukoaminochrome o-semiquinone radical, which is extremely reactive with oxygen (Baez et al., 1995; Segura-Aguilar et al., 1998). Leukoaminochrome-o-semiquinone radical reduces oxygen to superoxide radicals resulting in the formation of superoxide radicals and aminochrome, generating a redox cycling between aminochrome and leukoaminochrome o-semiquinone radical with concomitant formation of hydrogen peroxide, hydroxyl radicals, depletion of β-nicotinamide adenine dinucleotide, reduced (NADH) and O2 required for energy production, and depletion of β-Nicotinamide adenine dinucleotide phosphate, reduced (NADPH) required for the reduction of oxidized glutathione catalyzed by glutathione peroxidase.
Previously, we have reported evidence that aminochrome induces acute neurotoxicity, mitochondria damage, apoptosis, and formation of autophagic vacuoles, but these studies were performed by using different oxidizing agents to form aminochrome (Arriagada et al., 2004; Fuentes et al., 2007; Paris et al., 2001, 2005a, 2009). Aminochrome formed with manganese3+ induces acute neurotoxicity in 2 h in the presence of DIC (Arriagada et al., 2004), whereas in this study, we have observed that purified aminochrome induces cell death at 48 h. The explanation to this enormous difference in the rate of cell death is the presence of manganese in the cell culture medium used as an oxidizing agent that under dopamine oxidation is reduced to manganese2+ that can be transported into the cell by DMT1 that is expressed in RCSN-3 cells (Paris et al., 2008). Manganese2+ inside the cell can be oxidized by superoxide to manganese3+ and participates in oxidation reactions inside the cell. Inhibition of VMAT-2 with reserpine to induce dopamine autooxidation to aminochrome in the presence of oxygen as a consequence of an increase in intracellular dopamine exhibited an apoptotic cell death in the presence of DIC (Fuentes et al., 2007), suggesting that aminochrome induces an apoptotic cell death. In the present study, we show that purified aminochrome induces a slow cell death at 48-h incubation with early caspase-3 activation at 24 h when DT-diaphorase is inhibited by DIC, suggesting that aminochrome induces a cell death with initial features apoptotic. Another interesting feature of aminochrome cell death is the cell shrinkage induced in cells incubated with aminochrome and DIC (see Fig. 5D), characterized by the apparent cell volume reduction and a spherical shape contrasting the elongated shape observed in control cells or in cells incubated with aminochrome or DIC alone (Figs. 5A–C). These morphological changes are a consequence of disruption of actin that becomes condensed around the cell membrane when the cells were incubated with aminochrome and DIC (Paris et al., 2010). Actin is an important component of the cellular cytoskeleton, which is essential for sculpting and maintaining cell shape (Cingolani and Goda, 2008). However, apoptotic volume decrease leading to cell shrinkage seems to be a core event in the course of apoptosis (Humphrey et al., 2005; Núñez et al., 2010; Yagami et al., 2010), supporting the idea that aminochrome in the presence of DIC induces a cell death with features apoptotic. Interestingly, the cell shrinkage observed in cells treated with aminochrome and DIC coexisted with LC3-positive fluorescence, although autophagy determined with LC3-positive fluorescence was also present in cell treated with aminochrome alone.
It is seems to be plausible that aminochrome in the presence of DIC induces the mitochondrial (intrinsic) pathway where mitochondrial outer membrane permeability allows cytochrome C release and activation of caspase-3 inducing apoptosis and cell death (Green and Kroemerm, 2004). However, aminochrome alone also induce caspase-3 activation without cell shrinkage and disruption of mitochondria membrane potential or cytochrome C release. One possible explanation is that caspase-3 activation is not mediated by mitochondrial pathway but by death receptor (extrinsic) pathway where caspase-3 activation is dependent on caspase-8 activation (Schulze-Osthoff et al., 1998). Another possible explanation is that aminochrome induces endoplasmic reticulum stress with caspase-3 activation mediated by caspase-12 activation (Hitomi et al., 2004; Jin et al., 2010) or that caspase-3 can be pseudoactivated via calcium dependent proteolysis which also is independent cytochrome C release (Pelletier et al., 2005). The activation of caspase-3 mediated by both intrinsic and extrinsic pathway should lead to cell death. However, it is possible that the observed aminochrome-induced caspase-3 activation is not enough to trigger apoptosis, explaining why the cells treated with aminochrome induces a moderate cell death and they don’t show cell shrinkage. This idea is supported by the finding that in the embryonic epidermis, the number of cells undergoing classical apoptosis is as low as after birth despite the widespread expression and activation of caspase-3, suggesting that elevated caspase-3 levels in these cells are not sufficient to trigger apoptosis. Caspase-3 activation result instead in a selective targeting of substrates that are involved in the keratinocyte differentiation process including activation of protein kinase C-δ (Okuyama et al., 2004). Another possible explanation why the observed aminochrome-induced caspase-3 activation cannot trigger apoptosis is that activated caspase-3 can be degraded by autophagy (Yang et al., 2008). The caspase-3 activation seems to be prior to the formation autophagic vacuoles (Martin and Baehrecke, 2004). It is possible that autophagy is being utilized to maintain cell viability after the activation of caspases. Interestingly, nonapoptotic functions for caspase-3 activation have been reported where caspase-3 plays a regulatory role in synaptic plasticity, neurogenesis, and also in cellular responses to stroke, such as reactive astrogliosis and the infiltration of macrophages (D’Amelio et al., 2010; Wagner et al., 2011).
Another interesting feature of aminochrome cell death is the formation of autophagic vacuoles. Autophagy can lead to cell death in response to cellular stress (Jo et al., 2010; Shen and Codogno, 2011). However, autophagy is also an intracellular bulk degradation process whereby cytoplasm proteins and organelles are degraded and recycled through lysosomes (Kuma and Mizushima, 2010; Matsui et al., 2008; Tettamanti et al., 2008). It plays an important role in the elimination of damaged organelles such as mitochondria, peroxisomes and endoplasmic reticulum. There are several reports supporting the idea that autophagy plays a protective role in Parkinson’s disease (Rubinsztein et al., 2005) but also exist others reports that involves in cellular death (Cheng et al., 2011; Choi et al., 2010). Inhibition of chaperone-mediated autophagy (CMA) leads to an accumulation of soluble high molecular weight and detergent-insoluble species of alpha-synuclein (Vogiatzi et al., 2008) and expression of mutant A53T induces CMA dysfunction mediating aberrant alpha-synuclein toxicity (Xilouri et al., 2009). The pathogenic A53T and A30P α-synuclein mutants inhibit their own degradation and that of other substrates (Cuervo et al., 2004). Aminochrome in the presence of DIC induces the formation of autophagic vacuoles with double membrane determined with electron microscopy. The protein LC3 has been used as a specific marker for quantification of autophagosomes and over expression of GFP-LC3 is a well-accepted, straightforward, and specific assessment of autophagosome formation that does not perturb autophagosome number or function (Tanida and Waguri, 2010). RCSN-3 cells transfected with GFP-LC3 and incubated with aminochrome and DIC showed that a significant increase (16-fold) in number of cells with autophagic vacuoles in comparison with control cells. Autophagy is a process that occurs in several steps as follows: formation of phagophores, formation of mature autophagosomes, targeting and trafficking of autophagosomes to lysosomes, formation of autolysosomes by fusion between autophagosomes and lysosomes, and finally degradation of the autophagic bodies within the lysosomes (Yamamoto et al., 2010). Vinblastine, that induces autophagosome accumulation by blocking the fusion of these organelles with endosomes and lysosomes (Seglen and Brinchmann, 2010), showed a significant increase in the cell death induced by aminochrome in the presence or absence of DIC, suggesting that autophagy is a very important protective mechanism against aminochrome-induced cell death. The protective role of autophagy against aminochrome-induced cell death was supported by the significant decrease of cell death in cells preincubated with rapamycin because this compound inactivate mTor pathway inducing a dramatic upregulation in macroautophagy (Schmelzle and Hall, 2000). Autophagy induction with rapamycin protects dopamine neurons down regulating p53 and its related apoptotic pathways and by inducing autophagy to degrade aggregated proteins. Therefore, rapamycin might be protective against neuronal injury in the Parkinson’s disease (Du et al., 2009). Induction of autophagy mediated by rapamycin also prevents rotenone-dependent apoptosis in SH-SY5Y cells (Pan et al., 2009). Autophagy activation restores the mitochondrial membrane potential impaired by rotenone in SH-SY5Y cell lines overexpressing alpha-synuclein (Dadakhujaev et al., 2010). Aminochrome induces both apoptosis features and autophagy where apoptosis leads to the cell death and autophagy is working as a rescue mechanism to recycle damaged mitochondria in order to prevent the cell death. Therefore, it seems to be plausible that inhibition of the fusion of autophagy vacuoles with lysosomes in the presence of vinblastine that increase aminochrome-induced cell death can be associated to an increase of apoptosis as it has been reported before (Tang et al., 2011) . Autophagy and apoptosis are not mutually exclusive pathways, they could to act in synergy where both pathways are inter-connected. It is possible that both autophagy and apoptosis form part of the same mechanism where autophagy acts as an antagonist to block apoptosis promoting cell survival (Eisenberg-Lerner et al., 2009). However, the data presented in this work are not enough to support or discard this idea.
Aminochrome in the presence of DIC induces a significant disruption in mitochondrial membrane potential determined by JC-1 technique, as a consequence of mitochondrial outer membrane permeabilization leading to the release of proteins such as cytochrome C that normally are found in the space between outer and inner membranes. Disruption of the mitochondrial membrane potential has been reported in apoptosis and autophagy (Bustamante et al., 2004; Cui et al., 2011). It is possible that mitochondrial membrane potential disruption is a consequence of mitochondrial transition pores formation or others pores (He and Lemasters., 2002; Tharakan et al., 2009). Over expression of alpha-synuclein induces activation of mitochondrial transition pore (Zhu et al., 2011). There are reports indicating that mitochondrial transition pores inhibitors prevented collapse in mitochondrial membrane potential and also cytochrome c release (Tharakan et al., 2009). Several studies showed that the mitochondrial cytochrome c release occurs when exist mitochondrial depolarization, although also is possible in their absence (Cai et al., 2011; Krohn et al., 1999). Our results with JC-1 are in agreement with cytochrome C release from mitochondria observed in cells incubated with aminochrome together with DIC. Interestingly, no significant change in membrane potential was observed when the cells were treated with aminochrome alone that is in agreement with the lack of cytochrome C release from mitochondria. Dopamine o-quinone, the transient precursor of aminochrome, alters mitochondrial respiration (Berman and Hastings, 1999). Dopamine o-quinone is the precursor of aminochrome and is a transient compound at physiological pH levels because its amino chain undergoes cyclization to form aminochrome (Segura-Aguilar and Lind, 1989). Dopamine o-quinone also induces covalent modification in various mitochondrial proteins, including proteins from the tricarboxylic acid cycle and subunits of complex I and III (Van Laar et al., 2009). Mitochondrial dysfunction and energy collapse seem to be a mechanism involved in aminochrome neurotoxicity because aminochrome induces covalent modifications of mitochondrial proteins, mtDNA damage, and generates a redox cycling between aminochrome and leukoaminochrome o-semiquinone radical (reactions 3 and 4, Fig. 8) (Baez et al., 1995; Segura-Aguilar et al., 1998) when aminochrome is reduced by flavoenzymes that use NADH as electron donor. This redox cycling depletes NADH and oxygen required for mitochondrial respiration and ATP formation.
FIG. 8.

Possible mechanisms for cell death induced by aminochrome. Our results suggest that aminochrome can induce cell death by two different mechanisms: (A) Incubation of aminochrome in the presence of DIC an inhibitor of DT-diaphorase (reaction 1). Aminochrome is one electron reduced by flavoenzymes that use β-Nicotinamide adenine dinucleotide, reduced (NADH) as electron donator to leukoaminochrome o-semiquinone radical (reaction 3) that reduce oxygen to superoxide radicals (reaction 4) with subsequent formation of hydrogen peroxide the precursor of hydroxyl radical. This reaction generates a redox cycling between aminochrome and leukoaminochrome o-semiquinone radical (reactions 3 and 4) that continues until NADH or oxygen are depleted, affecting the electron transport chain in the mitochondria that is not able to produce ATP with subsequent cell energy collapse. Flavoenzymes that use β-Nicotinamide adenine dinucleotide phosphate, reduced (NADPH) as electron donors are also able to reduce aminochrome according reaction 3, resulting in the depletion of NADPH requires for the reduction of oxidized glutathione catalyzed by glutathione reductase. This mechanism induces autophagy, apoptosis, formation of clusters of condensed chromatine, mtDNA damage, and finally cell death. However, aminochrome is also able to form adducts with proteins such as mitochondria proteins (35) (reaction 2) and mtDNA. Aminochrome is also able to induce cell shrinkage by forming adducts with actin and α/β-tubulin of cytoskeleton (21), explaining the change of elongated to spherical shape as a consequence of disruption of cell cytoskeleton. In this mechanism, autophagy try to rescue the cell by recycling damaged mitochondria, but this is not enough to prevent completely the cell death, explaining why the cell death is increased when vinblastine inhibited the fusion of autophagic vacuoles with lysosomes. (B) Incubation of cells with aminochrome alone. Under these conditions, DT-diaphorase is catalyzing the two electron reduction of aminochrome by using both NADH and NADPH available in the cell preventing that aminochrome participates in neurotoxic reaction, such as formation of adducts with proteins or its one electron reduction (reactions 2 and 3 in A). However, aminochrome alone induces a moderate cell death despite the fact that DT-diaphorase is very active enzyme and should prevent that aminochrome participates in neurotoxic reactions. It seems to be plausible that DT-diaphorase capacity to reduce aminochrome was surpassed due to the used concentration in our experiments was too high that free molecules of aminochrome were able to participate in other reactions. Under these conditions, it is not possible that aminochrome can be one one electron reduced because this reaction requires NADH or NADPH that DT-diaphorase was using both as electron donors. Therefore, it seems plausible that amnochrome forms adducts with proteins or nDNA beceause it has been reported that aminochrome induces covalent modification of subunits of complex I and III of mitochondria (35). Aminochrome induces the formation of alpha-synuclein protofibrils, but RCSN-3 cell line doesn’t have constitutive expression of alpha-synuclein (17). Aminochrome takes up by dopamine transporter into RCSN-3 cells, and the majority of aminochrome is reduced by DT-diaphorase (reaction 1) but the excess of aminochrome, that DT-diaphorase is not able to reduce, forms adducts with mitochondrial proteins (reaction 2) inducing autophagy (reaction 3) that prevent the cell death. However, inhibition of the fusion of autophagic vacuoles with lysosomes in the presence of vinblastine results in the induction of cell death.
Our results support the proposed antioxidant and neuroprotective role of DT-diaphorase because aminochrome neurotoxicity is dependent on DT-diaphorase inhibition. DT-diaphorase (EC.1.6.99.2) is a unique flavoenzyme, which catalyzes the reduction of two electrons of quinones to hydroquinone and can use both NADH and NADPH as electron donators. DT-diaphorase immunoreactivity has been found in neurons of both substantia nigra and ventral tegmental area, and its colocalization with tyrosine hydroxylase-like immunoreactivity has also been observed. DT-diaphorase constitutes 97% of the total quinone reductase activity in rat substantia nigra cytosol. DT-diaphorase immunoreactivity has also been found in Bergmann glia, astrocytes, and tanycytes (Schultzberg et al., 1988). The neuroprotective role of DT-diaphorase against aminochrome in the presence of manganese is supported by studies done by using the inhibitior DIC (Arriagada et al., 2004). Although, DIC is a specific inhibitor of DT-diaphorase because DT-diaphorase activity is defined as DIC sensitive quinone reductase activity, we cannot discard an unspecific effect of DIC. The use of short interfering RNA (siRNA) against DT-diaphorase supports the neuroprotective role of this enzyme against aminochrome neurotoxicity because the siRNA is acting on DT-diaphorase mRNA silencing its expression and inducing aminochrome neurotoxicity (Lozano et al., 2010). However, this publication with siRNA did not include the results presented in the present work related to caspase-3 activation, mtDNA damage, disruption of mitochondria membrane potential, cytochrome C release, mitochondria damage, and the role of autophagic vacuoles with purified aminochrome when DT-diaphorase is inhibited.
Aminochrome-induced cell death results presented in Figures 1 and 6 are different because in Figure 1a, concentration of 50μM aminochrome was used in the presence of DIC inducing 62 ± 3% cell death, whereas in Figure 6 B, induces only 43 ± 2% cell death. This discrepancy can be explained by the fact that in Figure 6 was done in the presence of bovine serum that provide some protection because aminochrome is able to form adducts with proteins (LaVoie et al., 2005; Norris et al., 2005; Paris et al., 2010; Van Laar et al., 2009; Whitehead et al., 2001; Xu et al., 1998) resulting in a lower intracellular concentration and subsequent lower cell death. The important is that in both experiments, we have the same tendency where aminochrome in the presence of DIC are more toxic than aminochrome alone.
In conclusion, our results suggest the existence of two different mechanisms for aminochrome-dependent cell death: (1) One mechanism involves aminochrome alone in the absence of DT-diaphorase inhibitors. DT-diaphorase prevents that aminochrome can be one electron reduced or form adducts with proteins at lower concentrations such as 30μM but at higher concentration such as 50μM DT-diaphorase capacity to reduce aminochrome with two electrons was surpassed inducing cell death. However, the cell death observed in our experiments in the presence of aminochrome alone is probably dependent on its ability to form adducts with proteins, such as complex I and III of mitochondrias electron chain (Van Laar et al., 2008) because one electron reduction cannot proceed due to NADH or NADPH are used by DT-diaphorase that is very active and it is not inhibited. Aminochrome alone induces a moderate cell death with caspase-3 activation, but no significant disruption of mitochondrial membrane potential, mtDNA damage, and yet the presence of nDNA damage. Transmission electron microscopy showed a normal mitochondria coexisting with an autophagic vacuole containing a mitochondria-like organelle inside, and LC3-GFP technique showed a significant increase in cells with autophagic vacuoles. The presence of vinblastine in cells incubated with aminochrome alone induced a significant cell death at the same level that aminochrome induced in the presence of DIC, suggesting that aminochrome alone induced cell death is depending on inhibition of degradation and recycling of damaged organelles. Vinblastine prevents the fusion of autophagic vacuoles with endosomes and lysosomes and the increase in cell death induced by aminochrome alone suggests that autophagy plays an important protective role prolonging the survival by degrading damaged organelles and recycled through lysosomes. This idea is supported by the effect of rapamycin that induces autophagy, resulting in a significant decrease in the cell death and (2) the other mechanism involves one electron reduction of aminochrome when DT-diaphorase is inhibited, inducing significant cell death, caspase activation, release of cytochrome C, disruption of mitochondria membrane potential, and mitochondria damage. This cell death is accompanied by the formation of autophagic vacuoles that antagonize aminochrome neurotoxicity by degrading and recycling damaged organelles through lysosomes. Therefore, our results suggest that autophagy is an important protective mechanism against aminochrome-induced cell death both in the absence and in the presence of DT-diaphorase inhibitor DIC.
FUNDING
FONDECYT (1061083, 1100165, 1F32ES019008-01 to L.H.S., NS059806); American Parkinson Disease Association (J.T.G).
References
- Arriagada A, Paris I, Sanchez de las Matas MJ, Martinez-Alvarado P, Cardenas S, Castañeda P, Graumann R, Perez-Pastene C, Olea-Azar C, Couve E, et al. On the neurotoxicity of leukoaminochrome o-semiquinone radical derived of dopamine oxidation: mitochondria damage, necrosis and hydroxyl radical formation. Neurobiol. Dis. 2004;16:468–477. doi: 10.1016/j.nbd.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Ayala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van Houten B. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods. 2000;22:135–147. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- Baez S, Linderson Y, Segura-Aguilar J. Superoxide dismutase and catalase enhance autoxidation during one-electron reduction of aminochrome by NADPH-cytochrome P-450 reductase. Biochem. Mol. Med. 1995;54:12–18. doi: 10.1006/bmme.1995.1002. [DOI] [PubMed] [Google Scholar]
- Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J. Neurochem. 1999;73:1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson’s disease. Nat. Med. 2008;14:483–485. doi: 10.1038/nm0508-483. [DOI] [PubMed] [Google Scholar]
- Bustamante J, Caldas Lopes E, Garcia M, Di Libero E, Alvarez E, Hajos SE. Disruption of mitochondrial membrane potential during apoptosis induced by PSC 833 and CsA in multidrug-resistant lymphoid leukemia. Toxicol. Appl. Pharmacol. 2004;199:44–51. doi: 10.1016/j.taap.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Cai X, Zhang H, Tong D, Tan Z, Han D, Ji F, Hu W. Corosolic acid triggers mitochondria and caspase-dependent apoptotic cell death in osteosarcoma MG-63 Cells. Phytother. Res. 2011 doi: 10.1002/ptr.3422. Advance Access published on February 21, 2011; doi:10.1002/ptr.3422. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M, Wang C, During M, Talloczy Z, Tanaka K, et al. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J. Neurosci. 2011;31:2125–2135. doi: 10.1523/JNEUROSCI.5519-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KC, Kim SH, Ha JY, Kim ST, Son JH. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J. Neurochem. 2010;112:366–376. doi: 10.1111/j.1471-4159.2009.06463.x. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Cui T, Fan C, Gu L, Gao H, Liu Q, Zhang T, Qi Z, Zhao C, Zhao H, Cai Q, et al. Silencing of PINK1 induces mitophagy via mitochondrial permeability transition in dopaminergic MN9D cells. Brain Res. 2011 doi: 10.1016/j.brainres.2011.01.035. Advance Access published on January 21, 2011; doi:10.1016/j.brainres.2011.01.035. [DOI] [PubMed] [Google Scholar]
- Dadakhujaev S, Noh HS, Jung EJ, Cha JY, Baek SM, Ha JH, Kim DR. Autophagy protects the rotenone-induced cell death in alpha-synuclein overexpressing SH-SY5Y cells. Neurosci. Lett. 2010;472:47–52. doi: 10.1016/j.neulet.2010.01.053. [DOI] [PubMed] [Google Scholar]
- D'Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2010;17:1104–1114. doi: 10.1038/cdd.2009.180. [DOI] [PubMed] [Google Scholar]
- Díaz-Véliz G, Paris I, Mora S, Raisman-Vozari R, Segura-Aguilar J. Copper neurotoxicity in rat substantia nigra and striatum is dependent on DT-diaphorase inhibition. Chem. Res. Toxicol. 2008;21:1180–1185. doi: 10.1021/tx8001143. [DOI] [PubMed] [Google Scholar]
- Du Y, Yang D, Li L, Luo G, Li T, Fan X, Wang Q, Zhang X, Wang Y, Le W. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy. 2009;5:663–675. doi: 10.4161/auto.5.5.8377. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- Fuentes P, Paris I, Nassif M, Caviedes P, Segura-Aguilar J. Inhibition of VMAT-2 and DT-diaphorase induce cell death in a substantia nigra-derived cell line-an experimental cell model for dopamine toxicity studies. Chem. Res. Toxicol. 2007;20:776–783. doi: 10.1021/tx600325u. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemerm G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Taniguchi M, Honda A, Imaizumi K, Tohyama M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 2004;357:127–130. doi: 10.1016/j.neulet.2003.12.080. [DOI] [PubMed] [Google Scholar]
- Humphrey ML, Cole MP, Pendergrass JC, Kiningham KK. Mitochondrial mediated thimerosal-induced apoptosis in a human neuroblastoma cell line (SK-N-SH) Neurotoxicology. 2005;26:407–416. doi: 10.1016/j.neuro.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Jin CM, Yang YJ, Huang HS, Kai M, Lee MK. Mechanisms of L-DOPA-induced cytotoxicity in rat adrenal pheochromocytoma cells: implication of oxidative stress-related kinases and cyclic AMP. Neuroscience. 2010;170:390–398. doi: 10.1016/j.neuroscience.2010.07.039. [DOI] [PubMed] [Google Scholar]
- Jo YK, Park SJ, Shin JH, Kim Y, Hwang JJ, Cho DH, Kim JC. ARP101, a selective MMP-2 inhibitor, induces autophagy-associated cell death in cancer cells. Biochem. Biophys. Res. Commun. 2010;404:1039–1043. doi: 10.1016/j.bbrc.2010.12.106. [DOI] [PubMed] [Google Scholar]
- Krohn AJ, Wahlbrink T, Prehn JH. Mitochondrial depolarization is not required for neuronal apoptosis. J. Neurosci. 1999;19:7394–7404. doi: 10.1523/JNEUROSCI.19-17-07394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev. Biol. 2010;21:683–690. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005;11:1159–1161. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lozano J, Muñoz P, Nore BF, Ledoux S, Segura-Aguilar J. Stable expression of short interfering RNA for DT-diaphorase induces neurotoxicity. Chem. Res. Toxicol. 2010;23:1492–1496. doi: 10.1021/tx100182a. [DOI] [PubMed] [Google Scholar]
- Martin DN, Baehrecke EH. Caspases function in autophagic programmed cell death in Drosophila. Development. 2004;131:275–284. doi: 10.1242/dev.00933. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Kyoi S, Takagi H, Hsu CP, Hariharan N, Ago T, Vatner SF, Sadoshima J. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy. 2008;4:409–415. doi: 10.4161/auto.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J. Biol. Chem. 2005;280:21212–21219. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- Núñez R, Sancho-Martínez SM, Novoa JM, López-Hernández FJ. Apoptotic volume decrease as a geometric determinant for cell dismantling into apoptotic bodies. Cell Death Differ. 2010;17:1665–1671. doi: 10.1038/cdd.2010.96. [DOI] [PubMed] [Google Scholar]
- Okuyama R, Nguyen BC, Talora C, Ogawa E, Tommasi di Vignano A, Lioumi M, Chiorino G, Tagami H, Woo M, Dotto GP. High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev. Cell. 2004;6:551–562. doi: 10.1016/s1534-5807(04)00098-x. [DOI] [PubMed] [Google Scholar]
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164:541–551. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Paris I, Dagnino-Subiabre A, Marcelain K, Bennett LB, Caviedes P, Caviedes R, Olea-Azar C, Segura-Aguilar J. Copper neurotoxicity is dependent on dopamine-mediated copper uptake and one-electron reduction of aminochrome in a rat substantia nigra neuronal cell line. J. Neurochem. 2001;77:519–529. doi: 10.1046/j.1471-4159.2001.00243.x. [DOI] [PubMed] [Google Scholar]
- Paris I, Lozano J, Cardenas S, Perez-Pastene C, Saud K, Fuentes P, Caviedes P, Dagnino-Subiabre A, Raisman-Vozari R, Shimahara T, et al. The catecholaminergic RCSN-3 cell line: a model to study dopamine metabolism. Neurotox. Res. 2008;13:221–230. doi: 10.1007/BF03033505. [DOI] [PubMed] [Google Scholar]
- Paris I, Lozano J, Perez-Pastene C, Muñoz P, Segura-Aguilar J. Molecular and neurochemical mechanisms in PD pathogenesis. Neurotox. Res. 2009b;16:271–279. doi: 10.1007/s12640-009-9059-4. [DOI] [PubMed] [Google Scholar]
- Paris I, Martinez-Alvarado P, Cardenas S, Perez-Pastene C, Graumann R, Fuentes P, Olea-Azar C, Caviedes P, Segura-Aguilar J. Dopamine-dependent iron toxicity in cells derived from rat hypothalamus. Chem. Res. Toxicol. 2005b;18:415–419. doi: 10.1021/tx0497144. [DOI] [PubMed] [Google Scholar]
- Paris I, Martinez-Alvarado P, Perez-Pastene C, Vieira MN, Olea-Azar C, Raisman-Vozari R, Cardenas S, Graumann R, Caviedes P, Segura-Aguilar J. Monoamine transporter inhibitors and norepinephrine reduce dopamine-dependent iron toxicity in cells derived from the substantia nigra. J. Neurochem. 2005a;92:1021–1032. doi: 10.1111/j.1471-4159.2004.02931.x. [DOI] [PubMed] [Google Scholar]
- Paris I, Perez-Pastene C, Cardenas S, Iturriaga-Vasquez P, Muñoz P, Couve E, Caviedes P, Segura-Aguilar J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia-nigra-derived cell line. Neurotox. Res. 2010;18:82–92. doi: 10.1007/s12640-009-9148-4. [DOI] [PubMed] [Google Scholar]
- Paris I, Perez-Pastene C, Couve E, Caviedes P, Ledoux S, Segura-Aguilar J. Copper dopamine complex induces mitochondrial autophagy preceding caspase-independent apoptotic cell death. J. Biol. Chem. 2009a;284:13306–13315. doi: 10.1074/jbc.M900323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier M, Oliver L, Meflah K, Vallette FM. Caspase-3 can be pseudo-activated by a Ca2+-dependent proteolysis at a non-canonical site. FEBS Lett. 2005;579:2364–2368. doi: 10.1016/j.febslet.2005.02.079. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- Schultzberg M, Segura-Aguilar J, Lind C. Distribution of DT-diaphorase in the rat brain: biochemical and immunohistochemical studies. Neuroscience. 1988;27:763–766. doi: 10.1016/0306-4522(88)90181-9. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur. J. Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Brinchmann MF. Purification of autophagosomes from rat hepatocytes. Autophagy. 2010;6:542–547. doi: 10.4161/auto.6.4.11272. [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J, Diaz-Veliz G, Mora S, Herrera-Marschitz M. Inhibition of DT-diaphorase is a requirement for Mn(III) to produce a 6-OH-dopamine like rotational behaviour. Neurotox. Res. 2002;4:127–131. doi: 10.1080/10298420290015926. [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J, Lind C. On the mechanism of Mn3+ induced neurotoxicity of dopamine: prevention of quinone derived oxygen toxicity by DT-diaphorase and superoxide dismutase. Chem. Biol. Interact. 1989;72:309–324. doi: 10.1016/0009-2797(89)90006-9. [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J, Metodiewa D, Welch C. Metabolic activation of dopamine o-quinones to o-semiquinones by NADPH cytochrome P450 reductase may play an important role in oxidative stress and apoptotic effects. Biochim. Biophys. Acta. 1998;1381:1–6. doi: 10.1016/s0304-4165(98)00036-1. [DOI] [PubMed] [Google Scholar]
- Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7(5) doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- Tang Y, Jiang H, Nie D. The role of shorth-chain fatty acids in orchestrating two types of programmed cell death in colon cancer. Autophagy. 2011;7:235–237. doi: 10.4161/auto.7.2.14277. [DOI] [PubMed] [Google Scholar]
- Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010;648:193–214. doi: 10.1007/978-1-60761-756-3_13. [DOI] [PubMed] [Google Scholar]
- Tettamanti G, Saló E, González-Estévez C, Felix DA, Grimaldi A, de Eguileor M. Autophagy in invertebrates: insights into development, regeneration and body remodeling. Curr. Pharm. Des. 2008;14:116–125. doi: 10.2174/138161208783378716. [DOI] [PubMed] [Google Scholar]
- Tharakan B, Holder-Haynes JG, Hunter FA, Smythe WR, Childs EW. Cyclosporine A prevents vascular hyperpermeability after hemorrhagic shock by inhibiting apoptotic signaling. J. Trauma. 2009;66:1033–1039. doi: 10.1097/TA.0b013e31816c905f. [DOI] [PubMed] [Google Scholar]
- Van Laar VS, Mishizen AJ, Cascio M, Hastings TG. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol. Dis. 2009;34:487–500. doi: 10.1016/j.nbd.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008;283:23542–32356. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DC, Riegelsberger UM, Michalk S, Härtig W, Kranz A, Boltze J. Cleaved caspase-3 expression after experimental stroke exhibits different phenotypes and is predominantly non-apoptotic. Brain Res. 2011 doi: 10.1016/j.brainres.2011.01.041. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Whitehead RE, Ferrer JV, Javitch JA, Justice JB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J. Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Stokes AH, Roskoski R, Jr, Vrana KE. Dopamine, in the presence of tyrosinase, covalently modifies and inactivates tyrosine hydroxylase. J. Neurosci. Res. 1998;54:691–697. doi: 10.1002/(SICI)1097-4547(19981201)54:5<691::AID-JNR14>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Yagami T, Takase K, Yamamoto Y, Ueda K, Takasu N, Okamura N, Sakaeda T, Fujimoto M. Fibroblast growth factor 2 induces apoptosis in the early primary culture of rat cortical neurons. Exp. Cell Res. 2010;316:2278–2290. doi: 10.1016/j.yexcr.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Suzuki SO, Himeno M. The effects of dynein inhibition on the autophagic pathway in glioma cells. Neuropathology. 2010;30:1–6. doi: 10.1111/j.1440-1789.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, Nixon RA. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am. J. Pathol. 2008;173:665–681. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Bellei C, Costi P, Albertini A, Monzani E, Casella L, Gallorini M, Bergamaschi L, Moscatelli A, Turro NJ, et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc. Natl. Acad. Sci. U.S.A. 2008;105:17567–17572. doi: 10.1073/pnas.0808768105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Fariello R, Riederer P, Sulzer D, Gatti A, Tampellini D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002;510:216–220. doi: 10.1016/s0014-5793(01)03269-0. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Duan C, Lü L, Gao H, Zhao C, Yu S, Uéda K, Chan P, Yang H. α-Synuclein overexpression impairs mitochondrial function by associating with adenylate translocator. Int. J. Biochem. Cell Biol. 2011;43:732–741. doi: 10.1016/j.biocel.2011.01.014. [DOI] [PubMed] [Google Scholar]