

Yck2, like many palmitoylation substrate proteins, lacks hydrophobicity for targeting to membranes and thus to its Golgi-localized palmitoyl-transferase. Perhaps accommodating this targeting need, the Yck2 palmitoylation signal is found to be large and complex, consisting of domains local to, and distant from, the modification site cysteines.
Abstract
The yeast kinase Yck2 tethers to the cytoplasmic surface of the plasma membrane through dual palmitoylation of its C-terminal Cys-Cys dipeptide, mediated by the Golgi-localized palmitoyl-transferase Akr1. Here, the Yck2 palmitoylation signal is found to consist of three parts: 1) a 10-residue-long, conserved C-terminal peptide (CCTP) that includes the C-terminal Cys-Cys dipeptide; 2) the kinase catalytic domain (KD); and mapping between these two elements; and 3) a 176-residue-long, poorly conserved, glutamine-rich sequence. The CCTP, which contains the C-terminal cysteines as well as an important Phe-Phe dipeptide, likely serves as an Akr1 recognition element, because CCTP mutations disrupt palmitoylation within a purified in vitro palmitoylation system. The KD contribution appears to be complex with roles for both KD activity (e.g., Yck2-mediated phosphorylation) and structure (e.g., Akr1 recognition elements). KD and CCTP mutations are strongly synergistic, suggesting that, like the CCTP, the KD may also participate at the Yck2-Akr1 recognition step. The long, glutamine-rich domain, which is located between the KD and CCTP, is predicted to be intrinsically disordered and may function as a flexible, interdomain linker, allowing a coupled interaction of the KD and CCTP with Akr1. Multipart palmitoylation signals may prove to be a general feature of this large class of palmitoylation substrates. These soluble proteins have no clear means of accessing membranes and thus may require active capture out of the cytoplasm for palmitoylation by their membrane-localized transferases.
INTRODUCTION
Protein palmitoylation is one of three different lipid modifications of proteins that function to tether proteins to cytosolic membrane surfaces (for palmitoylation reviews, see Smotrys and Linder, 2004; Conibear and Davis, 2010; Fukata and Fukata, 2010; Salaun et al., 2010). The other two lipid modifications are myristoylation and prenylation. Protein palmitoylation is the attachment of fatty acyl moieties (often, the saturated, 16-carbon palmitate) in thioester linkage to select protein cysteines. Palmitoylation has been documented for a broad array of proteins, including many membrane-tethered signaling proteins; well-known examples include H- and N-Ras, the Gα components of many heterotrimeric G proteins, endothelial nitric oxide synthase, nonreceptor tyrosine kinases like Lck and Fyn, and the synaptic scaffolding protein PSD95. Palmitoylation also is frequently found to modify many transmembrane proteins; examples include surface signaling receptors, integrin-like proteins that mediate cell-cell interaction, as well as transporters and channels. For such transmembrane proteins, which embed in membranes through hydrophobic transmembrane domains (TMDs), the palmitoylation-tethering function seems superfluous, and thus roles for the modification either in altering the protein's transmembrane architecture or in modulating in-bilayer segregation to membrane microdomains (e.g., lipid rafts and caveolae) often are invoked. Perhaps the most intriguing palmitoylation characteristic is its potential reversibility. Obviously, the possibility of both adding and removing lipid tethers provides a powerful means of regulating protein localization. Such regulation, however, has been documented to date only for a relatively small collection of palmitoyl proteins (Conibear and Davis, 2010).
A variety of obstacles have slowed research on palmitoylation. Its occurrence within a wide variety of peptidyl contexts has precluded the development of robust consensuses, making prediction problematic. Furthermore, it is only in recent years that the enzymology and cell biology of both palmitoylation and depalmitoylation have begun to emerge. The first palmitoylation enzymes (i.e., the protein acyl-transferases [PATs]), were discovered only eight years ago through work in yeast (Lobo et al., 2002; Roth et al., 2002). The PATs are multipass transmembrane proteins with four to six predicted TMDs and their defining, 50-residue-long, zinc finger–like DHHC cysteine-rich domain (named for its core Asp-His-His-Cys tetrapeptide), the putative active site, is displayed within a cytoplasmically oriented inter-TMD loop (Politis et al., 2005). Yeast has seven DHHC PATs, and mammals have 23. Beyond their DHHC cysteine-rich domains, the DHHC PATs show remarkably little sequence conservation with one another. This enzymatic diversity likely evolved to support the extensive diversity of proteins that undergo palmitoylation, and the individual PATs do indeed show both distinct and overlapping substrate specificities (Fukata et al., 2004; Roth et al., 2006; Huang et al., 2009).
Because the PATs are membrane proteins, the palmitoylation substrate proteins need some means of accessing the cell's endomembrane system. Palmitoyl proteins can be roughly divided into three classes: 1) transmembrane proteins; 2) proteins, lipidated by either myristoylation or prenylation, in addition to palmitoylation; and 3) hydrophilic proteins that rely exclusively on palmitoylation for membrane interaction. For the first two categories, there are obvious mechanisms that are expected to lead the substrate protein to membranes. Transmembrane proteins, of course, are stitched into the endoplasmic reticulum (ER) membrane cotranslationally by the ER translocation apparatus. Presumably, their search for their cognate PAT occurs as part of their subsequent trafficking through the cellular membrane network. For proteins dually lipidated by either N-terminal myristoylation or C-terminal prenylation and then palmitoylation, interaction with membranes presumably is provoked following addition of the primary lipid modification (i.e., either the myristoylation or the prenylation), which is added either during, or soon after, the biosynthesis of the substrate protein. For the third class of palmitoyl proteins, i.e., the hydrophilic proteins that associate with membranes solely through their palmitoyl modifications, the mechanism by which access to their membrane-localized PAT is gained remains unclear. The work in this article focuses on one example of this third class, the yeast plasma membrane–localized, type 1 casein kinase (CK1) Yck2.
Yck2, together with its close homologue Yck1, participates in numerous cell surface processes, including endocytosis, cell morphogenesis, mRNA localization, and nutrient sensing (Robinson et al., 1993; Panek et al., 1997; Hicke et al., 1998; Feng and Davis, 2000; Abdel-Sater et al., 2004; Moriya and Johnston, 2004; Spielewoy et al., 2004; Pal et al., 2008; Toshima et al., 2009; Suchkov et al., 2010). Together, the two kinases provide yeast with an essential function (yck1Δ yck2Δ cells are inviable). Yck1 and Yck2 both are dually palmitoylated on C-terminal Cys-Cys dipeptides with palmitoylation being mediated by the Golgi-localized DHHC PAT Akr1 (Roth et al., 2002, 2006; Babu et al., 2004). The orthologous mammalian kinases, CK1γ1, -γ2, and -γ3, which play a critical role in Wnt signaling (Davidson et al., 2005), also likely are palmitoylated as these were detected in a large-scale proteomic analysis of protein palmitoylation in neurons (Kang et al., 2008); the likely palmitoyl acceptors for these mammalian CK1s are clusters of two to three cysteines that map near the C terminus.
The analysis shown here is a mutational dissection of the Yck2 palmitoylation signal. Like the signals for other posttranslational modifications, we expected that the Yck2 palmitoylation signal would map to the local sequence context of the modification site residues, the C-terminal Cys-Cys. Prior work on Yck2 identified a relatively large C-terminal domain (CTD), the C-terminal 47 residues as being the minimal domain that is sufficient to specify palmitoylation (Babu et al., 2002, 2004). While confirming this finding, we find substantial additional complexity, uncovering a novel role for the kinase catalytic domain (KD) in palmitoylation. Overall, our results support a tripartite model for the Yck2 palmitoylation signal, in which two of the three parts—a conserved, 10-residue-long sequence that includes the Cys-Cys palmitoyl acceptors and the Yck2 kinase domain—are likely to represent Akr1 recognition elements. The third essential part—a long, poorly conserved domain that is predicted to be intrinsically disordered—may act by flexibly accommodating the concomitant interaction of the two recognition elements to separate domains of the Akr1 PAT.
RESULTS
Yck2 structure
Based on the structures of the KDs of Schizosaccharomyces pombe Cki1 and rat CK1δ (Xu et al., 1995; Longenecker et al., 1996), the Yck2 KD can be precisely assigned to residues 72–360 (Figure 1). Beyond the KD, Yck2 has relatively large N- and C-terminal domains of 70 and 187 residues, respectively. The CTD, which terminates with the Cys-Cys palmitoylation acceptors, is quite poorly conserved overall, but does contain three segments of homology with Yck1, these being the 36 residues immediately downstream of the KD (residues 361–396), plus two short homology islands at residues 465–474 and at the Yck2 C terminus, residues 537–546. The Yck2 CTD also notably contains two relatively long intervals of low-complexity, glutamine-rich sequence (Figure 1). Secondary structure predictions for Yck2 find that the KD is replete with sequences that are predicted to adopt α-helical or β-sheet secondary structures as would be expected from the known KD structure (Xu et al., 1995; Longenecker et al., 1996). In contrast, few elements of secondary structure are predicted for either the N- or C-terminal domains (Figure 1). More striking is the output of algorithms that predict whether individual residues are buried or exposed within the folded protein structure (Petersen et al., 2009). Again, consistent with its known folded structure, much of the KD is predicted to be buried, whereas virtually no buried residues are predicted for either the N- or C-terminal domains. Finally, both the N- and C-terminal domains are strongly predicted to be intrinsically disordered—that is, neither is predicted to adopt a stably folded structure (Figure 1). Furthermore, low complexity, glutamine-rich sequences, such as those found within the CTD, are typical signatures of intrinsically unstructured protein regions (Dyson and Wright, 2005). Thus the CTD, the domain anticipated to be most directly involved in specifying palmitoylation, is both quite poorly conserved in evolution and strongly predicted to be intrinsically disordered.
FIGURE 1:

Yck2 structural predictions. Top, a Yck2 protein schematic indicating regions of Yck2/Yck1 sequence conservation (black segments) as well as the two glutamine-rich sequences (Q-rich). Bottom, the output of various sequence analysis tools. The entry labeled “homology (Yck1)” shows a representation of Basic Local Alignment Search Tool (BLAST) results between Saccharomyces cerevisiae Yck2 and Yck1, with the number of Yck2-Yck1 identities within each 10-residue-long sequence interval reported in graded shades of green (a fully conserved segment with 10 identities is true green, and a segment with no identities is black). To exclude the contribution of the low complexity, glutamine-rich CTD sequences, glutamine identities over the Yck2 C-terminal 150 residues were not included. β-sheet and α-helical secondary structures, as well as “buried residues,” were predicted by NetSurfP (Petersen et al., 2009). For “buried residues,” the probability that the individual Yck2 residues were likely to be buried within the folded protein or exposed to solvent at the protein surface were predicted. The number of buried residues predicted per each 10-residue segment are reported as graded shades of red (a fully buried 10-residue-long segment would be true red, and a fully exposed segment would be black). At the bottom is shown for Yck2 output from DISOPRED2 (Ward et al., 2004) an algorithm that predicts intrinsically disordered protein domains (sequences that are most strongly predicted to be disordered receive a value of 1.0).
Minimal palmitoylation domain (MPD)
To delineate the Yck2 sequences that direct palmitoylation, we analyzed a series of fusion proteins that attach C-terminal segments of Yck2 to the C terminus of Ura3 (Figure 2A), an enzyme within the uracil biosynthesis pathway that resides in the cytoplasm (Figure 2B). These fusion constructs, tagged at the Ura3 N terminus with both HA and FLAG epitopes, were introduced into yeast cells on single-copy centromeric plasmids, with expression being driven from the inducible GAL1 promoter, which affords ∼10-fold overproduction relative to the YCK2 promoter (Supplemental Figure S2). This GAL1P-driven overproduction, which is used for most all of the analysis reported herein, facilitates detection, enabling both immunofluorescent localization and the biochemical analysis of palmitoylation. Although this overproduction may skew results somewhat, this analysis should still reveal the core elements needed for palmitoylation.
FIGURE 2:

Ura3-Yck2 fusion proteins delineate MPD. Ura3-Yck2 constructs, derived by fusing the indicated C-terminal portions of Yck2 to the C terminus of N-terminally FLAG/HA-tagged Ura3, were expressed from single-copy CEN/ARS plasmids via a 2-h, galactose-induced expression period. (A) Ura3-Yck2 fusion protein schematic. The Yck2 portion of each fusion protein is shown, with amino acid coordinates of the added Yck2 indicated at right. (B) IIF microscopy of Ura3-Yck2 fusion proteins. Fusion proteins were detected via their N-terminal HA epitope tag, using anti HA.11 mAb. The amino acid coordinates of the attached Yck2 portion for each fusion are indicated. Top, the localizations of GAL1P-driven copies of the Ura3 and Yck2 parental proteins. (C) Ura3-Yck2 palmitoylation. Denatured protein extracts prepared from cells expressing the indicated Ura3-Yck2 fusions were subjected to ABE, which replaces thioester-linked acyl modifications with biotin (see Materials and Methods). Subsequently, fusion proteins were immunoprecipitated using anti-FLAG mAb and then subjected to Western analysis either with anti-biotin antibody or with anti-HA.11 mAb. (D) Effect of various internal in-frame deletions on the MPD-driven palmitoylation of the Ura3-Yck2(505-546) fusion. Left, the positions of the different MPD deletions are indicated. Right, the ABE analysis of these constructs is shown.
Ura3-Yck2 fusions were analyzed for subcellular localization by indirect immunofluorescence (IIF) microscopy (Figure 2B) and for relative palmitoylation levels by the acyl-biotinyl exchange (ABE) methodology, which replaces thioester-linked acyl modifications with easily detectible biotinyl moieties (Figure 2C). IIF analysis shows that Ura3 and Yck2, the two component proteins of our fusions, localize as expected: Yck2(wt) is mainly surface-localized, and Ura3 is smoothly distributed through the cytoplasm, but excluded from the membrane-enclosed vacuole (Figure 2B). Furthermore, the fusion with the entirety of Yck2 attached to the Ura3 C terminus [i.e., Ura3-Yck2(2-546)], localizes like Yck2(wt) to the surface (Figure 2B). Ura3-Yck2(2-546) also is robustly palmitoylated (Figure 2C). Thus the attachment of Ura3 to the Yck2 N terminus does not grossly impair either palmitoylation or localization.
Analyzing fusions with progressively smaller C-terminal portions of Yck2, we find a significant breakpoint between Yck2 residues 505 and 512, with palmitoylation being detected for Ura3-Yck2(505-546), but not for Ura3-Yck2(512-546) (Figure 2C). The IIF microscopy shows correlated localizations: Yck2(512-546) is fully cytoplasmic, whereas Yck2(505-546) shows some surface localization (Figure 2B). Thus we define the Yck2 42 C-terminal residues to be the minimal palmitoylation domain (MPD). Neither the C-terminal cysteines nor the 10-residue-long, conserved, C-terminal peptide (CCTP) alone suffice to direct any discernible palmitoylation (Figure 2C).
Although the MPD is a sufficient palmitoylation signal, it directs a palmitoylation that is substantially less robust than that directed by the entire Yck2 protein; this is starkly evident in the comparison of Ura3-Yck2(505-546) and Ura3-Yck2(2-546) localizations (Figure 2B), where a substantial subpopulation of Ura3-Yck2(505-546), perhaps the majority, is found to be mislocalized to the cytoplasm (Figure 2B). Many of the analyzed fusions show similar cytoplasmic mislocalizations (Figure 2B). Indeed, robust cell surface localizations are seen only for three of the fusions: Ura3-Yck2(2-546), Ura3-Yck2(32-546), and Ura3-Yck2(60-546) (Figure 2B). Interestingly, these three fusions all retain an intact KD (Figure 2A). Fusions with the KD partially truncated [i.e., Ura3-Yck2(122-546) and Ura3-Yck2(209-546)] show partial cytoplasmic mislocalizations similar in magnitude to those seen for fusions that lack the KD altogether, i.e., like Ura3-Yck2(397-546), Ura3-Yck2(497-546), and Ura3-Yck2(505-546) (Figure 2B). This difference between fusions that retain an intact KD and those that do not is also evident in the ABE palmitoylation analysis, where a substantially stronger palmitoylation is clearly evident for the three fusions that retain the KD (Figure 2C). Thus, in addition to the MPD, it appears that the KD may also have an important palmitoylation role. This KD palmitoylation role is further explored later in this article (Figure 4).
FIGURE 4:

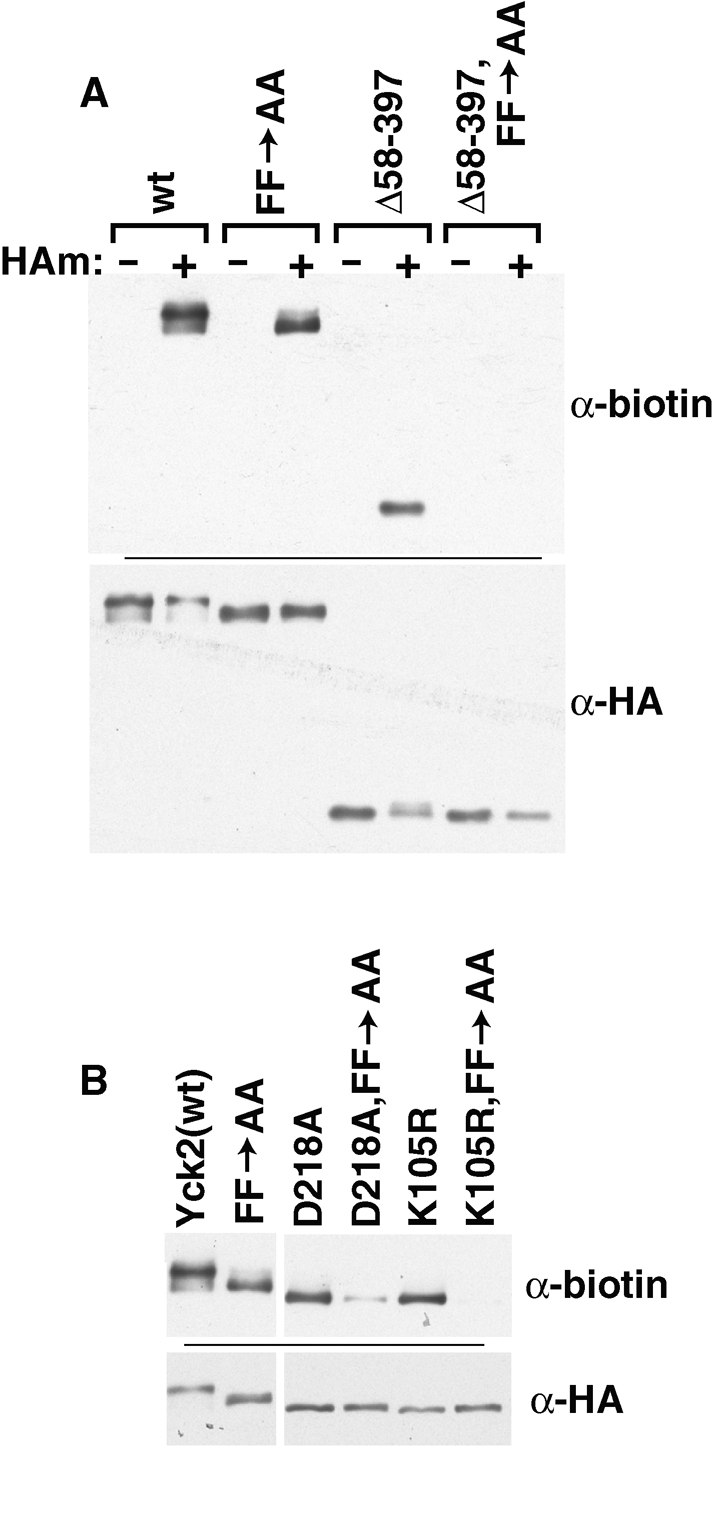
Effects of KD mutations on Yck2 palmitoylation and localization. (A) Schematic of two KD-deletion mutants that were tested. (B) Localization of kinase mutants was assessed by IIF microscopy as described for Figure 2B. (C) ABE palmitoylation analysis of KD-deletion and kinase-inactive mutants was performed as described for Figure 2C. (D) Analysis of the gel mobility shift induced by ABE for Yck2(Δ58-397). Left, extracts from cells expressing either Yck2(Δ58-397) or the nonpalmitoylatable mutant Yck2(Δ58-397,CC->SS) were subject to ABE processing either in the presence (+) or absence (–) of HAm, with the samples finally subjected to anti-HA Western blotting. The middle panel shows the results of treating the +HAm samples with β-mercaptoethanol (βME) before SDS–PAGE. Yck2(Δ396-496) and Yck2(Δ396-511) were processed through the + and –HAm ABE workups in parallel as controls. (E) Effects of the two kinase-inactive mutations on palmitoylation were assessed within the context of Yck2(Δ396-496) via the ABE mobility shift assay. The panel at left examines the gel mobilities of Yck2(Δ396-496) and of the nonpalmitoylatable Yck2(Δ396-496,CC->SS) following + and –HAm ABE workups of extracts from expressing cells. At right, mobilities of these same two proteins are compared with Yck2(Δ396-496,K105R) and Yck2(Δ396-496,D218A) following +HAm ABE workup. (F) Hyperphosphorylation of kinase mutants of Yck2 in trans by overproduced Yck2(wt). Plasmids constitutively expressing N-terminally HA epitope-tagged versions of the indicated mutant Yck2 proteins from the YCK2 promoter were transformed into wt BY4741 cells carrying either a centromeric GAL1-YCK2 plasmid (untagged Yck2) or the equivalent empty plasmid control. Following a 2-h galactose induction period, protein extracts were prepared and subjected to SDS–PAGE and then anti-HA Western blotting.
The MPD, defined earlier in the text as the minimal sequence that is able to serve as a sufficient palmitoylation signal, consists of the 10-residue-long CCTP (residues 537–546) plus an additional 32 residues of adjoining CTD sequence (residues 505–536). Based on both its evolutionary conservation (Figure 1 and Supplemental Figure S1) and its proximity to the cysteinyl acceptors, we anticipated that the CCTP likely would be an important part of the Yck2 palmitoylation signal. The contribution of the adjacent, 32-residue-long stretch seems more curious because the sequence is both poorly conserved and strongly predicted to be intrinsically disordered. To analyze MPD substructure, a series of short, in-frame deletions were introduced across the MPD within the Ura3-Yck2(505-546) fusion protein (Figure 2D). Each of the four deletions fully abolished palmitoylation (Figure 2D), indicating that required elements distribute throughout the 505–546 interval. One possible explanation is that the MPD may need to be properly folded into domain structure to be recognized by Akr1. The prediction of intrinsic disorder for much of this domain, however, argues against a key role for folded structure.
Within the holo-Yck2 context, the MPD is largely dispensable
We also examined the MPD contribution within the holo-Yck2 context, where both the KD as well as the bulk of the CTD are retained. For this examination, a series of in-frame deletions were introduced into the CTD of FLAG/HA epitope-tagged Yck2(wt), with expression again driven by the GAL1P (Figure 3A). Localization of these Yck2 mutants was assessed by two different measures, by IIF microscopy and by their level of phosphorylation. We have found that the second measure (i.e., the phosphorylation level) provides a good measure of surface localization. Our prior work found that surface-localized forms of Yck2, when overexpressed from the GAL1 promoter, are hyperphosphorylated, showing a well-discerned gel mobility shift, whereas mutant forms of Yck2 that are mislocalized either to the cytoplasm or to the cell's endomembrane system show a much less prominent shift, consistent with hypophosphorylation (Roth et al., 2002; Politis et al., 2005; Papanayotou et al., 2010).
FIGURE 3:

Analysis of in-frame CTD deletions (holo-Yck2 context). (A) Summary of results. Left, below a schematic of the C-terminal portion of Yck2, the removed Yck2 segment for each in-frame deletion is indicated (dotted line). Right, the results of three different analyses: 1) IIF microscopic localization, 2) phosphorylation level, and 3) yck1Δ yck2-ts complementation. For both the IIF and the phosphorylation analysis, HA/FLAG-tagged deletion mutants, carried on centromeric plasmids, were expressed from the GAL1P for 2 h. For IIF analysis, cells were fixed and anti-HA stained as described earlier in the text (Figure 2B). The subcellular localizations were judged as being fully surface-localized (surface), fully mislocalized (cyto), or mixed (surface + cyto). The “cyto (trace surface)” designation indicates that the overwhelming bulk of the mutant protein was judged to be cytoplasmically mislocalized, with just a small amount of staining being evident at the surface of some cells. Phosphorylation status was judged by the magnitude of the gel mobility shift induced following phosphatase treatment (panel C, bottom, provides an example of such analysis). Mutants were classed as being either predominantly hyperphosphorylated (hyper), predominantly hypophosphorylated (hypo), or a mixture of the two phospho forms (hyper + hypo). For the complementation analysis, the indicated deletion mutants were expressed at native levels (on centromeric plasmids; YCK2 promoter) within the yck1Δ yck2-ts cell context (Babu et al., 2002). Cell growth both at 30°C (permissive temperature) and at 34°C (nonpermissive temperature). Results were scored as either full complementation (+) or no complementation (–); no instances of partial complementation (reduced cell numbers or reduced colony size at nonpermissive temperature) were seen for any of the tested mutants. See Supplemental Figure S4 for supporting data and additional experimental background. nd, not determined. (B) IIF microscopic analysis for the indicated Yck2 deletion mutants. Cells were prepared and anti-HA stained as described for Figure 2B. Cells expressing both Yck2(wt) and the nonpalmitoylatable Yck2(SS) mutant, which has the C-terminal Cys-Cys dipeptide replaced by Ser-Ser coding (SS) were included as controls. (C) The phosphorylation status was assessed for the indicated Yck2 deletion mutants. Denatured protein extracts, prepared from expressing cells, were incubated either with (+) or without (–) phosphatase and then subjected to SDS–PAGE and anti-HA Western blotting. Again, extracts from Yck2(wt)- and Yck2(SS)-expressing cells were included as controls.
Experiments validating the use of hyper-/hypophosphorylation as a metric of surface localization are presented within the supplement (Supplemental Figures S2 and S3). Supplemental Figure S2 explores the underlying biology of Yck2 hyperphosphorylation, showing its dependence on Yck2 overproduction and also the requirement that the overproduced Yck2 be kinase-active, indicating this phosphorylation to be an autophosphorylation (Supplemental Figure S2, A and B). Supporting the conclusion that the hyperphosphorylation is mediated by Yck2 itself (i.e., autophosphorylation), Yck2(wt), but not the kinase-inactive mutant Yck2(D218A), is hyperphosphorylated when overproduced in Escherichia coli (Roth et al., 2002). A second conclusion that we draw from these analyses is that Yck2 hyperphosphorylation requires that Yck2 be properly delivered to the surface plasma membrane. Consistent with our prior report (Roth et al., 2002), the plasma membrane–localized Yck2 mutant [Yck2(CCIIS)] is hyperphosphorylated, whereas cytoplasmic and endomembrane-localized mutants [Yck2(SS) and Yck2(SCIIS), respectively] are not (Supplemental Figure S2C). A requirement for surface localization also is supported by the finding that the hyperphosphoryl gel shift is blocked by sec9-ts–mediated blockade of Yck2(wt) delivery to the plasma membrane (Supplemental Figure S2D). Our current thinking is that the excess CK1 activity that accumulates at the surface of overproducing cells serves to hyperphosphorylate newly synthesized Yck2 as it is delivered to the plasma membrane. In addition, we have validated the hyper-/hypophosphorylation metric against a well-characterized series of Yck2 deletion mutants that have graded palmitoylation defects (Supplemental Figure S3). Mutants that are fully defective for palmitoylation are found to be exclusively hypophosphorylated, whereas mutants that are partially defective (i.e., that have both palmitoylated and nonpalmitoyated subpopulations) present as doublets of hyper- and hypophosphorylated species, where distribution into the two phospho-forms nicely reflects the magnitude of the palmitoylation defect (Supplemental Figure S3). This capacity to simultaneously see both the hyper- and hypo-phospho-forms provides a useful means of assessing relative surface localization and thus, indirectly, relative palmitoylation status.
Together with our standard IIF microscopy, we have used this phosphorylation analysis to analyze a number of in-frame deletion mutants. First, we considered several deletion mutants that center around the MPD (Figure 3A). Yck2(Δ495-542) removes more than 90% of the MPD, retaining just the four C-terminal Yck2 residues that conclude with the C-terminal Cys-Cys. IIF microscopy finds Yck2(Δ495-542) to be severely mislocalized to the cytoplasm, with just trace surface staining being seen (Figure 3, A and B). Assessments of Yck2(Δ495-542) phosphorylation were made by analyzing the gel mobility shift induced by phosphatase digestion (Figure 3C). Whereas Yck2(wt) is hyperphosphorylated [consistent with its surface localization (Figure 3B)], Yck2(Δ495-542) shows only the modest shift indicative of hypophosphorylation (Figure 3C). Thus, by both these analyses, IIF microscopy and phosphorylation, Yck2(Δ495-542) clearly is quite severely mislocalized, indicating a severe palmitoylation defect. Likewise, Yck2(Δ535-542), which has a small, eight-residue-long deletion centering on the CCTP portion of the MPD, also is severely mislocalized (Figure 3, A–C), indicating that the CCTP is required for efficient palmitoylation within the holo-Yck2 context. Yck2(Δ495-536), however, which removes 76% of the supposedly inviolable MPD (Figure 2D), surprisingly shows both wild-type (wt)-like surface localization and wt-like hyperphosphorylation, indicating that it is fully palmitoylation competent (Figure 3, A–C). Interestingly, the MPD portion removed by the Δ495–536 deletion precisely corresponds to the 32-residue-long segment (residues 505–536) that is both poorly conserved and predicted to be structurally disordered (Figure 1 and Supplemental Figure S1). Thus, within this holo-Yck2 context, the portion of the MPD that is predicted to be unstructured can be deleted with little or no impact on palmitoylation. Our hypothesis, which is further developed later in this article (see Discussion), is that the 134 residues of the CTD that are retained by Yck2(Δ495-536) (residues 361–494), for which intrinsic disorder also is predicted (Figure 1), likely compensate for the unstructured MPD segment (residues 505–536) that is removed.
Essential, but redundant contribution made by unstructured CTD
To test if CTD elements beyond the CCTP also may play significant roles in palmitoylation, we constructed and tested Yck2(Δ361-536) mutant; this mutant retains the CCTP, while the 176-residue-long, putatively unstructured portion of the CTD has been precisely removed (Figure 3A). Yck2(Δ361-536) appears to be fully palmitoylation deficient, replicating the nonpalmitoylatable Yck2(SS) mutant (Ser-Ser replacement of Cys-Cys palmitoyl acceptors) with regard to its cytoplasmic mislocation, its absence of hyperphosphorylation, and its inability to complement growth of the yck1Δ yck2-ts strain at nonpermissive temperature (Figure 3A). Thus it appears that the 361–536 interval does contain some essential palmitoylation element. To further localize this element, the 361–536 interval was subdivided with two additional deletion mutants, Yck2(Δ361-397) and Yck2(Δ396-536) (Figure 3A). Yck2(Δ361-397) shows both wt surface localization and hyperphosphorylation, indicating that it is not required for palmitoylation, whereas Yck2(Δ396-536) appears to be fully defective (Figure 3A). Continuing the search, we then subdivided the 396–536 interval by Yck2(Δ396-496) and Yck2(Δ495-536). Here, surprisingly, both mutants appear to be fully wt (Figure 3A). Thus, although Δ396–536 fully abolishes palmitoylation function, neither of the two component deletions (i.e., neither Δ396–496 nor Δ495–536) show any discernible effect. Clearly, this result is incompatible with the premise that the essential 396–536 interval contains just a single essential palmitoylation element. It could, however, be compatible with models in which multiple redundant elements distribute through the CTD with some mapping to the 396–496 interval and some to the 496–536 interval. The alternative possibility that we favor and that we further explore later in the text (see Discussion) is that the CTD, which is predicted to be largely unstructured (Figure 1), is required as a flexible linker domain, connecting the CCTP and the KD. From this perspective, the redundancy documented here may indicate that efficient palmitoylation requires a linker length that is somewhat shorter than the length supplied by the entire CTD. Yck2(Δ396-496) and Yck2(Δ495-536) both retain substantial portions of the putatively unstructured CTD and thus both may retain linker domains of sufficient length to fulfill this essential role.
KD role
The Ura3-Yck2 analysis (Figure 2, B and C) suggested a role for Yck2 KD in palmitoylation. To explore the KD role, we have examined two mutants which delete the entire KD, Yck2(Δ58-397) and the more precise KD deletion, Yck2(Δ70-361) (Figure 4A), as well as two point mutations within the KD that are expected to fully inactivate kinase activity, a K105R and D218A mutation. K105R mutates the invariant KD lysine that is typically mutated for Ser/Thr kinase inactivation; this lysine anchors and orients the α and β phosphates of ATP (Hanks and Hunter, 1995). D218A mutates the Asp of the invariant subdomain VII DFG tripeptide within the kinase magnesium binding loop (Hanks and Hunter, 1995). Consistent with kinase inactivation, both mutant alleles fail to complement the yck1Δ yck2-ts strain (Supplemental Figure S4), and both mutant proteins show no discernible autophosphorylation (Supplemental Figure S2B).
First, subcellular localizations of the different KD mutants were assessed by IIF microscopy (Figure 4B). The two KD deletion mutants, Yck2(Δ58-397) and Yck2(Δ70-361), both are significantly mislocalized to the cytoplasm. The severity of the mislocalization defects for the two mutants seems roughly equivalent, indicating that the defect results from the loss of elements that map within the KD, and not from the loss of elements, mapping within the KD-proximal domains that are included within the larger Δ58–397 deletion. For the two kinase-inactive mutants, Yck2(K105R) and Yck2(D218A), less severe mislocalization defects are seen, with Yck2(D218A) reproducibly showing a less severe defect than that seen for Yck2(K105R) (Figure 4B).
In addition, the palmitoylation of the different KD mutants was assessed by ABE (Figure 4C). Consistent with the substantial subpopulations of each mutant that are found to be surface-localized (Figure 4B), each also shows significant levels of palmitoylation. A rough assessment of per protein palmitoylation levels for each protein, made by normalizing the anti-biotin palmitoylation signal for each protein to each protein's anti-HA signal, indicates Yck2(Δ58-397) palmitoylation to be reduced by 55% relative to Yck2(wt), whereas Yck2(K105R) and Yck2(D218A) show palmitoylation reductions of less certain significance, 30 and 18% reductions, respectively.
Although the IIF and phosphorylation assays provide only indirect assessments of palmitoylation, both do have the advantage of allowing simultaneous visualization of both the palmitoylated and nonpalmitoylated subpopulations. For instance, mutants that are more defective for palmitoylation show in our phosphorylation analyses not only a decreased level of the hyper-phospho-form, but also an increased level of the hypo-phospho-form (Supplemental Figure S3). This capacity to see both forms is quite useful when comparing the relative palmitoylation defect severity imposed by different mutations. Typically, analysis of palmitoylation by ABE analyses lacks this advantage, as the severity of the palmitoylation defect is inferred only from the intensity of the anti-biotin Western blot signal (as an example, see Figure 4C). To obtain a more robust measure, we have developed an approach based on the ABE-induced gel mobility shifts such as those that were noted in our prior ABE analysis of the Δ396 deletion series (Supplemental Figure S3A). This analysis showed that the ABE chemical replacement of thioester-linked acyl modifications with the biotinyl moiety, donated from N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide (HPDP-biotin), is efficient, with close to 100% of the palmitoylated subpopulation shifting as a result of the ABE chemical workup (Supplemental Figure S3A). An analogous ABE-induced gel mobility shift also may be discerned in the ABE analysis of Yck2(Δ58-397) (Figure 4C). Note that ABE-processed Yck2(Δ58-397) presents as a doublet in the anti-HA blot, but as a singlet when just the palmitoylated subpopulation is detected using anti-biotin antibody. The anti-biotin singlet comigrates with the upper band of the anti-HA doublet, indicating that this upper doublet band corresponds to the biotinylated component (Figure 4C). Additional analysis shows that this upper species is not seen when the hydroxylamine (HAm) thioester cleavage step is omitted, nor when the nonpalmitoylatable mutant, Yck2(Δ58-397,SS), which has the Cys-Cys palmitoyl acceptors replaced by the Ser-Ser dipeptide, is used (Figure 4D). Thus the upper doublet band is indeed the result of ABE replacement of acyl modifications by the HPDP-biotin moiety (430 Da per added HPDP-biotin moiety). Furthermore, consistent with disulfide linkage of HPDP-biotin to cysteine, β-mercaptoethanol causes the upper doublet component to collapse to the lower, faster migrating position (Figure 4D). Two CTD deletions, Yck2(Δ396-496) and Yck2(Δ396-512), were included in this analysis of Yck2(Δ58-397) palmitoylation as controls, because our prior analysis found quantitative shifting of the palmitoylated subpopulations of these two mutants (Supplemental Figure S3A). Again here, for Yck2(Δ396-496), which has unimpaired palmitoylation (Figure 3A), the ABE treatment results in a clear shift for the bulk of the mutant protein whereas, for the partially palmitoylated Yck2(Δ396-511), just the hyperphosphorylated component (i.e., the palmitoylated component) shifts (Figure 4D), indicating that ABE replacement in this analysis to be again efficient. Thus the finding of <50% shifting for Yck2(Δ58-397) indicates that <50% of this protein population is palmitoylated, a finding that is nicely consistent with the previously mentioned analyses (Figure 4, B and C).
We also wished to apply this powerful ABE mobility shift approach to assess palmitoylation of the two kinase-inactive mutants, Yck2(K105R) and Yck2(D218A). Unfortunately, we have been unable to discern ABE-induced mobility shifts within the full-length Yck2 context (unpublished data). Because, however, ABE shifts are seen for the fully palmitoylated and fully surface-localized Yck2(Δ396-496) mutant (Figure 4D and Supplemental Figure S3A), we opted to test the kinase-inactive mutations instead within the Δ396–496 context. Thus we compared the gel mobility of ABE-treated Yck2(Δ396-496) both with the nonpalmitoylatable mutant Yck2(Δ396-496,SS) and with the two kinase-inactive mutant versions, Yck2(Δ396-496,K105R) and Yck2(Δ396-496,D218A) (Figure 4E). To eliminate confounding effects of phosphorylation on gel mobility, all samples were treated with phosphatase before electrophoresis. Comparing plus- and minus-HAm ABE workup conditions for Yck2(Δ396-496), we see that productive ABE treatment (+HAm) produces a well-discerned gel shift that is not seen for the equivalent CC->SS mutant, indicating that the ABE-induced upward shift is in fact due to the biotinylation of palmitoylated Yck2(Δ396-496). Applying this same analysis to the two kinase-inactive mutants finds significant underpalmitoylation for both, with a significant portion of each mutant population (∼30%) escaping palmitoylation (Figure 4E).
We also hoped to use Yck2 hyperphosphorylation as a metric of surface localization for the different kinase mutants. Being an autophosphorylation, however, hyperphosphorylation is not seen for kinase-inactive mutants (Supplemental Figure S2B). To try to circumvent this obstacle, we contrived a situation in which GAL1-driven overexpression of an untagged, wt Yck2 is used for in trans hyperphosphorylation of the surface-localized subpopulation of the HA epitope–tagged, kinase-mutated Yck2 proteins. The epitope-tagged test proteins were expressed from their native YCK2 promoter either in control cells or in cells overexpressing untagged Yck2(wt) from the GAL1 promoter. Because hyperphosphorylation is not seen for Yck2(wt) at native expression levels (Supplemental Figure S2A), neither Yck2(wt) nor any of the tested mutant Yck2 proteins show any hyperphosphorylation within control cells that lack the GAL1-YCK2 construct (Figure 4F). In the Yck2-overproducing context, however, Yck2(wt), consistent with its surface localization, is found to be efficiently hyperphosphorylated (Figure 4F), indicating that hyperphosphorylation can be mediated in trans. For Yck2(K105R) and Yck2(D218A), which were examined in parallel, ∼40–50% of the mutant protein escapes such hyperphosphorylation (Figure 4F); presumably, this underphosphorylated subpopulation corresponds to the subpopulation that is not palmitoylated and, therefore, not localized to the plasma membrane. For Yck2(Δ58-397), an even smaller fraction of population is hyperphosphorylated (Figure 4F), indicating that an even smaller portion of this mutant likely is surface localized. Thus this indirect measure of localization provides results that are again consistent with the previously mentioned analyses (Figure 4, B–E): Whereas the two kinase-inactive mutants both clearly are impaired for palmitoylation, a more severe impairment is seen when the KD is fully deleted. Thus, although kinase activity appears to play some positive role in palmitoylation, it clearly is not the whole story. Additional elements that map within the KD, that remain undefined, also play an important role in driving Yck2 palmitoylation. In other words, the KD contribution to Yck2 palmitoylation is complex, with a role not only for Yck2-mediated phosphorylation, but for other elements as well, perhaps KD structural elements that may be directly recognized by Akr1.
The Phe-Phe dipeptide is a critical palmitoylation determinant
To further investigate the CCTP role in palmitoylation, six di-alanine replacement mutations, in which adjacent residue pairs were replaced by the Ala-Ala dipeptide, were introduced across the CCTP interval (Figure 5A). The di-alanine mutations were tested first within the holo-Yck2 context (Figure 5, B and C). Considering the evolutionary conservation of this sequence, it is a bit surprising to find that five of the six replacement mutants show no phenotype with regard to either localization (Figure 5B) or hyperphosphorylation (Figure 5C); each is indistinguishable from Yck2(wt). The one exception is the replacement of the Phe539-Phe540 dipeptide (i.e., Yck2(FF->AA)), which stands out as being defective, being both severely mislocalized (Figure 5B) and severely underphosphorylated (Figure 5C). Assessments of Yck2(FF->AA) palmitoylation both by ABE and by the newly developed click-based methodology (Hang et al., 2007; Charron et al., 2009), find Yck2(FF->AA) palmitoylation to be substantially reduced relative to Yck2(wt) (Figure 5D).
FIGURE 5:

Analysis of CCTP di-alanine substitution mutants. Consecutive residues across the C-terminal Yck2 14 amino acids were replaced with Ala-Ala. The di-alanine substitution mutations were compared with the unsubstituted, wt sequence and to the nonpalmitoylatable CC->SS replacement for effects on palmitoylation, phosphorylation, and localization within both the holo-Yck2 context as well as within the KD-deleted Yck2(Δ58-397) context. (A) Yck2 C-terminal sequence with the residues that were substituted by Ala-Ala are underlined. (B) Effects of di-alanine substitutions on localization assessed within the holo-Yck2 context by IIF microscopy. (C) Hyperphosphorylation of di-alanine substitution mutants assessed within the holo-Yck2 context by anti-HA Western blotting. (D) The effect of the FF->AA mutation on palmitoylation assessed within the holo-Yck2 context, using either the ABE or click-based detection methodologies (see Materials and Methods). (E) Palmitoylation effects of di-alanine mutations were tested within the partially impaired Yck2(Δ58-397) context using the click-based detection methodology (see Materials and Methods). (F) Palmitoylation effects of di-alanine mutations were tested in an in vitro system, using purified Akr1 and purified, di-alanine-substituted Yck2 substrate proteins (see Materials and Methods). Palmitoylation competence was assessed by the acceptance of [3H]-palmitate label donated from labeled palmitoyl-CoA. Note that the purified substrate proteins tested here all have the kinase-inactivating D218A mutation in addition to the mutations indicated. (G) Relative contribution of individual CCTP dipeptides.
In hopes of exposing additional, more subtle CCTP contributions to palmitoylation, the di-alanine replacements also were tested within the palmitoylation-compromised, KD-deleted Yck2(Δ58-397) context. Again, a click-based palmitoylation analysis finds that the most severe defect was associated with the FF->AA mutation (Figure 5E). In addition, however, within this palmitoylation-compromised context, clear palmitoylation defects also are evident for several of the other mutants, namely for the replacements of the Leu543Gly544, Lys537Gly538, and Ser535Ser536 dipeptides (Figure 5E).
We also have examined the effects of the different di-alanine substitutions within an in vitro palmitoylation system that relies on purified Akr1 enzyme and Yck2 substrate proteins (Roth et al., 2002). For technical reasons, the Yck2 substrate proteins that are used in our in vitro analyses all harbor the kinase-inactivating D218A mutation (Roth et al., 2002). Just as the palmitoylation defects of the di-alanine mutations are accentuated within the palmitoylation-compromised Yck2(Δ58-397) context (Figure 5E), similar accentuation might be anticipated in vitro, within this palmitoylation-compromised Yck2(D218A) context. In this in vitro analysis, Yck2(FF->AA) again stands out; like the nonpalmitoylatable Yck2(CC->SS) mutant, Yck2(FF->AA) shows no palmitoylation (Figure 5F). Furthermore, consistent with the results of in vivo palmitoylation analysis within the Yck2(Δ58-397) context (Figure 5E), partial palmitoylation defects are again highlighted for the KG->AA and LG->AA mutations. The relative magnitudes of the defects associated the different di-alanine substitution mutations extrapolated both from the in vivo Yck2(Δ58-397) context data (Figure 5E) and from the in vitro system (Figure 5F) are depicted in Figure 5G with the size of the replaced dipeptide reflecting their relative palmitoylation contributions. Finally, we draw one important conclusion: Because defects are clearly being seen for these CCTP mutations in vitro (Figure 5F), where Akr1 and Yck2 effectively are the only two proteins that are present, we conclude that the CCTP functions directly in the interaction of Yck2 with Akr1.
To examine the contributions of the individual Phe539Phe540 phenylalanines, Yck2(F539A) and Yck2(F540A) were constructed; both show clear localization defects that are somewhat less severe than those observed for Yck2(FF->AA) (Figure 6A). Consistent with these partial mislocalizations, both mutants also show intermediate phosphorylation phenotypes, with both hyper- and hypophosphorylated species being prominently evident (Figure 6B). Thus both Phe-539 and Phe-540 participate in directing palmitoylation.
FIGURE 6:

Analysis of the Phe-Phe dipeptide palmitoylation contribution. (A) Effects of the individual Phe-Phe mutations F539A and F540A on Yck2 localization assessed by IIF microscopy. (B) Effects of the F539A and F540A mutations on Yck2 hyperphosphorylation were examined by assessing the effects of phosphatase treatment on gel mobility. (C) Effects of repositioned Phe-Phe dipeptides on Yck2 localization assessed by IIF microscopy. (D) Effects of the repositioned Phe-Phe dipeptides on Yck2 hyperphosphorylation.
In addition, we have also tested the importance of the native spacing of the Phe-Phe dipeptide with respect to the C-terminal cysteines, with the Phe-Phe dipeptide being repositioned either two residues closer to, or two residues farther away from, the C-terminal Cys-Cys. The mutant with the Phe-Phe repositioned closer to the C terminus [i.e., Yck2(KGAAFFLGCC)] shows strong surface localization that is indistinguishable from Yck2(wt), whereas the mutant with the Phe-Phe repositioned two residues farther away from the C terminus [i.e., Yck2(FFAASKLGCC)] is unable to support proper localization (Figure 6C). Correlated effects on phosphorylation are again seen, with reduced hyperphosphorylation being seen for the mutant having the Phe-Phe dipeptide positioned two residues farther away from the C terminus, but not for the mutant with the the Phe-Phe dipeptide positioned closer to the palmitoyl acceptors (Figure 6D). As controls, we also tested two mutants that have four consecutive phenylalanines [i.e., Yck2(FFFFSKLGCC) and Yck2(KGFFFFLGCC)]. In essence, these constructs retain Phe-Phe in its native position while adding a second Phe-Phe that is positioned as it is for either Yck2(FFAASKLGCC) or Yck2(KGAAFFLGCC). These two control constructs both show wt surface localization and wt hyperphosphorylation, indicating that the Yck2(FFAASKLGCC) defect is indeed due to the inability of the more distantly positioned Phe-Phe sequence to direct palmitoylation at the C-terminal cysteines, rather than being the consequence of an active disruption of CCTP function, imposed by the repositioned phenylalanines.
Synergy of KD mutations with FF->AA mutation
We also have tested the consequences of combining different KD mutations with the FF->AA mutation. Alone, the KD mutations and the FF->AA mutation both result in partial palmitoylation defects (Figures 4 and 5). When the FF->AA mutation is combined into the KD-deleted Yck2(Δ58-397) context, however, we see that palmitoylation is fully abolished (Figure 7A). Likewise, similar synergy is seen when FF->AA is combined with the two kinase-inactive missense mutations, K105R and D218A (Figure 7B). Earlier in this article, we suggested that the CCTP mutations, based on their in vitro defects (Figure 5F), likely were disrupting the direct interaction of Yck2 with Akr1. The synergy documented here for the KD mutations with FF->AA suggests that, like the CCTP, the KD also may play a role in mediating the direct interaction of Yck2 with Akr1.
FIGURE 7:
Synergistic defect for double mutants having the FF->AA mutation in combination with kinase inactivating or deletion mutations. The palmitoylation levels of the indicated double mutants were compared with single mutant levels by ABE. The panel A experiment includes –HAm controls; the panel B experiment does not.
Akr1 specificity
In the wt cell context, Yck2 palmitoylation relies exclusively on Akr1 (Roth et al., 2002, 2006). Recent work, however, has shown that, when overproduced, other, noncognate yeast PATs also can palmitoylate Yck2 (Hou et al., 2009), indicating that other PATs, beyond Akr1, potentially have the capacity to recognize and palmitoylate Yck2. Furthermore, a recent characterization of palmitoylation requirements for yeast Vac8 found a relaxed specificity for some Vac8 mutants, in which these mutants could be palmitoylated by a broader array of PATs than normally acts upon Vac8(wt) (Nadolski and Linder, 2009). One important new insight that was provided by this Vac8 work is that specificity can be conferred not only by substrate features that promote interaction with the cognate modification enzyme, but also in some cases by substrate features that restrict interaction with noncognate enzymes. To see whether the residual palmitoylation seen for many of our Yck2 mutants might reflect palmitoylation by noncognate PATs, a variety of mutants were tested for Akr1 dependence by comparing their palmitoylation in wt and akr1Δ cells (Figure 8). Like Yck2(wt), each of the mutants remained wholly dependent on Akr1 for palmitoylation, indicating that the residual palmitoylation that occurs for these partially impaired mutants is not due to relaxed specificity. Note the striking mobility shifts seen for Yck2(wt) as well as some of the mutant Yck2 proteins within the akr1Δ cell context (Figure 8, bottom anti-HA panel). Presumably, these shifts are due to impaired phosphorylation, secondary to the impaired localization and palmitoylation caused by the lost Akr1 function.
FIGURE 8:

Residual palmitoylation of Yck2 deletion mutants remains Akr1-dependent. ABE methodology was used to test palmitoylation of the indicated Yck2 mutants in both the wt and the isogenic akr1Δ context.
DISCUSSION
Tripartite Yck2 palmitoylation signal
For most posttranslational modifications, the substrate signal that attracts the modification enzymes to the modification site typically resides locally, within the sequence surround of the modification site residue. By this criterion, the best candidate Yck2 palmitoylation signal would be the conserved, 10-residue-long CCTP sequence that includes the Cys-Cys palmitoyl acceptors. Indeed, the CCTP clearly is important. Nonetheless, it alone is not sufficient as other Yck2 domains play important roles as well. Indeed, we find that sequences important for palmitoylation are spread across the entire Yck2 sequence, being composed of three discrete parts—the KD, the CCTP, and between the two, a long, poorly conserved domain that is predicted to be intrinsically disordered. Figure 9 provides a model showing how these three parts of the Yck2 palmitoylation signal might collaborate to effect palmitoylation. The CCTP is shown as interacting directly with the putative Akr1 active site, which is the conserved DHHC cysteine-rich domain and is displayed within an inter-TMD cytosolic loop. The KD is shown as contributing to recognition through an interaction with the Akr1 ankyrin repeat domain. The intervening CTD sequences are shown as contributing by acting as a flexible tether that allows the comcomitant interaction of the KD and the CCTP with the separate Akr1 domains. Below, we discuss the evidence supporting these different contributions of the different Yck2 domains.
FIGURE 9:
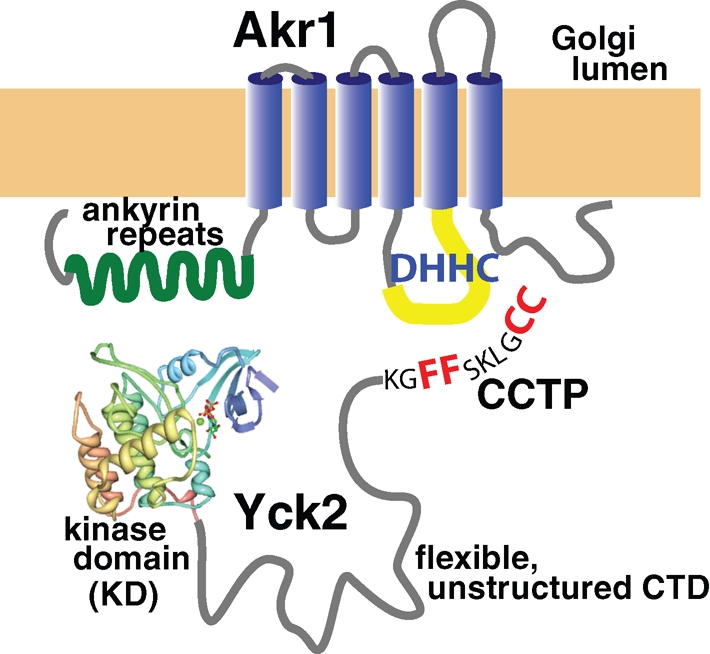
A model for how the three Yck2 palmitoylation domains may collaborate to promote Akr1-mediated palmitoylation. The KD and CCTP are hypothesized to interact with two separate Akr1 domains, the ankyrin repeats and the active site DHHC loop, respectively. The poorly conserved CTD, which is predicted to be intrinsically disordered is suggested to be a flexible linker that facilitates the simultaneous interaction of the KD and the CCTP with Akr1.
CCTP
The CCTP is critical not only in vivo, but also within our in vitro palmitoylation system. This in vitro role, in which only Yck2 and Akr1 are present, indicates that the CCTP functions directly in the Akr1-Yck2 enzyme-substrate recognition, not, for instance, in some hypothetical transport process that might deliver cytoplasmic Yck2 to Golgi-localized Akr1. An analysis of the CCTP fine structure finds a paramount role for the central Phe-Phe, with lesser roles being played by the surrounding conserved residues. Our prior work indicated that the C-terminal Cys-Cys dipeptide, in addition to serving as the palmitoyl acceptor, also is a critical recognition determinant, because the mutational removal of single cysteines was found to severely reduce palmitoylation at the retained cysteine (Papanayotou et al., 2010). Because the Cys-Cys dipeptide is part of the CCTP, we believe that the CCTP is likely to be recognized by the Akr1 active site, which is thought to map primarily to the conserved, 50-residue-long DHHC cysteine-rich domain. The finding that the Phe-Phe can be mutationally repositioned two residues closer to the Cys-Cys acceptors without apparent palmitoylation defect suggests substantial flexibility within the Akr1 substrate-binding pocket.
The kinase domain
The prominent role uncovered here for the KD is surprising. Deletion of this domain results in a substantial, >50% palmitoylation loss. We considered two hypotheses regarding how the KD might be contributing. Structural recognition elements embedded within the KD, we thought, might mediate either the direct interaction with or, possibly, the delivery to Akr1. Alternatively, kinase activity might be important, with Yck2-mediated phosphorylation potentially playing a positive role. To address these possibilities, we compared the palmitoylation of kinase-inactive mutants with kinase-deletion mutants. Unfortunately, the results of this analysis proved somewhat unsatisfying, not leading to a solid black or white conclusion. The kinase-inactive mutants that were analyzed both showed clear palmitoylation defects, yet these defects were substantially lower in magnitude than those seen for the KD-deleted mutants (Figure 4). Although our analysis indicates that both the Yck2(K105R) and Yck2(D218A) mutants are fully defective for kinase function [neither shows any functional complementation (Supplemental Figure S4), nor the characteristic Yck2 autophosphorylation (Supplemental Figure S2B)], it is possible that they retain, while the KD-deleted Yck2(Δ58-397) lacks, some low residual phosphorylation activity that suffices to stimulate palmitoylation. Plausibly, therefore, the positive KD palmitoylation role may be exerted solely through its phosphorylation function. The alternative extreme is that the KD palmitoylation role is fully determined by KD structural features, with the KD being directly recognized by either Akr1 or perhaps some ancillary factor that might aid in delivering Yck2 to the Golgi-localized Akr1. In this scenario, the partial impairment of the kinase-inactive mutants might be due to a partial misfolding of the KD. Such misfolding would not be surprising because conserved kinase residues mutated here are both involved in triangulating ATP within the kinase-binding pocket (Hanks and Hunter, 1995). Mutation of these residues may perturb nucleotide binding and thereby the final folded Yck2 structure.
The synergy of the KD mutations with the CCTP FF->AA mutation (Figure 8) suggests that, like the CCTP, the KD may also directly participate at the level of the Yck2-Akr1 interaction. If both the CCTP and KD contribute to the recognition of Yck2 by Akr1, then separate mutation of the two domains might be expected to lead to reduced interaction and reduced palmitoylation, whereas simultaneous comutation of the two domains might result in the interaction being fully abolished. Anecdotal support for a direct KD role in Yck2-Akr1 recognition comes from prior experiments aimed at establishing an in vitro palmitoylation assay for Akr1 (Roth et al., 2002). Whereas the full-length Yck2(D218A) proved to be an effective Akr1 substrate, an 150-residue-long, C-terminal Yck2 fragment, Yck2(396-546), which lacked the KD, did not (A. Roth and N. Davis, unpublished data). In light of our current data, this failure of the CTD fragment to act as an in vitro substrate might indicate a critical KD role in the direct interaction of Yck2 with Akr1. Our suggestion that the KD interaction site on Akr1 is the ankyrin repeat domain (Figure 9) currently has no experimental support. Ankyrin repeats, which consist of both conserved and variable elements, are thought to function as protein-protein interaction platforms, with binding specificity being determined, much as for immunoglobulins, by the variable portion of each repeat (Li et al., 2006). The surfaces on target proteins that bind ankyrin repeats generally are part of folded domains, and, consequently, the ankyrin-interacting residues typically are widely dispersed through the target protein's primary sequence. Perhaps it is relevant that the folded KD is among the wide array of targets that have been documented for ankyrin repeat domains, a classic example being the inhibition of the cell division cycle kinase CDK4 by the INK4 family of ankyrin repeat domain proteins (Li et al., 2006). Lending further credence to our suggestion that the Yck2 KD may interact with the Akr1 ankyrin repeats is recent work on the mammalian ortholog of Akr1,HIP14 (ZDHHC17) (Huang et al., 2009). The HIP14 ankyrin repeat domain was found to be an important mediator of enzymatic specificity, with substrate specificity being transferred from HIP14 to a second DHHC PAT, namely DHHC3, with the transplantation of the HIP14 ankyrin repeat domain to DHHC3.
Is the poorly conserved, intrinsically unstructured portion of the CTD an interdomain linker?
Sited between the KD (residues 70–360) and the CCTP (residues 536–546) is a 172-residue-long sequence stretch that is both poorly conserved and predicted to be intrinsically disordered (residues 361–536; Figure 1). This nondescript sequence is required for Yck2 palmitoylation: Both Yck2(Δ361-536) and Yck2(Δ396-536), which delete the bulk of this segment, are fully defective for palmitoylation and are unable to complement the yck1Δ yck2-ts mutations (Figure 3A). How might this large, poorly conserved, unstructured domain contribute? Intrinsically disordered sequences often function as flexible linkers to separate globular protein domains and allow their independent action (Dyson and Wright, 2005). Thus we propose that the CTD also may be functioning here, for palmitoylation, as a flexible linker, facilitating the concomitant binding of CCTP and KD to separate Akr1 domains (Figure 9). This model nicely fits with the curious redundancy that we have documented for this domain (Figure 3A). Recall that whereas deletion of the 396–536 interval fully abolishes palmitoylation, shorter deletions (the two component deletions that subdivide this interval, namely Δ396–496 and Δ495–536) show no palmitoylation defect (Figure 3A). Furthermore, a variety of large CTD deletions show intermediate palmitoylation defects (Figure 3A). For these mutants, the retained CTD segments that presumably facilitate the residual palmitoylation often do not overlap [ Figure 3A; see Yck2(Δ361-496) and Yck2(Δ461-536)]. Thus separate, nonoverlapping portions of the CTD are able to supply this essential palmitoylation function. Palmitoylation, we suggest, may require that the CCTP and KD be separated by some minimal length of flexible linker, with the required length perhaps being shorter than the 176-residue-long length present within wt Yck2.
If the required linker length for palmitoylation is substantially shorter than the 176-residue-long domain that is present within Yck2(wt), then one wonders about the evolutionary pressures that shaped emergence of such a long, unstructured domain. The explanation likely resides in Yck2's mature function as a plasma membrane–tethered kinase. A large, flexible tether might allow the membrane-anchored kinase to phosphorylate substrates within a large, juxtamembrane space. Without a flexible linker, the kinase domain directly attached to the bilayer would be able to access only a limited set of substrate phosphorylation sites.
Implications for other palmitoylation substrates
How might our Yck2 and Akr1 results generalize to other palmitoylation substrates and enzymes? Palmitoylation is incredibly diverse with a huge variety of substrates being modified by a relatively large collection of enzymes, the DHHC PATs. Thus we do not expect that palmitoylation will be governed by a simple set of generalizable rules. By narrowing our focus to just Akr1 and its mammalian ortholog HIP14 (DHHC17), however, a few trends may be beginning to emerge.
Akr1 has been shown to participate in the palmitoylation of several yeast proteins beyond Yck1 and Yck2, these being the following: the sphingosine kinase Lcb4; the membrane trafficking protein Meh1; the vacuolar CK1 Yck3; Env7, a protein involved in late endosome-vacuole trafficking; as well as three proteins of unknown function—Sna4, Anr2, and Ypl199c (Kihara et al., 2005; Roth et al., 2006). Unfortunately, our inspection of this set of Akr1 substrate proteins for local, CCTP-like sequences does not reveal a general Akr1 consensus motif. We do not, for instance, find Phe-Phe dipeptides spaced appropriately relative to the putative cysteinyl acceptors. One feature of the Akr1 substrates that is notable, however, is that seven of these nine substrate proteins (namely, Yck1, Yck2, Yck3, Lcb4, Env7, Anr2, and Ypl199c) fall into that class of palmitoyl proteins that associate with membranes solely through their palmitoyl modifications. Thus Akr1 may play a major role in recognizing this class of hydrophilic substrate proteins for palmitoylation.
Akr1 also stands out for its possession of an ankyrin repeat domain (Figure 9). Only one of the six other yeast DHHC proteins has an ankyrin repeat domain, this being Akr2, a DHHC protein not yet linked to any palmitoylation. Two of the 23 mammalian DHHC PATs have N-terminal ankyrin repeat domains; these are HIP14 (DHHC17) and the less studied HIP14L (DHHC13). As discussed earlier in the text, the HIP14 ankyrin repeats have been implicated as playing a key part in substrate recognition (Huang et al., 2009). Interestingly, like the Akr1 substrates, the proteins identified to date as likely HIP14 substrates also fall predominantly into the hydrophilic class of proteins that associate with proteins solely through their attached acyl moieties. Included among these putative HIP14 substrates are the postsynaptic receptor scaffolding protein PSD-95, the synaptic t-SNARE SNAP25, the disease protein for Huntington Disease Htt, and the cysteine string protein, a presynaptic chaperone (Huang et al., 2004, 2009; Greaves et al., 2008, 2009).
Might the ankyrin repeat domains of Akr1 and HIP14 function to capture hydrophilic substrates from the cytosol for palmitoylation? This substrate class has special needs regarding their accessing of membrane-localized PATs. As discussed in the Introduction, for the other two general classes of palmitoyl proteins, namely the palmitoylated transmembrane proteins and the large set of proteins in which palmitoylation occurs secondarily to either myristoylation or prenylation, there are clear, palmitoylation-independent mechanisms providing these proteins with access to cellular membrane systems. Such proteins likely access their PATs from within the membrane plane. The third class of substrates (i.e., the large set of purely hydrophilic substrates that includes Yck2) does not have this advantage—palmitoylation of these proteins requires their active retrieval from the cytoplasm. The fact that ankyrin repeats are found just on the subset of PATs that appear to be devoted to the hydrophilic substrate class suggests a possible role for the ankyrin repeat domain in this capture process.
Relevant to this discussion is a recent report on the SNAP25 palmitoylation signal (Greaves et al., 2009). Three different mammalian DHHC PATs (HIP14, DHHC3, and DHHC7) have been implicated as palmitoylating SNAP-25 (Fukata et al., 2004; Huang et al., 2004; Greaves et al., 2009). Recognition of SNAP25 by DHHC3 and DHHC7 relies on a signal mapping to the local sequences surrounding the acyl-accepting cysteine cluster. Palmitoylation by HIP14, however, requires, in addition to this local recognition element, a second SNAP25 recognition element, 20–30 residues removed from the cysteine cluster (Greaves et al., 2009). Furthermore, HIP14 palmitoylation of SNAP25 is blocked not only by mutations within these two recognition elements, but also by mutations that shorten the sequences intervening between the two (Greaves et al., 2009). Interestingly, our analysis of the SNAP25 protein sequence finds that intrinsic disorder is also strongly predicted for this putative linker region (unpublished data). Thus, like the Yck2 palmitoylation signal, the SNAP25 palmitoylation signal also appears to be tripartite, again two recognition elements separated by a required linker. It will be interesting to see if dual recognition elements prove to be a general feature of this class of hydrophilic substrate proteins that are modified by ankyrin repeat domain-containing PATs, like Akr1 and HIP14.
MATERIALS AND METHODS
Yeast strains
Most of the work reported later in the text uses the wt MATα ura3-52 leu2 his3 strain LRB759 (Panek et al., 1997) to track epitope-tagged Yck2 proteins expressed from plasmid vectors. Other strains that were used include NDY1405 (Roth et al., 2002), which is isogenic to LRB759 except for an unmarked akr1Δ allele, BY4741 (Brachmann et al., 1998), which is MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 strain (used only for Figure 9F), LRB934 (Babu et al., 2002), which is MATα ura3-52 leu2 his3 sec9-4, and LRB757, which is MATα ura3-52 leu2 his3 yck1-Δ1 yck2-2ts (Robinson et al., 1993). Finally, NDY1864, a strain that constitutively expresses the artificial ER marker protein GFP-HDEL from the TPI1P, was constructed by integrating a Stu I-cut, URA3-based integrating plasmid harboring the GFP-HDEL expression allele at the ura3-52 locus of LRB759. The GFP-HDEL integrating plasmid was derived by swapping the TPI1P-KAR2-GFP-HDEL fragment from YIplac204-GFP-HDEL (Rossanese et al., 2001) onto the URA3-based integrating vector pRS306 (Sikorski and Hieter, 1989). A similar strategy was then used to integrate alleles for GAL1P-driven expression of 6xHis/FLAG/HA-tagged versions of Yck2(wt) and Yck2(Δ396-528) into the leu2 locus of NDY1864. For these integrations, the two YCK2 constructs were first subcloned onto the LEU2-based integrating vector pRS305 (Sikorski and Hieter, 1989), which was linearized with BstXI to direct specific integration.
Plasmids
Both the Ura3-Yck2 fusion proteins and the in-frame Yck2 deletion mutants, which together provide the main focus for the present work, derive from a series of XhoI restriction site mutations that were introduced into Yck2 coding sequence all with the same reading frame, such that coding for two consecutive amino acid residues is replaced by the CTCGAG hexanucleotide sequence, encoding the Leu-Glu dipeptide sequence. XhoI site replacements were made at sequences encoding the following 13 Yck2 dipeptides, using standard methods for site-directed mutagenesis (Kunkel et al., 1987): Ser30Asn31, Ala58Ser59, Tyr120Arg121, Gly207Gln208, Lys361Leu362, Ser396Lys397, Arg461Glu462, Lys495Gln496, Asn503Gly504, Pro510Tyr511, Ala527Gln528, Ser535Ser536, and Ser541Lys542. In addition, a Leu-Glu-encoding XhoI site was inserted between the Yck2 codons for Met1 and Ser2.
Ura3-Yck2 fusion constructs
The Ura3-Yck2 constructs, which have various C-terminal portions of Yck2 attached to the C terminus of Ura3, all are carried on the LEU2/CEN/ARS vector plasmid pRS315 (Sikorski and Hieter, 1989). These fusion proteins are expressed from the inducible GAL1 promoter and, for detection, have the tripartite 6xHis/FLAG/HA epitope tag (Sun et al., 2004) at the Ura3 N terminus. They were derived from the GAL1P-6xHis/FLAG/HA-URA3(Sal I) plasmid pND645, which has a Val-Asp–encoding SalI site added to the URA3 open reading frame, immediately before the translational termination codon. This URA3 SalI site is engineered to be in the same reading frame as the previously mentioned YCK2 XhoI sites (SalI and XhoI sticky ends are compatible), thus allowing in-frame fusions of Ura3 to the various C-terminal Yck2 portions. To construct the Ura3-Yck2 fusions, the 1.7 kb SacI to SalI GAL1P-6xHis/FLAG/HA-URA3 fragment from pND645 was used to replace the upstream SacI to XhoI fragment within the appropriate YCK2 XhoI site mutant (SacI is sited within the upstream polylinker). For each Ura3-Yck2 fusion, the fusion joint encodes a Val-Glu dipeptide from the joined SalI/XhoI restriction site.
Yck2 in-frame deletion mutants
In-frame deletion mutations were constructed within YCK2 by ligating the relevant fragments through their XhoI sites. For instance, the Δ58–397 is derived by ligating the XhoI site at codons 58–59 to the site at codons 396–397. Thus this mutant, like all of these deletion mutants, has the deleted codons replaced by an XhoI site–encoded Leu-Glu dipeptide. The deletion mutants were constructed into two different URA3/CEN/ARS pRS316-based formats (Sikorski and Hieter, 1989), with expression being driven either by the GAL1 promoter or by the native YCK2 promoter. Expression from the GAL1 promoter affords a 10-fold overexpression of Yck2, facilitating both IIF microscopy and analysis of palmitoylation. The YCK2P-driven series was used primarily for testing complementation. The GAL1P-driven deletion mutants all derive from the Yck2(wt) construct, pND1427, which has the 6xHis/FLAG/HA epitope tri-tag at the Yck2 N terminus. The YCK2P-driven series derive from the Yck2(wt) construct, pND1987, which has a dual FLAG/HA epitope tag at the Yck2 N terminus.
Other Yck2 mutants
Derivatives of pND1427 that mutate the C-terminal Cys-Cys dipeptide to Ser-Ser [Yck2(SS)], to Cys-Cys-Ile-Ile-Ser [Yck2(CCIIS)], and to Ser-Cys-Ile-Ile-Ser [Yck2(SCIIS)] have been previously described (Roth et al., 2002). The di-alanine substitution mutants and the kinase-inactive mutants were derived from pND1427 via standard methods for site-directed mutagenesis (Kunkel et al., 1987), with the fidelity of the introduced mutation being confirmed by DNA sequencing. The expression plasmids for overexpression and purification of the di-alanine–subsituted Yck2 were derived from pND1483, the pET-based expression plasmid for expression of the kinase-inactive 6xHis/FLAG/HA-Yck2(D218A), originally used in our in vitro palmitoylation analyses (Roth et al., 2002).
Yeast cultures
For GAL1P-driven expression, plasmid-transformed yeast cells were inoculated from selective plates into YP-Raf medium (1% yeast extract, 2% peptone, 2% raffinose) for overnight log-phase growth. The next day, following appropriate culture dilution and an additional 2-h period of log-phase growth, cultures were subjected to a 2-h galactose-induction period, initiated with the addition of 2% galactose to YP-Raf cultures. For cultures intended for IIF microscopy, the 2-h galactose-induction period was terminated with the addition of 3% glucose, with cells harvested 30 min later. The final glucose repression period is intended to “chase” newly synthesized proteins to their final endpoint localizations.
IIF microscopy
Cells were fixed, solubilized, and processed for antibody staining as previously described (Sun et al., 2004), using a 1-h room temperature incubation with a 1:1000 dilution of HA.11 mAb (Covance, Princeton, NJ) as the primary antibody, followed by 1 h with Cy3-conjugated goat anti–mouse immunoglobulin G (IgG) secondary antibody. Images were captured and simultaneously deconvolved using a Zeiss Axioplan (Thornwood, NY) equipped with an ApoTome structured illumination module.
Palmitoylation analyses
Palmitoylation was assessed for the different Yck2 mutants by using one of two different approaches, either 1) by ABE methodology or 2) by metabolic labeling with the alkyne-tagged palmitic acid analog, 17-octadecynoic acid (Cayman Chemicals, Ann Arbor, MI), followed by an in vitro click reaction with azido-AlexaFluor647.
ABE detection of palmitoylation
A scaled-down version of the proteomic ABE protocol was used as has been previously described (Politis et al., 2005; Roth et al., 2006). In brief, denatured protein extracts, prepared from yeast cells expressing the FLAG/HA-tagged proteins of interest, were processed through the three ABE steps: 1) blockade of free thiols with N-ethylmaleimide, 2) cleavage of the thioester-linked acyl modifications with neutral pH hydroxylamine, and 3) marking of the newly uncovered acylation site cysteinyl thiols with the thiol-specific biotinylation reagent HPDP-biotin (Thermo Fisher Scientific, Pittsburgh, PA). Finally, the protein of interest is purified by anti-FLAG-agarose (Sigma-Aldrich, St. Louis, MO) immunoprecipitation, then subjected to Western blotting both with anti-biotin–horseradish peroxidase (Sigma-Aldrich) to detect palmitoylation and with anti-HA–horseradish peroxidase (Roche, Basel, Switzerland) to assess protein recovery and expression.
Click detection of palmitoylation
The Charron et al. click labeling protocol (Charron et al., 2009) was adapted to yeast as follows. Log-phase yeast cultures expressing FLAG/HA-epitope tagged from the GAL1 promoter were labeled with 25 μM 17-octadecynoic acid [added from 25 mM stock solution in dimethyl sulfoxide (DMSO); stored at –20°C] for the second hour of the 2-h galactose induction (see Yeast cultures earlier in the text). At the end of the labeling period, 108 labeled cells were harvested by centrifugation and resuspended into 100 μl of cold LB (50 mM Tris/Cl, 150 mM NaCl, 5 mM EDTA, pH 7.4) containing 2 mM phenylmethylsulfonyl fluoride (PMSF) and 2× PI (1 × PI: 250 μg/ml each of pepstatin, antipain, chymostatin, and leupeptin). The cell suspension was then lysed with five 45-s intervals of vortexing with a 100 μl volume of acid-washed glass beads (212- to 300-μm diameter; Sigma-Aldrich). Lysate was decanted away to a second tube, with the beads being washed with an additional 150 μl of LB, containing 1 mM PMSF and 1× PI. This second supernatant then was pooled with first lysate, with lysate membranes then being solubilized with the addition of Triton X-100 1% and a 30-min incubation at 4°C with gentle rotation. Unbroken cells then were removed by centrifugation (6000 × g, 30 s), and protein from 150 μl of the cleared lysate was then collected by chloroform-methanol precipitation (Wessel and Flugge, 1984). The resulting precipitate was dissolved into 25 μl of 4% SDS, 50 mM Tris/Cl, 5 mM EDTA, pH 7.4, with a 10-min incubation at 37°C. FLAG/HA-tagged proteins then were purified from 20 μl of denatured protein by immunoprecipitation (1 h, 4°C incubation with anti-FLAG agarose in 800 μl of LB, containing 0.2% Triton X-100, 1 mM PMSF, and 1× PI). After three washes with 1 ml of LB containing 0.2% Triton X-100 and 0.1% SDS, bound protein was eluted (5 min, 65°C) into 25 μl of 4% SDS, 50 mM triethanolamine, pH 7.4. Twenty microliters of the eluted protein was adjusted to 47 μl with the addition of 27 μl of CB (50 mM triethanolamine, 150 mM NaCl, 0.5% Triton X-100) with 1 mM PMSF and 1× PI. To initiate the click reaction, 3 μl of a mix composed of 5 μl of 10 mM azido-Alexa Fluor 647 (prepared in DMSO; Invitrogen, Carlsbad, CA), 5 μl of 10 mM Tris[(1-benzyl-1H-1,2,3-triazol-4y1)methyl-1]amine prepared in DMSO, 10 μl of 50 mM Tris(2-carboxyethyl)phosphine hydrochloride (freshly prepared), and 10 μl of 50 mM CuSO4/5H2O (freshly prepared). Following a 1-h incubation at room temperature, 100 μl of CB was added, and the reaction was terminated by chloroform-methanol precipitation (Wessel and Flugge, 1984). The protein precipitate was dissolved in 25 μl of SB (8 M urea, 5% SDS, 40 mM Tris/Cl, 0.1 mM EDTA, 0.4 mg/ml bromophenol blue, 1% β-mercaptoethanol) and heated at 65°C for 5 min prior to SDS–PAGE. Alexa Fluor 647 labeling was detected by fluorescent scans of the gel using a Typhoon 9200 (GE HealthCare). An identical second gel was analyzed by using anti-HA Western blotting to assess the expression of the different mutant proteins.
Phosphatase treatments
Harvested cell pellets, corresponding to 1.5 × 107 cells, were rapidly resuspended in 180 μl of cold 1.4 M sorbitol, 25 mM Tris/Cl. Forty-five microliters of 85% TCA was added, and samples were then flash frozen on dry ice (samples may be stored at –80°C). Then, a 200-μl volume of acid-washed glass beads was added, with cell suspension then being thawed on ice before disruption with 10 min of autovortexing at room temperature. After the addition of 250 μl of 5% TCA, the lysate was decanted away from the glass beads to a fresh tube. The glass beads were washed with 300 μl of 5% TCA, which was pooled together with the initial lysate. After 20 min at 0°C, precipitated protein was collected from the samples by centrifugation (10 min, 16,000 × g). After washing with 1 ml of acetone, the protein pellet was dessicated and then dissolved in 100 μl SB for 10 min at 37°C. Finally, to digest with phosphatase, 2-μl aliquots of the final protein extracts were diluted to 1 ml with the addition of PB (20 mM citrate/Na, 50 mM NaCl, pH6.0), containing 1 mM PMSF and 1× PI. Then, 30 μg of potato acid phosphatase (Roche) was added, and samples were incubated at room temperature for 1 h. Finally, 2 μg of bovine serum albumin was added as carrier, and the digested proteins were TCA precipitated, desiccated, and dissolved into 20 μl of SB for 10 min at 65°C, before SDS–PAGE and anti-HA Western analysis.
Complementation analysis
Single colonies of LRB757 yeast carrying URA3/CEN/ARS plasmids for YCK2P-driven expression of different Yck2 mutants were picked from selective, minus-uracil plates into 1 ml of YPD medium (1% yeast extract, 2% peptone, 2% glucose). Three microliter aliquots of the 10−1, 10−2, and 10−3 dilutions were spotted onto two YPD plates that were subsequently incubated for 2 d at 30°C (permissive temperature) and at 34°C (nonpermissive temperature).
In vitro palmitoylation
The in vitro palmitoylation analysis used eight different Yck2 substrate proteins, including the previously used, kinase-inactive Yck2(D218A) and its nonpalmitoylatable Yck2(D218A,SS) derivative, which lacks the C-terminal cysteines (Roth et al., 2002), as well as six new di-alanine-substituted mutants, which were also constructed within the kinase-inactive D218A Yck2 protein context. The kinase-inactivating D218A eliminates an extreme hyperphosphorylation that occurs in E. coli, which precludes palmitoylation within the in vitro system (unpublished data). The N-terminally 6xHis/FLAG/HA-tagged proteins were overexpressed in E. coli using the pET expression system (Novagen, Madison, WI) and isolated by Ni-NTA-agarose (Qiagen, Valencia, CA) affinity chromatography from clarified cell lysates (Roth et al., 2002). C-terminally 3×HA/FLAG/6xHis-tagged, but otherwise wt Akr1 was affinity-purified from yeast as previously described (Roth et al., 2002). The in vitro reaction, which monitored transfer of the [3H]palmitate label from [3H]palmitoyl-CoA to the Yck2 substrate protein was performed as previously described (Roth et al., 2002).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) R01 GM65525 (to N.G.D.). We thank Lucy Robinson (Louisiana State University, Shreveport) for providing yck1 yck2 and sec9 strains, Ben Glick (University of Chicago) for the GFP-HDEL construct, and Liz Conibear (University of British Columbia) for her insightful critique of the manuscript. For assistance with the microscopy, we thank Mary Olive, Charlie Harkins, and the Microscopy, Imaging and Cytometry Resources Core, which is supported by NIH Center grant P30CA22453 to The Karmanos Cancer Institute, Wayne State University, and the Perinatology Research Branch of the National Institute of Child Health and Development, Wayne State University.
Abbreviations used:
- ABE
acyl-biotinyl exchange
- CCTP
conserved C-terminal peptide
- CK1
type 1 casein kinase
- CTD
C-terminal domain
- DMSO
dimethyl sulfoxide
- ER
endoplasmic reticulum
- HPDP-biotin
N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide
- IIF
indirect immunofluorescence
- KD
kinase catalytic domain
- MPD
minimal palmitoylation domain
- PAT
protein acyl-transferase
- PMSF
phenylmethylsulfonyl fluoride
- TMD
transmembrane domain
- wt
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-02-0115) on June 8, 2011.
REFERENCES
- Abdel-Sater F, El Bakkoury M, Urrestarazu A, Vissers S, Andre B. Amino acid signaling in yeast: casein kinase I and the Ssy5 endoprotease are key determinants of endoproteolytic activation of the membrane-bound Stp1 transcription factor. Mol Cell Biol. 2004;24:9771–9785. doi: 10.1128/MCB.24.22.9771-9785.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu P, Bryan JD, Panek HR, Jordan SL, Forbrich BM, Kelley SC, Colvin RT, Robinson LC. Plasma membrane localization of the Yck2p yeast casein kinase 1 isoform requires the C-terminal extension and secretory pathway function. J Cell Sci. 2002;115:4957–4968. doi: 10.1242/jcs.00203. [DOI] [PubMed] [Google Scholar]
- Babu P, Deschenes RJ, Robinson LC. Akr1p-dependent palmitoylation of Yck2p yeast casein kinase 1 is necessary and sufficient for plasma membrane targeting. J Biol Chem. 2004;279:27138–27147. doi: 10.1074/jbc.M403071200. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- Conibear E, Davis NG. Palmitoylation and depalmitoylation dynamics at a glance. J Cell Sci. 2010;123:4007–4010. doi: 10.1242/jcs.059287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–872. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Feng Y, Davis NG. Akr1p and the type I casein kinases act prior to the ubiquitination step of yeast endocytosis: Akr1p is required for kinase localization to the plasma membrane. Mol Cell Biol. 2000;20:5350–5359. doi: 10.1128/mcb.20.14.5350-5359.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Greaves J, Prescott GR, Fukata Y, Fukata M, Salaun C, Chamberlain LH. The hydrophobic cysteine-rich domain of SNAP25 couples with downstream residues to mediate membrane interactions and recognition by DHHC palmitoyl transferases. Mol Biol Cell. 2009;20:1845–1854. doi: 10.1091/mbc.E08-09-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Salaun C, Fukata Y, Fukata M, Chamberlain LH. Palmitoylation and membrane interactions of the neuroprotective chaperone cysteine-string protein. J Biol Chem. 2008;283:25014–25026. doi: 10.1074/jbc.M802140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL. Chemical probes for the rapid detection of fatty-acylated proteins in mammalian cells. J Am Chem Soc. 2007;129:2744–2745. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hicke L, Zanolari B, Riezman H. Cytoplasmic tail phosphorylation of the alpha-factor receptor is required for its ubiquitination and internalization. J Cell Biol. 1998;141:349–358. doi: 10.1083/jcb.141.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, John Peter AT, Meiringer C, Subramanian K, Ungermann C. Analysis of DHHC acyltransferases implies overlapping substrate specificity and a two-step reaction mechanism. Traffic. 2009;10:1061–1073. doi: 10.1111/j.1600-0854.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- Huang K, Sanders S, Singaraja R, Orban P, Cijsouw T, Arstikaitis P, Yanai A, Hayden MR, El-Husseini A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, et al. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Kang R, et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Kurotsu F, Sano T, Iwaki S, Igarashi Y. Long-chain base kinase Lcb4 Is anchored to the membrane through its palmitoylation by Akr1. Mol Cell Biol. 2005;25:9189–9197. doi: 10.1128/MCB.25.21.9189-9197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Li J, Mahajan A, Tsai MD. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry. 2006;45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- Lobo S, Greentree WK, Linder ME, Deschenes RJ. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2002;277:41268–41273. doi: 10.1074/jbc.M206573200. [DOI] [PubMed] [Google Scholar]
- Longenecker KL, Roach PJ, Hurley TD. Three-dimensional structure of mammalian casein kinase I: molecular basis for phosphate recognition. J Mol Biol. 1996;257:618–631. doi: 10.1006/jmbi.1996.0189. [DOI] [PubMed] [Google Scholar]
- Moriya H, Johnston M. Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proc Natl Acad Sci USA. 2004;101:1572–1577. doi: 10.1073/pnas.0305901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadolski MJ, Linder ME. Molecular recognition of the palmitoylation substrate Vac8 by its palmitoyltransferase Pfa3. J Biol Chem. 2009;284:17720–17730. doi: 10.1074/jbc.M109.005447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal G, Paraz MT, Kellogg DR. Regulation of Mih1/Cdc25 by protein phosphatase 2A and casein kinase 1. J Cell Biol. 2008;180:931–945. doi: 10.1083/jcb.200711014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panek HR, Stepp JD, Engle HM, Marks KM, Tan PK, Lemmon SK, Robinson LC. Suppressors of YCK-encoded yeast casein kinase 1 deficiency define the four subunits of a novel clathrin AP-like complex. EMBO J. 1997;16:4194–4204. doi: 10.1093/emboj/16.14.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanayotou I, Sun B, Roth AF, Davis NG. Protein aggregation induced during glass bead lysis of yeast. Yeast. 2010;27:801–816. doi: 10.1002/yea.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis EG, Roth AF, Davis NG. Transmembrane topology of the protein palmitoyl transferase Akr1. J Biol Chem. 2005;280:10156–10163. doi: 10.1074/jbc.M411946200. [DOI] [PubMed] [Google Scholar]
- Robinson LC, Menold MM, Garrett S, Culbertson MR. Casein kinase I-like protein kinases encoded by YCK1 and YCK2 are required for yeast morphogenesis. Mol Cell Biol. 1993;13:2870–2881. doi: 10.1128/mcb.13.5.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Feng Y, Chen L, Davis NG. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol. 2002;159:23–28. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, 3rd, Davis NG. Global analysis of protein palmitoylation in yeast. Cell. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun C, Greaves J, Chamberlain LH. The intracellular dynamic of protein palmitoylation. J Cell Biol. 2010;191:1229–1238. doi: 10.1083/jcb.201008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smotrys JE, Linder ME. Palmitoylation of intracellular signaling proteins: regulation and function. Annu Rev Biochem. 2004;73:559–587. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- Spielewoy N, Flick K, Kalashnikova TI, Walker JR, Wittenberg C. Regulation and recognition of SCFGrr1 targets in the glucose and amino acid signaling pathways. Mol Cell Biol. 2004;24:8994–9005. doi: 10.1128/MCB.24.20.8994-9005.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchkov DV, DeFlorio R, Draper E, Ismael A, Sukumar M, Arkowitz R, Stone DE. Polarization of the yeast pheromone receptor requires its internalization but not actin-dependent secretion. Mol Biol Cell. 2010;21:1737–1752. doi: 10.1091/mbc.E09-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Chen L, Cao W, Roth AF, Davis NG. The yeast casein kinase Yck3p is palmitoylated, then sorted to the vacuolar membrane with AP-3-dependent recognition of a YXXPhi adaptin sorting signal. Mol Biol Cell. 2004;15:1397–1406. doi: 10.1091/mbc.E03-09-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima JY, Nakanishi J, Mizuno K, Toshima J, Drubin DG. Requirements for recruitment of a G protein-coupled receptor to clathrin-coated pits in budding yeast. Mol Biol Cell. 2009;20:5039–5050. doi: 10.1091/mbc.E09-07-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, Mc Guffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Xu RM, Carmel G, Sweet RM, Kuret J, Cheng X. Crystal structure of casein kinase-1, a phosphate-directed protein kinase. EMBO J. 1995;14:1015–1023. doi: 10.1002/j.1460-2075.1995.tb07082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.