

Abstract
Diabetes results in enhanced chemical modification of proteins by advanced lipoxidation end products (ALEs) and advanced glycation end products (AGEs) precursors. These modifications have been linked to the development of several secondary diabetic complications. Our previous studies showed that aldose reductase (AR; AKR1B3) catalyzes the reduction of ALEs and AGEs precursors; however, the in vivo significance of this metabolic pathway during diabetes and obesity has not been fully assessed. Therefore we examined the role of AR in regulating ALEs and AGEs formation in murine models of diet-induced obesity and streptozotocin-induced diabetes. In comparison with wild-type (WT) and AR-null mice fed normal chow, mice fed a high-fat (HF) diet (42% kcal fat) showed increased accumulation of AGEs and protein–acrolein adducts in the plasma. AGEs and acrolein adducts were also increased in the epididymal fat of WT and AR-null mice fed a HF diet. Deletion of AR increased the accumulation of 4-hydroxy-trans-2-nonenal (HNE) protein adduct in the plasma and increased the expression of the AGE receptor (RAGE) in HF fed mice. No change in AGEs formation was observed in the kidneys of HF-fed mice. In comparison, renal tissue from AR-null mice treated with streptozotocin showed greater AGE accumulation than streptozotocin-treated WT mice. These data indicated that AR regulated the accumulation of lipid peroxidation derived aldehydes and AGEs under conditions of severe, but not mild, hyperglycemia and that deletion of AR increased RAGE-induction via mechanisms that were independent of AGEs accumulation.
Keywords: Aldose reductase, Advanced glycation end products, Advanced lipoxidation end products, RAGE
1. Introduction
Post translational modification of lysine and arginine residues of proteins by reactive carbonyl species results in the formation of advanced glycation end products (AGEs) and advanced lipoxidation end products (ALEs). Protein modification due to the formation of AGEs or ALEs has been suggested to play a major role in exacerbating secondary diabetic complications [1,2]. AGEs and ALEs are formed by condensation reactions of the carbonyl group of sugar or reactive aldehydes. The N-terminus of the free amino acid of proteins forms a Schiff’s base that undergoes re-arrangement to generate Amadori adducts. AGEs and ALEs are generated normally as a result of aerobic metabolism and their tissue accumulation increases upon aging. The formation and the accumulation of AGEs and ALEs is further increased in conditions of diabetes and obesity because of an increase in the generation of several reactive carbonyl compounds such as methylglyoxal, deoxyglucosone, 4-hydroxy-trans-2-nonenal (HNE) and acrolein [3,4].
Previous studies have shown that interactions of AGEs and ALEs epitopes with the signal transduction receptor RAGE activates NADPH oxidase and increases the production of reactive oxygen species (ROS). This stimulates the phosphorylation of the extracellular signal-regulated kinase (ERK1/2) and results in the activation of the pro-inflammatory transcription factor – NF-κB [5–7]. Activation of the RAGE by AGEs and ALEs has been shown to induce inflammation and trigger pro-inflammatory events. It has been shown that binding of AGEs with RAGE promotes the progression of atherosclerosis in apoE null diabetic mice and that the deletion of RAGE attenuates the development of atherosclerotic lesion formation [8]. In addition, endothelial dysfunction associated with atherosclerosis has been attributed to the signaling mechanisms involving the interaction of AGEs with RAGE that result in the upregulation of VCAM1 and MMPs levels in aortic endothelial cells (ECs). In contrast the deletion of RAGE has been reported to decrease levels of VCAM1 and MMPs, thereby preventing tissue activation and adhesion [8]. Collectively these findings suggest that the interaction of AGEs and ALEs with RAGE plays an important role in eliciting immune responses and in causing tissue inflammation.
Studies from our laboratories have shown that several carbonyl products generated either from the oxidation of lipids, phospholipids or sugars are reduced by aldose reductase (AR). This enzyme has broad substrate specificity and recognizes short chain sugar derived carbonyls (glycoaldehyde, methylglyoxal), medium chain hydrophobic aldehydes (4-hydroxynonenal, hexenal) and phospholipid aldehydes (1-palmitoyl-2-oxo valeroyl phosphatidylcholine, POVPC). The enzyme also catalyzes the reduction of the glutathione conjugates of HNE and acrolein. Based on this substrate specificity, we proposed that the enzyme is involved in the metabolism and detoxification of AGE and ALE precursors and therefore it prevents tissue injury and inflammation associated with ALEs and AGEs accumulation. In agreement with this hypothesis we have found that haplo-insufficiency of these enzymes increases AGEs and ALEs formation in murine models of atherosclerosis and diabetes and promotes the formation of atherosclerotic lesions [9]. To further assess the role of AR in the reduction of AGEs precursors under different metabolic conditions and to determine whether the enzyme regulates the AGE–RAGE axis, we examined the AGEs and ALEs accumulation in two different models of diabetes. Our results show that AR regulates the formation of AGEs and ALEs in the plasma and the absence of the enzyme can induce RAGE expression.
2. Materials and methods
Monoclonal AGE and RAGE antibodies were purchased from Cosmo Bio Ltd. and R&D systems respectively. Polyclonal antibodies against KLH (Keyhole-Limpet Haemocyanin)–HNE (KLH–4-HNE) and KLH–acrolein were raised and tested as previously described [10,11].
2.1. High fat feeding and induction of diabetes in WT and AR null mice
The animals were maintained in a temperature-controlled room (22 °C) on a 12-h light-dark cycle. Mice were divided into two groups and were fed either a high-fat (HF) diet (Teklad Harlan) or received continuous feeding of a normal diet for up to 12 weeks. On caloric basis, the high-fat diet consisted of 42% fat from anhydrous milk fat, 42.7% carbohydrate, and 15.2% protein, whereas the normal diet contained 13.4% fat, 58.986% carbohydrate, and 27.55% protein. Food intake and body weights were taken once a week. Diabetes was induced in 6 weeks old male WT (n = 7) and akr1b3-null (n = 7) mice by repeated low dose streptozotocin (STZ; 65 mg/kg/day for 6 consecutive days, i.p.) treatment as described by Park et al. [12]. Control mice were treated with vehicle only (0.05 mM sodium citrate, pH 4.5). The study was approved by the Animal Ethics Committee at University of Louisville.
2.2. Western blotting for the quantification of AGEs ALEs and RAGE
For the identification and measurement of circulating AGEs and ALEs in the plasma of NC and HF fed WT and akr1b3-null mice plasma samples were separated by SDS–PAGE. AGEs, acrolein, HNE adducts and RAGE expression were identified by Western analysis using monoclonal anti-AGE and anti-RAGE, polyclonal anti-acrolein and anti-HNE antibodies, diluted 1:3000 and 1:1000 respectively in 5% (v/v) blocking solution in TBS at room temperature. Band intensity was quantified by using Image Quant TL software (Amersham Biosciences) and bands were normalized to Amido black staining.
2.3. Statistical analysis
Data are expressed as mean ± S.E.M. Student’s t-test was used for the data analysis when the data were restricted to two groups. Statistical significance was accepted at P < 0.05 level.
3. Results
3.1. High-fat feeding increases the generation of plasma AGEs acrolein and HNE adducts
To examine how diet-induced obesity regulates the formation of AGEs and ALEs, WT and AR-null mice were fed with Western diet (42% fat) and normal chow (NC) (14% fat) for 12 weeks. HF fed mice showed marked hyperglycemia, and the levels of plasma glucose measured after 12 weeks, were slightly higher in the AR-null than WT mice (data not shown). After 12 weeks on normal chow or high-fat, the mice were euthanized and the plasma was used to measure AGEs formation using Western analysis. As shown in Fig. 1, multiple AGE adducts were detected in the plasma of WT and AR-null mice that were fed normal chow. The intensity of most of the bands in the WT mice was similar to that in the AR-null mice, indicating that deletion of AR does not affect AGEs formation at baseline in mice fed normal chow. However, high-fat diet was associated with a significant increase in the intensity of several immunopositive bands. Quantification of clearly resolved bands at 25 kDa and 30 kDa showed that the overall intensity of these bands was higher in HF fed WT and AR-null mice when compared with NC fed WT or AR-null mice (Fig. 1A). Similarly the Western analysis of the plasma with anti-acrolein antibodies (Fig. 1B) showed that there was a 2–3-fold increase in the intensity of a protein–acrolein adduct (200 kDa band) in WT and AR-null mice that were fed a HF diet. Collectively, these observations suggest that high-fat diet increases AGEs and protein–acrolein adduct formation and that the abundance of these adducts in the plasma is not regulated by AR.
Fig. 1.

Increased accumulation of AGEs and acrolein adducts in the plasma of HF fed mice. Western blots of plasma from NC and HF fed WT and akr1b3-null mice were probed with (A) anti-AGEs and (B) anti-acrolein antibodies. Bar graphs show the intensity of indicated anti-AGEs and anti-acrolein positive bands normalized to Amido-Black stained blots. Data are presented as mean ± S.E.M. *P < 0.01 vs WT (NC).
In addition to acrolein, peroxidation of membrane lipids also increases the formation of HNE. To evaluate whether AR regulates the formation of protein–HNE adducts in HF-fed mice, we measured the abundance of these adducts in the plasma of WT and AR-null mice fed NC or HF-diet. The most prominent protein–HNE adduct in the plasma corresponded to a molecular weight of 25 kDa (Fig. 2). The levels of this adduct in the plasma of NC-fed AR-null mice were slightly higher than that in the NC fed WT mice, however, this difference was not statistically significant. Feeding HF diet led to a small, but statistically insignificant increase in the protein–HNE adducts in WT mice, however, significantly greater levels of these adducts were detected in the plasma of HF-fed AR-null mice. These observations suggest that deletion of AR results in a greater accumulation of protein–HNE adducts in the plasma of HF-fed mice.
Fig. 2.
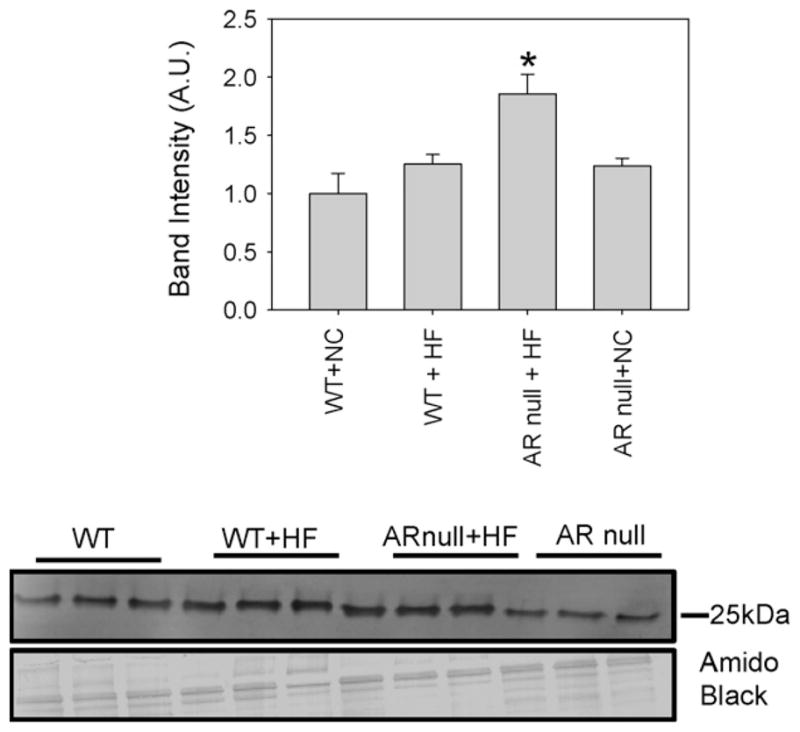
HNE adduct formation is enhanced in the plasma of akr1b3-null mice. Western blots of plasma from NC and HF fed mice from WT and akr1b3-null mice were probed with anti-HNE antibodies. NC fed WT and akr1b3-null plasma served as respective controls. Western blots were developed using anti-HNE antibodies. Bar graphs show the intensity of the indicated anti-HNE normalized to Amido Black stains. Data are presented as mean ± S.E.M. *P < 0.01 vs WT NC and HF fed mice.
3.2. Increased accumulation of ALEs and AGEs in epididymal fat of HF fed mice
To assess whether in addition to plasma, AGEs and ALEs also accumulate in tissues, we measured the levels of these adducts in the epididymal fat. Western analysis of epipidymal fat from HF-fed WT and AR-null mice showed an increased accumulation of acrolein adducts (200 kDa) compared with the NC fed WT mice (Fig. 3A). Similarly, HF feeding increased the formation of AGEs in both the WT and AR-null mice with higher intensity of bands at 200 kDa and 150 kDA bands in the epididymal fat analyzed with anti-AGE antibodies (Fig. 3B). These observations indicate that HF-feeding increases the abundance of AGEs and protein–acrolein adducts in fat tissue, however, the levels of these adducts were not affected by the absence of AR.
Fig. 3.

AGEs and acrolein adduct formation is increased in the epididymal fat of HF fed mice. Cytosolic fractions of epididymal fat from WT and akr1b3-null HF and NC fed mice were probed with (A) anti-AGE antibody to identify AGEs and (B) anti-acrolein antibody. The membranes were visualized by chemiluminescence. Western blots were normalized by Amido Black stains *P < 0.05 compared with WT (NC).
3.3. AR regulates AGEs formation in diabetic kidneys
Although HF-fed mice are useful model of human metabolic syndrome, the resultant phenotype is rather mild. Hence to examine the role of AR under more severe conditions of hyperglycemia, we studied changes in AGEs and ALEs formation in low dose STZ-treated mice. Treatment with multiple low doses of STZ results in profound hyperglycemia and dyslipidemia. Our previous studies [9] show that both WT and AR-null mice respond similarly although the AR-null mice show slightly higher levels of blood glucose (744 ± 55 mg/dl) than the WT mice (566 ± 30 mg/ml). Similarly, levels of total cholesterol were higher in the AR-null mice (131 ± 7 mg/dl) than those in WT mice (84 ± 5 mg/dl). These observations suggest that deletion of AR exacerbates diabetes-induced hyperglycemia and hyperlipidemia.
To examine whether AR regulates the formation of AGEs and ALEs in diabetic tissue, we examined the abundance of these adducts in the kidney. In renal tissue, the uptake of glucose is not regulated by insulin and therefore, plasma hyperglycemia results in an increase in the intracellular concentration of glucose. As a result, kidney is the major target of hyperglycemia, and nephropathy is a leading cause of death in type 1 diabetics. Measurement of AGEs in the kidney showed detectable levels of these adducts in WT non-diabetic mice. Interestingly, significantly higher levels of AGEs were observed in kidneys from AR-null mice. As shown in Fig. 4, there were significant increases in AGEs with masses of 30 and 40 kDa. These observations suggest that even in the absence of diabetes or hyperglycemia, AGEs formation in kidneys is regulated by AR. In comparison with vehicle-treated mice, higher AGE levels were detected in kidneys of STZ-treated mice. Moreover, the abundance of AGEs was much higher in diabetic AR-null mice. No significant difference in the expression of AR was observed between diabetic and non-diabetic mice (Fig. 4). In addition, there was no significant difference in AGE accumulation in the kidneys of WT and AR-null mice fed a HF-diet (data not shown).
Fig. 4.

Increase of AGEs in diabetic akr1b3-null kidneys. Cytosolic fractions from kidney homogenates of WT and akr1b3-null non-diabetic and diabetic mice were probed with anti-AGE antibody to identify modified proteins. Lower panels represent Western blots for aldose reductase. The membranes were visualized by chemiluminescence. Intensity of the AGEs was normalized by normalizing the bands to actin loading. *P < 0.05 vs WT and #P < 0.05 vs WT diabetic.
3.4. Deletion of AR increases the RAGE expression
Although the formation of covalent adducts with aldehydes and AGE precursors could directly impair protein function, AGEs and ALEs also serve ligands of the pattern-recognition receptor RAGE. Hence, we evaluated whether an increase in the formation of AGEs and ALEs during diet-induced obesity results in an up regulation of RAGE and whether this is regulated by AR. As shown in Fig. 5, only low basal levels of the RAGE protein were expressed in the hearts of mice fed a normal chow diet. In comparison hearts from AR-null mice showed significantly greater expression of RAGE, indicating that even in healthy non-diabetic mice, deletion of AR increases RAGE expression. Mice fed a HF diet showed higher levels of RAGE than those fed normal chow (Fig. 5); however, RAGE induction by HF diet was greater in AR-null mice. The extent of induction by high fat feeding in AR null was lower compared to WT high fat fed mice due to the basal expression of RAGE. From these data we conclude that lack of AR upregulates RAGE expression. Because in comparison with WT mice AR-null mice do not show excessive accumulation of AGEs, these results indicate that the increase in RAGE in AR-null mice is not secondary to AGEs accumulation.
Fig. 5.
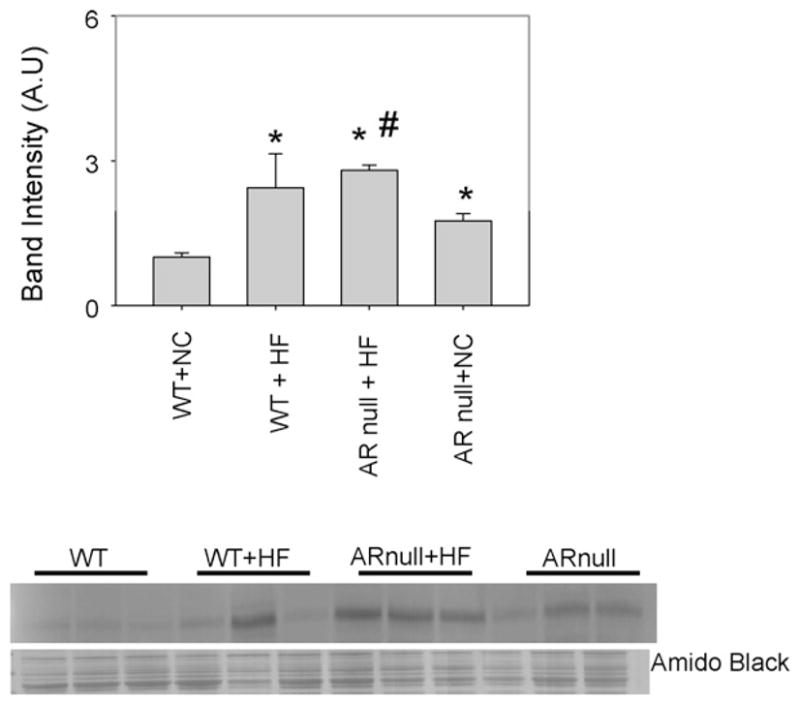
Genetic ablation of akr1b3 increases RAGE expression in HF fed mice. Cytosolic fractions of heart homogenates from NC and HF fed WT and akr1b3-null mice were immuno-blotted with anti-RAGE antibody. Bar graphs show the intensity of indicated anti-RAGE positive bands normalized to Amido-Black stained blots. *P < 0.05 vs WT NC and #P < 0.05 vs WTHF.
4. Discussion
The major findings of current study are that diet-induced obesity increases tissue and plasma accumulation of AGEs and ALEs. Significant increases were observed in AGEs, protein–acrolein and protein–HNE adducts suggesting that obesity is associated with an increase in the formation of lipid peroxidation-derived aldehydes as well as AGE precursors. The metabolism of these carbonyls in HF-fed mice was not significantly affected by the presence of AR; however, deletion of AR increased AGE formation during severe hyperglycemia induced by STZ. Moreover, deletion of AR resulted in an increase in RAGE expression. Because the increase in RAGE expression was observed under conditions in which the formation of protein–HNE, but not AGEs, was increased by deletion of AR, we speculate that the expression of RAGE is regulated by oxidative stress and protein–HNE formation in AR-null mice.
Several previous studies show that the metabolites generated from glucose oxidation form covalent adducts with plasma and tissue proteins (AGEs) [13]. The accumulation of AGEs is increased during aging and AGEs formation has been linked to protein cross-linkage in senescent tissue [14]. Increased accumulation of AGEs has also been reported in diseased tissues as well as plasma proteins [15–19]. AGE protein adducts have been detected in atherosclerotic lesions of euglycemic LDL receptor-deficient rabbits [20], suggesting that increased accumulation of AGEs may be indicative of tissue inflammation and lipid oxidation. The accumulation of AGEs is further increased in diabetes and high levels of AGE-modified protein have been detected in the circulation and tissues from diabetic subjects [15–19]. However, the current study shows for the first time, excessive accumulation of AGEs in the plasma and tissue of obese mice. This observation is consistent with the notion that diet-induced obesity is a pro-inflammatory state [21] that results in the activation of inflammatory signaling due to JNK1 and IKKβ [22] and an increase in plasma cytokine production due in part to the activation of the TLR4 receptors [23,24]. In addition to AGEs, we also observed in the formation of protein–acrolein adducts. Acrolein is generated endogenously as a product of lipid oxidation [25] or by myeloperoxidase-catalyzed threonine oxidation [26]. Increased accumulation of protein–acrolein adducts have been detected in the oxidized lipoproteins and atherosclerotic lesions of mice and humans [27,28]. Our current observation that both protein–acrolein and protein–AGE adducts was increased in high-fat fed mice suggesting that both lipid peroxidation and glucose oxidation were increased during obesity. In vitro and in vivo analyses of oxidized LDL suggest that there is a direct relationship between the levels of AGEs and lipid oxidation products [16]. The formation of lipid–AGE adducts in LDL has been shown to increase the oxidation of unsaturated lipids, demonstrating a close association between lipid peroxidation and AGE accumulation [16]. Hence, the contemporaneous increase in both AGEs and lipid peroxidation products in tissue to obese mice indicate an on-going and interlinked process of oxidation and glycosylation.
Protein-linked AGEs are generated by the addition of non-enzymatic oxidation products of sugars such as glucose and glucose-generated metabolites such as methylglyoxal and glyoxal to primary amino groups of proteins. Our studies show that aldose reductase (AR) is an efficient catalyst for the reduction of AGE precursors including methylglyoxal, glyoxal, furfural, 3-hydoxyfurfural and deoxyglucosone and that the reduction of AGE precursors is diminished in AR-null mice. Moreover, diabetic AR-null mice that are devoid of reductive metabolism in different tissues accumulate more AGEs in the plasma and the hearts than WT mice and deletion of AR increases AGE accumulation and atherosclerotic lesion formation in apoE-null mice [9]. In agreement with these observations we found that deletion of AR increased the AGEs accumulation in kidneys of STZ-treated mice [9]. These observations suggest that AR plays an important role in removing AGE precursors in diabetic kidneys and are in contrast to several previous reports that showed that inhibition of AR prevents early diabetic nephropathy in rats. In STZ-treated rats, treatment with AR inhibitors has been shown to prevent diabetes-induced increase in vascular permeability in the kidney [29], urinary albumin excretion [30], and single nephron filtration rate [31]. However, most of the protective effects of AR inhibition were apparent during early stages of diabetes. AR inhibitors administered from the time of induction of diabetes [32] decrease urinary albumin excretion and early changes in glomerular hyper-filteration in diabetic rats for 6–12 weeks [33–35], but no protection was observed on long-term (>6 months) follow-up [36,37]. Moreover, a 5-year administration of AR inhibitor in diabetic dogs did not produce any beneficial effects on renal structure or albuminuria [38]. Similarly clinical trials with AR inhibitors show only marginal protection against diabetic nephropathy in patients with diabetic nephropathy [39,40]. Our current data showing that deletion of the AR gene increases AGEs accumulation in diabetic kidney provide one explanation for the mixed results obtained with AR inhibitors. These data suggest that even though inhibition of AR may be protective against early glomerular changes in diabetes, in the long run, inhibition of AR could exacerbate renal injury by promoting AGEs accumulation; hence the early protection provided by AR inhibitors could be eroded by gradual accumulation of AGEs and ALEs. It is now recognized that AR is a multifunctional enzyme that catalyzes the reduction of a range of AGE precursors [9] and lipid peroxidation products [41] and therefore inhibition of the enzyme could prevent the removal of these potentially toxic species. This view is consistent with the observation that transgenic overexpression of AR in the kidney paradoxically prevents diabetes-induced albuminuria [42] and decreases the accumulation of oxidative stress-induced dicarbonyls in renal tubules [43]. Hence, we propose that long-term inhibition of AR is unlikely to prevent the development of renal disease in diabetes because it promotes the accumulation of lipid peroxidation products and AGEs.
AGEs generated in diabetic or senescent tissue could induce tissue dysfunction by modifying structural or cytoskeletal proteins or proteins involved in metabolism and cell signaling. In addition AGEs could also trigger inflammation by binding to RAGE. RAGE is a surface receptor that belongs to the immunoglobulin superfamily of proteins [44]. Binding of AGEs to RAGE leads to the activation of signal transduction pathway that result in an increase in ROS production and NF-κB activation. Together these events increase cytokine production and establish a pro-inflammatory state. In addition to endogenously generated AGEs, AGEs present in the diet, food-derived AGEs such as pronyl-glycine are also RAGE ligands [45]. RAGE is widely expressed in many tissues [44] and its expression is further increased in diabetes [46]. Pharmacological inhibition of RAGE or treatment with soluble RAGE prevents diabetes-induced acceleration of atherogenesis in apoE-null mice [12]. Similar deletion of RAGE prevents structural and functional changes associated with diabetic nephropathy in mouse models of type 1 diabetes [47], and diabetic RAGE-null mice are protected against albuminuria, hyperfiltration, and glomerulosclerosis [48]. Our observation that HF-diet increases the expression of RAGE suggests that part of the inflammation induced by a high-fat diet may be mediated by the activation of RAGE. That there was a concurrent increase in AGEs in tissues of obese mice is consistent with the notion. Since both AGEs and RAGE are increased in obesity the AGE–RAGE axis could be an important component of the sequealae of pro-inflammatory events associated with obesity. Further investigations are required to examine the role of AGEs and RAGE in obesity, however, our current studies show that deletion of AR led to an increase in the expression of RAGE in mice-fed normal chow and upon feeding a high-fat diet, the RAGE expression in AR-null mice was significantly greater than in WT mice (Fig. 5). These results identify AR as a new regulator of RAGE expression and suggest the presence of significant cross-talk between AGEs and the polyol pathway. While the mechanism by which AR deficiency upregulates RAGE remains unknown, we speculate that this may be in response to an increase in the accumulation of lipid peroxidation products. While feeding a high-fat diet led to an increase in AGEs, there was no difference in AGE levels in WT and AR-null mice, suggesting that it is unlikely that RAGE expression is increased in response to an increase in AGEs. Previous work has shown that in addition to AGEs, other ligands also bind to RAGE and that RAGE expression is increased under conditions of inflammation and tissue injury. Clearly, much additional work is required to identify the mechanisms that regulate RAGE expression and how it may be regulated or dependent upon AR.
In summary, the results of this study show that diet-induced obesity is associated with an increase in the accumulation of AGEs and ALEs. The accumulation of ALEs, but not AGEs in AR-null mice fed a high-fat diet and that deletion of AR increases AGEs accumulation in diabetic kidney. We also found that AR-null mice express higher RAGE levels than their WT counterparts and that deletion of AR increases high-fed-induced RAGE up regulation. These results reveal a new relationship between the AGE–RAGE axis and AR and suggest that RAGE-dependent inflammation may be regulated in part by AR. Further studies are required to understand this relationship and to assess its impact on secondary complications of diabetes.
Acknowledgments
Travel for BSP was supported by USPHS NIH grant R13–AA019612 to present this work at the 15th International Meeting on Enzymology and Molecular Biology of Carbonyl Metabolism in Lexington, KY, USA.
Abbreviations
- AGE
advanced glycation end products
- ALE
advanced lipoxidation end products
- AR
aldose reductase
- HF
high fat
- HNE
4-hydroxy-trans-2-nonenal
- NC
normal chow
- RAGE
receptor for advanced glycation end products
- ROS
reactive oxygen species
- STZ
streptozotocin
Footnotes
Conflict of interest
None.
References
- 1.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 2.Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia. 2001;44(2):129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 3.Horie K, Miyata T, Maeda K, Miyata S, Sugiyama S, Sakai H, van Ypersole de Strihou C, Monnier VM, Witztum JL, Kurokawa K. Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions. Implication for glycoxidative stress in the pathogenesis of diabetic nephropathy. J Clin Invest. 1997;100(12):2995–3004. doi: 10.1172/JCI119853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest. 1997;99(3):457–468. doi: 10.1172/JCI119180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li JH, Wang W, Huang XR, Oldfield M, Schmidt AM, Cooper ME, Lan HY. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol. 2004;164(4):1389–1397. doi: 10.1016/S0002-9440(10)63225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med. 2009;87(3):235–247. doi: 10.1007/s00109-009-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280(5):E685–694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 8.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118(1):183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baba SP, Barski OA, Ahmed Y, O’Toole TE, Conklin DJ, Bhatnagar A, Srivastava S. Reductive metabolism of AGE precursors: a metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes. 2009;58(11):2486–2497. doi: 10.2337/db09-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S, Ramana KV, Tammali R, Srivastava SK, Bhatnagar A. Contribution of aldose reductase to diabetic hyperproliferation of vascular smooth muscle cells. Diabetes. 2006;55(4):901–910. doi: 10.2337/diabetes.55.04.06.db05-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conklin DJ, Haberzettl P, Prough RA, Bhatnagar A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am J Physiol Heart Circ Physiol. 2009;296(5):H1586–1597. doi: 10.1152/ajpheart.00867.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4(9):1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 13.Friedman EA. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care. 1999;22(Suppl 2):B65–71. [PubMed] [Google Scholar]
- 14.Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem. 1989;264(36):21597–21602. [PubMed] [Google Scholar]
- 15.Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, Friedman EA, Cerami A, Vlassara H. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325(12):836–842. doi: 10.1056/NEJM199109193251202. [DOI] [PubMed] [Google Scholar]
- 16.Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci USA. 1993;90(14):6434–6438. doi: 10.1073/pnas.90.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makita Z, Vlassara H, Cerami A, Bucala R. Immunochemical detection of advanced glycosylation end products in vivo. J Biol Chem. 1992;267(8):5133–5138. [PubMed] [Google Scholar]
- 18.Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci USA. 1991;88(24):11555–11558. doi: 10.1073/pnas.88.24.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayase F, Nagaraj RH, Miyata S, Njoroge FG, Monnier VM. Aging of proteins: immunological detection of a glucose-derived pyrrole formed during maillard reaction in vivo. J Biol Chem. 1989;264(7):3758–3764. [PubMed] [Google Scholar]
- 20.Palinski W, Koschinsky T, Butler SW, Miller E, Vlassara H, Cerami A, Witztum JL. Immunological evidence for the presence of advanced glycosylation end products in atherosclerotic lesions of euglycemic rabbits. Arterioscler Thromb Vasc Biol. 1995;15(5):571–582. doi: 10.1161/01.atv.15.5.571. [DOI] [PubMed] [Google Scholar]
- 21.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107(5):579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- 22.Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24(8):2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 23.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100(11):1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 25.Uchida K. Current status of acrolein as a lipid peroxidation product. Trends Cardiovasc Med. 1999;9(5):109–113. doi: 10.1016/s1050-1738(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 26.Anderson MM, Hazen SL, Hsu FF, Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive alpha-hydroxy and alpha,beta-unsaturated aldehydes by phagocytes at sites of inflammation. J Clin Invest. 1997;99(3):424–432. doi: 10.1172/JCI119176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato R, Mori C, Kitazato K, Arata S, Obama T, Mori M, Takahashi K, Aiuchi T, Takano T, Itabe H. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29(1):33–39. doi: 10.1161/ATVBAHA.108.164723. [DOI] [PubMed] [Google Scholar]
- 28.Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005;280(43):36386–36396. doi: 10.1074/jbc.M508169200. [DOI] [PubMed] [Google Scholar]
- 29.Williamson JR, Chang K, Tilton RG, Prater C, Jeffrey JR, Weigel C, Sherman WR, Eades DM, Kilo C. Increased vascular permeability in spontaneously diabetic BB/W rats and in rats with mild versus severe streptozocin-induced diabetes. Prevention by aldose reductase inhibitors and castration. Diabetes. 1987;36(7):813–821. doi: 10.2337/diab.36.7.813. [DOI] [PubMed] [Google Scholar]
- 30.McCaleb ML, Sredy J, Millen J, Ackerman DM, Dvornik D. Prevention of urinary albumin excretion in 6 month streptozocin-diabetic rats with the aldose reductase inhibitor tolrestat. J Diabetes Complicat. 1988;2(1):16–18. doi: 10.1016/0891-6632(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 31.Bank N, Mower P, Aynedjian HS, Wilkes BM, Silverman S. Sorbinil prevents glomerular hyperperfusion in diabetic rats. Am J Physiol. 1989;256(6 Pt 2):F1000–1006. doi: 10.1152/ajprenal.1989.256.6.F1000. [DOI] [PubMed] [Google Scholar]
- 32.Chang WP, Dimitriadis E, Allen T, Dunlop ME, Cooper M, Larkins RG. The effect of aldose reductase inhibitors on glomerular prostaglandin production and urinary albumin excretion in experimental diabetes mellitus. Diabetologia. 1991;34(4):225–231. doi: 10.1007/BF00405080. [DOI] [PubMed] [Google Scholar]
- 33.Tilton RG, Chang K, Pugliese G, Eades DM, Province MA, Sherman WR, Kilo C, Williamson JR. Prevention of hemodynamic and vascular albumin filtration changes in diabetic rats by aldose reductase inhibitors. Diabetes. 1989;38(10):1258–1270. doi: 10.2337/diab.38.10.1258. [DOI] [PubMed] [Google Scholar]
- 34.Goldfarb S, Ziyadeh FN, Kern EF, Simmons DA. Effects of polyol-pathway inhibition and dietary myo-inositol on glomerular hemodynamic function in experimental diabetes mellitus in rats. Diabetes. 1991;40(4):465–471. doi: 10.2337/diab.40.4.465. [DOI] [PubMed] [Google Scholar]
- 35.Donnelly SM, Zhou XP, Huang JT, Whiteside CI. Prevention of early glomerulopathy with tolrestat in the streptozotocin-induced diabetic rat. Biochem Cell Biol. 1996;74(3):355–362. doi: 10.1139/o96-038. [DOI] [PubMed] [Google Scholar]
- 36.Mauer SM, Steffes MW, Azar S, Brown DM. Effects of sorbinil on glomerular structure and function in long-term-diabetic rats. Diabetes. 1989;38(7):839–846. doi: 10.2337/diab.38.7.839. [DOI] [PubMed] [Google Scholar]
- 37.Reddi AS, Jyothirmayi GN. Aldose reductase inhibition by ponalrestat (statil) does not prevent proteinuria in long-term diabetic rats. J Diabetes Complicat. 1993;7(4):233–240. [PubMed] [Google Scholar]
- 38.Kern TS, Engerman RL. Aldose reductase and the development of renal disease in diabetic dogs. J Diabetes Complicat. 1999;13(1):10–16. doi: 10.1016/s1056-8727(98)00015-4. [DOI] [PubMed] [Google Scholar]
- 39.Passariello N, Sepe J, Marrazzo G, De Cicco A, Peluso A, Pisano MC, Sgambato S, Tesauro P, D’Onofrio F. Effect of aldose reductase inhibitor (tolrestat) on urinary albumin excretion rate and glomerular filtration rate in IDDM subjects with nephropathy. Diabetes Care. 1993;16(5):789–795. doi: 10.2337/diacare.16.5.789. [DOI] [PubMed] [Google Scholar]
- 40.Pedersen MM, Christiansen JS, Mogensen CE. Reduction of glomerular hyperfiltration in normoalbuminuric IDDM patients by 6 mo of aldose reductase inhibition. Diabetes. 1991;40(5):527–531. doi: 10.2337/diab.40.5.527. [DOI] [PubMed] [Google Scholar]
- 41.Srivastava S, Harter TM, Chandra A, Bhatnagar A, Srivastava SK, Petrash JM. Kinetic studies of FR-1, a growth factor-inducible aldo-keto reductase. Biochemistry. 1998;37(37):12909–12917. doi: 10.1021/bi9804333. [DOI] [PubMed] [Google Scholar]
- 42.Ng DP, Hardy CL, Burns WC, Muggli EE, Kerr N, McCausland J, Alcorn D, Adams TE, Zajac JD, Larkins RG, Dunlop ME. Prevention of diabetes-induced albuminuria in transgenic rats overexpressing human aldose reductase. Endocrine. 2002;18(1):47–56. doi: 10.1385/ENDO:18:1:47. [DOI] [PubMed] [Google Scholar]
- 43.Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S3–12. doi: 10.1046/j.1523-1755.2000.07702.x. [DOI] [PubMed] [Google Scholar]
- 44.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106(5):842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Somoza V, Lindenmeier M, Hofmann T, Frank O, Erbersdobler HF, Baynes JW, Thorpe SR, Heidland A, Zill H, Bek S, Huber J, Weigle T, Scheidler S, Busch AE, Sebekova K. Dietary bread crust advanced glycation end products bind to the receptor for AGEs in HEK-293 kidney cells but are rapidly excreted after oral administration to healthy and subtotally nephrectomized rats. Ann NY Acad Sci. 2005;1043:492–500. doi: 10.1196/annals.1333.056. [DOI] [PubMed] [Google Scholar]
- 46.Kislinger T, Tanji N, Wendt T, Qu W, Lu Y, Ferran LJ, Jr, Taguchi A, Olson K, Bucciarelli L, Goova M, Hofmann MA, Cataldegirmen G, D’Agati V, Pischetsrieder M, Stern DM, Schmidt AM. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2001;21(6):905–910. doi: 10.1161/01.atv.21.6.905. [DOI] [PubMed] [Google Scholar]
- 47.Reiniger N, Lau K, McCalla D, Eby B, Cheng B, Lu Y, Qu W, Quadri N, Ananthakrishnan R, Furmansky M, Rosario R, Song F, Rai V, Weinberg A, Friedman R, Ramasamy R, D’Agati V, Schmidt AM. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes. 2010;59(8):2043–2054. doi: 10.2337/db09-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan AL, Sourris KC, Harcourt BE, Thallas-Bonke V, Penfold S, Andrikopoulos S, Thomas MC, O’Brien RC, Bierhaus A, Cooper ME, Forbes JM, Coughlan MT. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2010;298(3):F763–770. doi: 10.1152/ajprenal.00591.2009. [DOI] [PubMed] [Google Scholar]