

Abstract
The mitochondrial DEAD-box proteins Mss116p of Saccharomyces cerevisiae and CYT-19 of Neurospora crassa are ATP-dependent helicases that function as general RNA chaperones. The helicase core of each protein precedes a C-terminal extension and a basic tail, whose structural role is unclear. Here we used small-angle X-ray scattering to obtain solution structures of the full-length proteins and a series of deletion mutants. We find that the two core domains have a preferred relative orientation in the open state without substrates, and we visualize the transition to a compact closed state upon binding RNA and adenosine nucleotide. An analysis of complexes with large chimeric oligonucleotides shows that the basic tails of both proteins are attached flexibly, enabling them to bind rigid duplex DNA segments extending from the core in different directions. Our results indicate that the basic tails of DEAD-box proteins contribute to RNA-chaperone activity by binding nonspecifically to large RNA substrates and flexibly tethering the core for the unwinding of neighboring duplexes.
Keywords: ribozyme, RNA folding, RNA helicase, RNA–protein interaction
DEAD-box proteins comprise the largest family of helicases and play critical roles in all aspects of RNA metabolism, including RNA splicing, translation, ribosome assembly, RNA degradation, and RNA transport (1, 2). Although their functions vary broadly, all DEAD-box proteins have a conserved helicase core consisting of two RecA-like domains (denoted domains 1 and 2) separated by a flexible linker and operate by a mechanism involving conformational changes within this core (3–6). These conformational changes couple cycles of ATP binding and hydrolysis to RNA binding and release by the core, enabling DEAD-box proteins to promote local RNA unwinding and remodeling of structured RNAs and RNA–protein complexes (3–5).
Structural and biochemical studies have given important insights into how DEAD-box proteins unwind RNA (2–4). The two helicase core domains are separated from each other in an open state in the absence of substrates, but they interact to form a compact, closed state upon binding of ATP and RNA. In this closed state, the interface between the two core domains forms a catalytic site for ATP hydrolysis and an RNA-binding cleft, which can accommodate a short region of a duplex strand (7). The tight binding of the RNA strand within this cleft results in a bend that is incompatible with partner-strand base pairing, leading to local RNA unwinding (7–10). ATP hydrolysis and dissociation of the Pi product are then necessary to release the bound single-stranded RNA (9, 11, 12).
Whereas the helicase core is central to the biochemical activities of DEAD-box proteins, most also have substantial extensions or additional domains at their N- and/or C-termini (1, 3). These extensions vary widely in size, composition, and function, but many are thought to interact with RNA or protein components of complexes to direct the DEAD-box proteins to desired sites of action. A common type of extension is a basic C-terminal sequence (C-tail), which is typically predicted to lack a defined structure, and in some cases, truncation of the C-tail has been shown to affect activities of DEAD-box proteins (13–16). Unstructured extensions, such as C-tails, are absent from the crystal structures of DEAD-box proteins reported to date, likely reflecting their flexibility or disorder, and thus there is little direct information about how they function together with the helicase core.
As general RNA chaperones, the DEAD-box proteins Mss116p of Saccharomyces cerevisiae and CYT-19 of Neurospora crassa are important model systems for studying the mechanisms of DEAD-box proteins (Fig. 1A). These proteins interact nonspecifically with RNA substrates and disrupt stable inactive structures that limit productive RNA folding (17–19). Mss116p is required for the efficient splicing of 13 mitochondrial (mt) group I and group II introns and also functions in translational activation and RNA end processing (20). It consists of a helicase core that is flanked on one side by an N-terminal extension (NTE) and on the other by a functionally important α-helical C-terminal extension (CTE) and a basic tail (C-tail; Fig. 1A). CYT-19 plays similar roles in mt RNA metabolism and is to a large degree functionally interchangeable with Mss116p in vitro and in vivo (17, 19–21).
Fig. 1.

DEAD-box proteins Mss116p and CYT-19 and nucleic acid substrates. (A) Schematic representations of the domain architectures of Mss116p, CYT-19, and deletion mutants. Mss116p consists of a mt targeting sequence, which is cleaved in vivo and absent in the constructs used here (white); an N-terminal extension, which corresponds to the N-terminus of the mature proteins (NTE; dark blue); a helicase core of two RecA-like domains (domain 1 and domain 2; light blue and green, respectively), which are joined by a flexible linker (gray); a structured C-terminal extension (CTE; orange); and a basic hydrophilic tail (C-tail; red). CYT-19 consists of the same elements but with a shorter NTE. (B) The crystal structure of the closed-state helicase core and the CTE of Mss116p [Protein Data Bank (PDB) ID code 3I6I (22)] with domains colored as in A. The bound single-stranded U10-RNA and the ATP-analogue 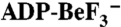 are shown in yellow and black, respectively. (C) Schematic representations of nucleic acid substrates. RNA and DNA nucleotides are shown in yellow and gray, respectively, and nucleic-acid secondary structure was predicted using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi).
are shown in yellow and black, respectively. (C) Schematic representations of nucleic acid substrates. RNA and DNA nucleotides are shown in yellow and gray, respectively, and nucleic-acid secondary structure was predicted using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi).
High-resolution X-ray crystal structures have been obtained for Mss116p in complex with a single-stranded U10-RNA oligonucleotide and three different ATP analogs (AMP-PNP,  , and
, and  ) (22). These structures show the entire closed-state helicase core (residues 88–505) and CTE (residues 506–596), and they reveal that the CTE packs against the core, stabilizing it and functioning in RNA unwinding (Fig. 1B). The helicase core induces a bend in the bound RNA, similar to that seen in other DEAD-box proteins, and the CTE produces a second bend resulting in RNA crimping. Because the C-tail was not present in the crystallized construct of Mss116p and the NTE was not visible, their structures and how they might contribute to RNA-chaperone function have remained unclear.
) (22). These structures show the entire closed-state helicase core (residues 88–505) and CTE (residues 506–596), and they reveal that the CTE packs against the core, stabilizing it and functioning in RNA unwinding (Fig. 1B). The helicase core induces a bend in the bound RNA, similar to that seen in other DEAD-box proteins, and the CTE produces a second bend resulting in RNA crimping. Because the C-tail was not present in the crystallized construct of Mss116p and the NTE was not visible, their structures and how they might contribute to RNA-chaperone function have remained unclear.
Based on sequence similarity and limited proteolysis studies, CYT-19 is predicted to have a helicase core, CTE, and basic C-tail similar to those of Mss116p (Fig. 1A). The C-tails of Mss116p and CYT-19 enhance RNA binding, and they have been suggested to contribute to RNA-chaperone activities by forming additional, nonspecific interactions with RNA substrates (13, 14). These interactions could tether the core to structured RNAs, either rigidly or with sufficient flexibility to allow it to sample neighboring regions and binding orientations while remaining near the substrate. However, the ability of C-tails to bind separate sites on nucleic acid substrates and the degree of flexibility between the C-tail and the helicase core upon RNA binding have not been determined for these or any other DEAD-box protein.
Here we use small-angle X-ray scattering (SAXS) to obtain solution structures of full-length Mss116p and CYT-19 and a series of deletion mutants both in the open and closed states. Our results reveal the locations of the NTE and the C-tail; enable us to visualize the conformational changes that occur upon binding of RNA and adenosine nucleotide; and indicate a mechanism in which the C-tail flexibly tethers the core to large, physiological RNA substrates.
Results
Experimental Strategy.
For both Mss116p and CYT-19, we collected SAXS data for full-length proteins and for constructs lacking the basic C-tails (Fig. 1A). Additionally, for Mss116p we used versions that lack the NTE, which is much smaller and not conserved in CYT-19. All of these truncations retain the helicase core and CTE regions present in the previous X-ray crystal structures of Mss116p (Fig. 1B). Below, we first present data for Mss116p by itself and in complex with nucleic acid substrates (Fig. 1C), and then we present corresponding data for CYT-19.
SAXS Reconstructions Reveal the Spatial Organization of Full-Length Mss116p.
The SAXS profiles of Mss116p constructs in solution are shown in Fig. 2A. Particle molecular weights were estimated from the scattering intensity at zero angle (I(0)) when calibrated against protein and RNA standards and indicated that all of the constructs are monomeric and free from aggregates under these solution conditions (Table S1). Values for the radius of gyration (Rg) ranged from 32.8 Å for the minimal Mss116p/ΔNTE + ΔC-tail construct to 38.0 Å for full-length Mss116p (Table S1) and were in good agreement with the hydrodynamic radii of the proteins measured by size-exclusion chromatography (SEC) (Table S2).
Fig. 2.

SAXS analysis of Mss116p in the open state without substrates. SAXS data are shown for full-length Mss116p (dark blue), Mss116p/ΔC-tail (light blue), Mss116p/ΔNTE (green), and Mss116p/ΔNTE + ΔC-tail (red). (A) Scattering profiles, which are displaced along the logarithmic axis for visualization, are shown as the logarithm of the scattering intensity, I (black dots), as a function of the momentum transfer, q = 4π sin(θ)/λ, where 2θ is the scattering angle and λ is the X-ray wavelength. The solid curves overlaying the SAXS data are the expected scattering profiles of the corresponding BUNCH models (see below). (B) Normalized distance distribution functions calculated from the scattering profiles using the program AUTOGNOM (27). (C–F) Ab initio and rigid-body SAXS reconstructions of the open state of full-length Mss116p (C), Mss116p/ΔC-tail (D), Mss116p/ΔNTE (E), and Mss116p/ΔNTE + ΔC-tail (F). Low-resolution envelopes calculated by DAMMIN are shown separately (Upper) and superposed onto atomic models determined by BUNCH (Lower). In this and other figures, protein domains are colored as in Fig. 1 and views are rotated by 90 ° about the vertical axis for each model.
The scattering profiles can be represented in real space by a distance distribution function, P(r), revealing two maxima for each protein construct (Fig. 2B). This pattern indicates the presence of a dumb-bell-like scattering macromolecule, as expected for the two separated helicase core domains in the open state (3). The first maximum, at approximately 30 Å for each construct, corresponds to the size of the domains, and the second at approximately 50 Å corresponds to their separation (23). Estimates of the maximum particle diameter, Dmax, for the different constructs vary from 115–135 Å (Table S1) and suggest that, in the absence of ligands, Mss116p adopts an elongated conformation in solution.
To obtain more specific information on solution structures of the helicase core and to evaluate the positions of the C-tail and NTE, we generated particle envelopes from the SAXS data using DAMMIN and GASBOR (24, 25) and atomic models using BUNCH (26) (Fig. 2, Fig. S1A, and Table S3). For the latter method, we used the domain structures determined by X-ray crystallography and modeled the C-tail and NTE as unstructured extensions, as predicted from their sequences (14) and supported by far ultraviolet circular dichroism (far-UV CD) spectra (Fig. S2 and Table S4). Reconstructed envelopes for all of the protein constructs revealed well-separated lobes corresponding to the two domains of the helicase core (Fig. 2), and the scattering data were well-described by single molecular models calculated using BUNCH (χ = 0.67–2.13; Fig. 2A and Table S3).
In addition to the core domains, the SAXS envelope for full-length Mss116p included two clear protrusions that were not seen in the NTE and/or C-tail deletion mutants (Fig. 2C). The locations of these protrusions indicate that the C-tail extends outward from domain 2 and the NTE extends from domain 1 on the opposite face of the protein. Although crystal structures of DEAD-box proteins in the absence of ligands have suggested that the two core domains are separated from each other and free to adopt different relative orientations (3), our reconstructions indicate that Mss116p core domains are restricted sufficiently that the extensions appear as discrete protrusions. This result implies that the two domains exist in a preferred relative orientation, even in the absence of substrates (see Discussion).
The best-fit molecular models for those constructs containing the C-tail depict the tail in a strongly bent conformation. Although the full conformational ensemble of the C-tail cannot be determined from these data, they raised the possibility of a conformational bias that predisposes the C-tail to being less than fully extended. To explore this question further, we performed rigid-body modeling with conformational sampling. Starting from the simplest model in which the C-tail and NTE are fully unstructured, an ensemble optimization method was used to select a group of conformers that best describes the scattering profile for each protein construct from a large pool of conformers that randomly samples the conformational space of unstructured polypeptides (28). The optimized ensembles for proteins that include the C-tail have Rg and Dmax distributions with lower averages than those of the pool of random structures (Fig. S3), suggesting that the C-tail is biased to be more compact than expected for a fully random chain. In contrast, the ensembles for the mutants lacking the C-tail have Rg and Dmax distributions similar to or slightly larger than those of the corresponding random pools (Fig. S3). Thus, we conclude that the C-tail has a conformational bias that predisposes it to be in proximity to domain 2.
The C-tail Is Positioned to Bind RNA Extending from the Helicase Core.
We next used SAXS to study the closed-state conformation of Mss116p by forming complexes with U10-RNA substrate and the nonhydrolyzable ATP analog ADP-BeFx (Fig. 3). Mss116p binds tightly to U10-RNA in the presence of ADP-BeFx under the SAXS buffer conditions (Fig. S4), and the formation of a stable complex was confirmed by SEC (Fig. S5 and Table S2). The Rg values for the U10-RNA bound proteins range from 26.3–33.8 Å, indicating a decrease of at least 4 Å upon formation of the closed state (Table S1). The experimental scattering profile and Rg values measured for closed-state Mss116p/ΔNTE + ΔC-tail agree with those calculated from the crystal structure of the equivalent complex (Fig. 3A and Table S3), indicating that the closed complex of Mss116p in solution is similar to that in the crystal.
Fig. 3.

SAXS analysis of Mss116p ternary complexes with ssRNA and a nonhydrolyzable ATP analog. (A) Scattering profiles and (B) normalized distance distribution functions for full-length Mss116p (dark blue), Mss116p/ΔC-tail (light blue), Mss116p/ΔNTE (green), and Mss116p/ΔNTE + ΔC-tail (red) bound to U10-RNA and ADP-BeFx. In (A), the scattering profiles are shown as black dots, and the solid curves overlaying the data are the expected scattering profiles of the corresponding BUNCH models, except for the Mss116p/ΔNTE + ΔC-tail complex where the overlay is the expected scattering profile calculated from the corresponding X-ray crystal structure using CRYSOL (29). (C–F) SAXS reconstructions of full-length Mss116p (C), Mss116p/ΔC-tail (D), Mss116p/ΔNTE (E), and Mss116p/ΔNTE + ΔC-tail (F) in the closed state. Low-resolution envelopes calculated by DAMMIN (Upper) are colored as in (A) and atomic models (Lower) are shown aligned inside the DAMMIN envelope (gray).
The P(r) functions for the U10-RNA complexes are bell-like and are typical of compact globular particles (23), indicating that the two core domains are no longer separated but instead behave as a single structural module (Fig. 3B), as seen in the crystal structure (Fig. 1B) (22). Therefore, we used this structure as a constraint in BUNCH reconstructions. Models generated using DAMMIN, GASBOR, and BUNCH gave good internal agreement, and the SAXS envelopes accommodated the two interacting core domains in the closed state (Fig. 3 C–F and Fig. S1B). Importantly, the reconstructions indicate that the NTE and C-tail protrude from the helicase core in positions similar to those in the open, unbound state, with no indication that the C-tail interacts with the RNA-binding cleft or other regions of the core (Fig. 3). The latter result provides evidence against models in which the C-tail communicates allosterically with the core. Rather, the disposition of the C-tail adjacent to domain 2 suggests that it is poised to interact with nucleic acid extending from the RNA-binding cleft of the core, a hypothesis that is probed further by experiments below.
The C-tail Interacts with Longer Nucleic Acid Substrates.
The position of the C-tail extending adjacent to the U10-RNA bound to the core is consistent with previous models in which the C-tails of Mss116p and CYT-19 interact with nearby RNA regions (13, 14). To test this model and to probe the degree of flexibility between RNA-binding interactions of the C-tail and the helicase core, we examined the solution structures of versions of Mss116p with or without the C-tail in complexes with larger chimeric substrates that contain a short ssRNA linked to a DNA duplex extension. These substrates were based on similar ones used in previous DEAD-box protein studies (8, 13, 30) and were composed of an 11-nt ssRNA covalently linked at either its 5′-end (RNA-DNA duplex 1) or 3′-end (RNA–DNA duplex 2) to a 10-bp hairpin DNA duplex (Fig. 1C). Such DNA extensions do not compete effectively with ssRNA for binding to the helicase core, which relies on interactions with 2′-OH groups of RNA, but can interact functionally with the C-tail, as demonstrated previously by enhancements in RNA unwinding (13). Thus, both substrates are expected to bind with the ssRNA in the RNA-binding cleft of the core and the rigid duplex extending out from one or the other end, arriving at very different spatial positions depending on its polarity. SAXS data and ab initio reconstructions for the substrates by themselves (Fig. S6) are consistent with them adopting the predicted secondary structure shown in Fig. 1C.
SAXS profiles were determined for constructs of Mss116p with or without the C-tail in complex with ADP-BeFx and either RNA–DNA duplex 1 or RNA–DNA duplex 2 (denoted substrates 1 and 2, respectively; Fig. 4A). The complexes were isolated by SEC prior to SAXS measurements to ensure sample homogeneity (Fig. S5). Although Mss116p and other DEAD-box proteins can potentially function as oligomers on large substrates (5, 14), the SEC and SAXS measurements indicate that Mss116p binds both substrates as a monomer under these conditions. Rg values were approximately 36 and 33 Å for complexes in the presence and absence of the C-tail, respectively, and P(r) functions displayed characteristics of globular species (Fig. 4B and Table S1). We used the multiphase bead-modeling program MONSA to reconstruct ab initio models that include two phases, protein and nucleic acid, by accounting for the different scattering intensities of these components (24, 31). Several independent runs gave reproducible models that had an average normalized spatial discrepancy of less than 0.65 and described the scattering data well (χ = 0.7–1.5) (Table S3).
Fig. 4.

Binding modes of Mss116p to large nucleic acid substrates. (A) Scattering profiles and (B) normalized distance distribution functions for Mss116p/ΔNTE (green) and Mss116p/ΔNTE + ΔC-tail (red) bound to ADP-BeFx and either RNA–DNA duplex 1 (solid lines) or RNA-DNA duplex 2 (dashed lines). In (A), the scattering profiles are shown by black dots, and the colored lines represent the fit of the ab initio model of the complex obtained by MONSA. (C–F) Ab initio multiphase reconstructions from the SAXS data in (A) of complexes of Mss116p/ΔNTE + ΔC-tail with either RNA–DNA duplex 1 (substrate 1; C) or RNA–DNA duplex 2 (substrate 2; D) and complexes of Mss116p/ΔNTE with either RNA–DNA duplex 1 (E) or RNA–DNA duplex 2 (F). Two-phase models of protein (colored as above) and nucleic acid (yellow) were reconstructed by MONSA (Upper). The ssRNA regions of these substrates were assumed to bind within the helicase core in the orientation observed in the crystal structure of the closed state of Mss116p (Fig. 1B). This information was used to place manually atomic models for protein and nucleic acid inside the corresponding SAXS envelopes (Lower).
The reconstructions for the minimal helicase core construct Mss116p/ΔNTE + ΔC-tail indicate that the polarity of the duplex region dictates its position and orientation. For RNA–DNA duplex 1, the DNA duplex region of the substrate protrudes at the 5′ end of the ssRNA, close to the CTE of Mss116p and along the long axis of the protein (Fig. 4C). This position is near the region occupied by the C-tail in the intact protein in solution (see Figs. 2 and 3 above), and indeed the reconstruction of the same duplex substrate in complex with the Mss116p construct that includes the C-tail shows additional volume that corresponds to protein in this region, indicating interaction of the C-tail with the duplex extension (Fig. 4E). In contrast, the duplex region of RNA–DNA duplex 2 extends from the 3′ end of the ssRNA, closer to the center of the helicase core and nearly perpendicular to the long axis of the protein (Fig. 4D). Strikingly, the reconstruction of this complex with Mss116p including the C-tail indicates that the changes in position and orientation of the duplex extension are matched by the C-tail, such that it remains associated with the duplex (Fig. 4F), presumably interacting by electrostatic interactions with the negatively charged nucleic acid. These results indicate that the C-tail and core bind different sites of large substrates and that the C-tail remains anchored to the extensions while moving relative to the core over a wide region of space.
CYT-19 Behaves Similarly to Mss116p in Solution.
To explore whether the behavior of Mss116p is characteristic of other DEAD-box protein RNA chaperones, we performed additional SAXS experiments with CYT-19, which performs similar functions to Mss116p (17). CYT-19 lacks a significant NTE but is similar to Mss116p in that the helicase core is followed by a homologous CTE and a C-tail, which is predicted to be unstructured (Fig. 1A). The C-tail of CYT-19 is shorter than that of Mss116p (approximately 49 residues based on partial proteolysis results compared to 68 residues for Mss116p), but it is more basic and has a higher proportion of arginine residues (13, 14).
We performed SAXS measurements on two versions of CYT-19, a wild-type construct and one in which the C-tail was removed by deleting the C-terminal 49 amino acids (Fig. 5 and Fig. S7) (13). As for Mss116p, the ab initio and rigid-body SAXS reconstructions indicate that CYT-19 in the absence of ligands has an extended structure with two distinct globular domains (Fig. 5 A–C) and that this open conformation compacts to a closed globular structure upon binding of ADP-BeFx and U10-RNA (Fig. 5 D–F). In both the closed and open conformations, BUNCH atomic models show that the C-tail emerges from domain 2 in a position to form additional RNA contacts similar to Mss116p. Importantly, the ternary complexes with the large RNA–DNA substrates 1 and 2 gave scattering profiles that are well-described by MONSA models in which CYT-19 binds as a monomer and the C-tail interacts with the duplex extension regardless of its polarity (Fig. 5 G–I). Thus, the position and flexibility of the C-tail are properties common to Mss116p and CYT-19.
Fig. 5.

SAXS analysis of CYT-19. (A–C) SAXS data for full-length CYT-19 (green) and CYT-19/ΔC-tail (red) in the absence of ligands. (A) Normalized distance distribution functions. (B) and (C) low-resolution envelopes calculated by DAMMIN (Upper) and BUNCH atomic models (Lower), which are colored as in Fig. 1 and aligned inside the DAMMIN envelope (gray). BUNCH models were generated using a homology model of CYT-19 that is based upon its sequence similarity to Mss116p (see SI Methods). (D–F) SAXS data for CYT-19 bound to U10–RNA and ADP-BeFx, shown in the same arrangement as in (A–C). For the minimal CYT-19/ΔC-tail complex, the DAMMIN envelope in (F) is aligned to the homology model for CYT-19. (G–I) SAXS data for full-length CYT-19 bound to large nucleic acid substrates. (G) Normalized distribution functions for CYT-19-ADP-BeFx bound to RNA-DNA-duplex 1 (solid green line) and RNA-DNA-duplex 2 (dashed green line). (H and I) Ab initio multiphase reconstructions of CYT-19 in complex with RNA–DNA duplex 1 (substrate 1) and RNA–DNA duplex 2 (substrate 2), respectively. Two-phase models of protein (green) and nucleic acid (yellow) were constructed by MONSA (Upper) and atomic models for protein and nucleic acid were manually placed inside the corresponding SAXS envelopes (Lower).
Discussion
Here, we used SAXS to obtain solution structures of full-length DEAD-box proteins bound to nucleic acid substrates. These structures give important insights into the architectures of the RNA-chaperone proteins Mss116p and CYT-19 and their interactions with RNA. They show directly that the two domains of the helicase core undergo a large-scale, global compaction to a “closed” conformation upon binding RNA and adenosine nucleotide in solution, supporting models proposed from crystallography and fluorescence measurements (3). Further, they reveal the locations of the basic tail and NTE in Mss116p, and provide insight into the function of the basic tail in binding to large substrates.
The SAXS results indicate that the NTE, which is attached to domain 1 of Mss116p, is on the opposite face of the protein from the bound RNA. It apparently does not contact RNA but may interact with protein partners, such as the mt RNA polymerase (32). The C-tail is attached to the CTE of domain 2. It does not appear to interact with the helicase core but is instead positioned adjacent to domain 2, where it extrudes into the solvent and is available to bind to structured RNAs.
The protrusions corresponding to the NTE and C-tail provide landmarks on each of the two core domains that enable us to infer their relative orientation in both the open and closed states. Surprisingly, we find that the two core domains of Mss116p adopt a preferred relative orientation even in the open state in the absence of substrates. This result was not expected from previous X-ray crystallographic studies of DEAD-box proteins in the absence of substrates, which showed that the two core domains could exist in multiple relative orientations and suggested that they were free to move independently of each other via the flexible linker (3). The preferred orientation of the two domains found for Mss116p could reflect that the linker is less flexible than was believed previously and/or that there are transient or unstable interactions between the domains in the open state. Although it is not known if all DEAD-box proteins behave similarly in this respect, prealignment of the two core domains prior to substrate binding would be advantageous in reducing the entropic cost of binding ATP and RNA at sites formed at the domain interface.
Importantly, the SAXS data of RNA-protein complexes provides information about the structure, function, and flexibility of the C-tail, which could not be obtained by X-ray crystallography. In complexes with nucleic acid substrates that include rigid duplex DNA extensions at either the 5′ or 3′ end of a short ssRNA segment, the C-tail interacts with the extension regardless of the polarity. Because the ssRNA portion of the substrate can bind the core in only one orientation, the attached duplex segments extend from the core in very different directions. The ability of the tail to contact both extensions indicates that it is sufficiently flexible to interact with neighboring parts of large substrates over a wide arc extending from the core (Fig. 6A).
Fig. 6.

The tethering range observed for the basic tail of Mss116p when bound to nucleic acid substrates. (A) Range of motion of Mss116p’s flexibly attached C-tail. The cone shows the lower limit for the region of space over which the C-terminal tail of Mss116p can bind nucleic acid, as indicated by MONSA reconstructions of complexes with chimeric substrates containing duplex DNA extensions. (B) Model of Mss116p interacting via the C-tail with the Tetrahymena group I intron ribozyme (http://www-ibmc.u-strasbg.fr/upr9002/westhof/index.html). The model shows that, when anchored by the flexible C-tail, the helicase core of Mss116p can act at numerous sites over a wide region of the RNA.
In the context of large RNAs that are the physiological substrates for these DEAD-box proteins, we envision the basic tail as being fixed by nonspecific, electrostatic interactions with the RNA and then functioning as a tether to allow the helicase core to interact with nearby single-stranded and double-stranded regions of the RNA (Fig. 6B). These regions can include segments that are adjacent in linear sequence, as well as those that are noncontiguous but close in tertiary structure (33). Binding of the core to ssRNA segments could enhance folding by sequestering these segments, stabilizing intermediates or preventing formation of kinetically trapped species, whereas binding to dsRNA segments disrupts kinetic traps as the strands are separated in an ATP-dependent unwinding reaction. The two strands are then released at different times, allowing them to find new partners and the RNA to refold (18, 19, 30, 34).
Our model suggests that RNA binding by the DEAD-box protein C-tail plays multiple roles in RNA-chaperone activity. First, the interactions of the C-tail increase the affinity for large RNA substrates, thereby enhancing activity. For CYT-19, the C-tail has been shown to increase the efficiency for unwinding RNA duplexes with extensions, implying that the C-tail binds the extension and that this interaction is maintained in the transition state for duplex unwinding (13). Second, the C-tail may remain bound to the RNA substrate after the core has undergone a cycle of RNA binding and release, thus allowing the protein to perform multiple rounds of unwinding while remaining tethered to the structured RNA. Under these circumstances, the flexibility of the tail relative to the core, as shown here, would be critical to allow the core to change positions and orientations during large-scale RNA conformational transitions of complex RNAs, such as group I and group II introns. An analogous mechanism may apply to DEAD-box proteins that use ancillary domains to target specific RNAs or RNA–protein complexes, such as the bacterial DbpA/YxiN DEAD-box proteins (35–38). In these cases, the RNA-unwinding activity of the helicase core would be limited to the spatial proximity of the specific tethering site. A final possibility is that the unstructured tails participate in disruption of RNA structure, as many positively charged proteins have been shown to possess RNA-chaperone activity, presumably by binding preferentially to unstructured regions of RNA (39). However, for CYT-19, Mss116p, and other DEAD-box proteins that have been tested, the principal unwinding activity arises from the helicase core, as only low levels of unwinding are observed in the absence of ATP, and much of this activity presumably reflects ATP-independent unwinding by the core (8).
Although tethering by the C-tail is likely to be broadly important for the RNA-chaperone activity, the available evidence suggests that its functional contribution can vary between DEAD-box proteins. The activity of CYT-19 appears to depend strongly upon the C-tail, as its removal compromises the RNA binding and RNA unwinding of extended duplexes by 7–20-fold (13). For Mss116p, however, deletion of the C-tail and a small neighboring region of the CTE has smaller effects, causing only a two- to fivefold decrease in splicing efficiency for group II introns aI5γ and bI1 in vitro and a modest decrease in levels of spliced COX1 and COB mRNAs in strains containing multiple group I and II introns in vivo (14). Thus, it is possible that the C-tail plays a greater role in the functions of CYT-19 than Mss116p. Mss116p is highly expressed (40) and nonspecific binding by the core in the absence of adenosine nucleotide appears to be stronger than for CYT-19 (14). Further, Mss116p and other DEAD-box proteins appear to function as oligomers under some conditions, raising the possibility that one protomer can provide an anchor for RNA binding by another protomer, analogous to the role of the C-tail (5, 14). These differences may lead to less dependence by Mss116p on the basic C-tail for RNA binding and unwinding.
Flexible basic tails are a common feature of DEAD-box proteins and are also found in the related SWI/SNF family of superfamily 2 helicase proteins, members of which interact with DNA and function in chromatin remodeling (1, 3, 6, 39, 41, 42). Interestingly, members of both groups of proteins bind nonspecifically to nucleic acid structures and promote local structural rearrangements. Basic polypeptide extensions are also found in other classes of RNA-binding proteins, raising the possibility that these tails are a hallmark of proteins that bind nonspecifically to RNAs of diverse structure. Consistent with this hypothesis is the observation that an unstructured, basic tail in the La protein participates in binding to a variety of RNAs (43). The unstructured and pliable nature of these extensions, as indicated by the results here, may be critical to allow the flexibility necessary for them to interact productively with the diverse shapes and binding surfaces presented by structured RNAs.
Methods
Proteins were expressed and purified as described (13, 22). Complexes with proteins were assembled at room temperature prior to SAXS measurements, and all proteins, substrates and complexes were purified to homogeneity by SEC in 20 mM Tris-HCl (pH 7.5), 500 mM KCl (200 mM KCl for CYT-19), 10% glycerol, 1 mM DTT, 5 mM MgCl2. Sample concentrations for SAXS were 1–3 mg/mL. SAXS data were collected at the Advanced Photon Source beamlines 12-ID-C and 18-ID-D with twenty 1 s exposures recorded for each sample (12 keV) at a sample-detector distance of 2 m. For each species, data were collected at three concentrations to test for monodispersity. Analysis of SAXS data, ab initio reconstructions, and molecular modeling were done as described in SI Methods. Structural figures were prepared using PyMOL (http://www.pymol.org/).
Supplementary Material
Acknowledgments.
We thank Michael Brenowitz and Eckhard Jankowsky for comments on the manuscript. This work was supported by National Institutes of Health Grants GM037951 (A.M.L.) and GM070456 (R.R.). A.L.M is a recipient an EMBO Long-Term Fellowship (ALTF 389-2010) and was supported, in part, by a Research Fellowship from St John’s College, Cambridge, UK.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109566108/-/DCSupplemental.
References
- 1.Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 2.Jankowsky E, Fairman ME. RNA helicases—one fold for many functions. Curr Opin Struct Biol. 2007;17:316–324. doi: 10.1016/j.sbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol Chem. 2009;390:1237–1250. doi: 10.1515/BC.2009.135. [DOI] [PubMed] [Google Scholar]
- 4.Jarmoskaite I, Russell R. DEAD-box proteins as RNA helicases and chaperones. WIREs: RNA. 2011;2:135–152. doi: 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jankowsky E. RNA helicases at work: Binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linder P. Dead-box proteins: A family affair—active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, et al. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc Natl Acad Sci USA. 2008;105:20203–20208. doi: 10.1073/pnas.0811075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F, Putnam A, Jankowsky E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc Natl Acad Sci USA. 2008;105:20209–20214. doi: 10.1073/pnas.0811115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Q, Del Campo M, Lambowitz AM, Jankowsky E. DEAD-box proteins unwind duplexes by local strand separation. Mol Cell. 2007;28:253–263. doi: 10.1016/j.molcel.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Henn A, et al. Pathway of ATP utilization and duplex rRNA unwinding by the DEAD-box helicase, DbpA. Proc Natl Acad Sci USA. 2010;107:4046–4050. doi: 10.1073/pnas.0913081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao W, et al. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-box proteins. J Mol Biol. 2011;409:399–414. doi: 10.1016/j.jmb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grohman JK, et al. Probing the mechanisms of DEAD-box proteins as general RNA chaperones: The C-terminal domain of CYT-19 mediates general recognition of RNA. Biochemistry. 2007;46:3013–3022. doi: 10.1021/bi0619472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohr G, et al. Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro. J Mol Biol. 2008;375:1344–1364. doi: 10.1016/j.jmb.2007.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bizebard T, Ferlenghi I, Iost I, Dreyfus M. Studies on three E. coli DEAD-box helicases point to an unwinding mechanism different from that of model DNA helicases. Biochemistry. 2004;43:7857–7866. doi: 10.1021/bi049852s. [DOI] [PubMed] [Google Scholar]
- 16.Yang Q, Jankowsky E. ATP- and ADP-dependent modulation of RNA unwinding and strand annealing activities by the DEAD-box protein DED1. Biochemistry. 2005;44:13591–13601. doi: 10.1021/bi0508946. [DOI] [PubMed] [Google Scholar]
- 17.Mohr S, Stryker JM, Lambowitz AM. A DEAD-box protein functions as an ATP-dependent RNA chaperone in group I intron splicing. Cell. 2002;109:769–779. doi: 10.1016/s0092-8674(02)00771-7. [DOI] [PubMed] [Google Scholar]
- 18.Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature. 2007;449:1014–1018. doi: 10.1038/nature06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Campo M, et al. Unwinding by local strand separation is critical for the function of DEAD-box proteins as RNA chaperones. J Mol Biol. 2009;389:674–693. doi: 10.1016/j.jmb.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang HR, et al. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc Natl Acad Sci USA. 2005;102:163–168. doi: 10.1073/pnas.0407896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohr S, Matsuura M, Perlman PS, Lambowitz AM. A DEAD-box protein alone promotes group II intron splicing and reverse splicing by acting as an RNA chaperone. Proc Natl Acad Sci USA. 2006;103:3569–3574. doi: 10.1073/pnas.0600332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Del Campo M, Lambowitz AM. Structure of the yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol Cell. 2009;35:598–609. doi: 10.1016/j.molcel.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svergun DI, Koch MHJ. Small-angle scattering studies of biological macromolecules in solution. Rep Prog Phys. 2003;66:1735–1782. [Google Scholar]
- 24.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J Appl Crystallogr. 2007;40:S223–S228. [Google Scholar]
- 28.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 29.Svergun D, Barberato C, Koch MHJ. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 30.Tijerina P, Bhaskaran H, Russell R. Nonspecific binding to structured RNA and preferential unwinding of an exposed helix by the CYT-19 protein, a DEAD-box RNA chaperone. Proc Natl Acad Sci USA. 2006;103:16698–16703. doi: 10.1073/pnas.0603127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svergun DI, Nierhaus KH. A map of protein-rRNA distribution in the 70 S Escherichia coli ribosome. J Biol Chem. 2000;275:14432–14439. doi: 10.1074/jbc.275.19.14432. [DOI] [PubMed] [Google Scholar]
- 32.Markov DA, et al. Identification of proteins associated with the yeast mitochondrial RNA polymerase by tandem affinity purification. Yeast. 2009;26:423–440. doi: 10.1002/yea.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Q, Jankowsky E. The DEAD-box protein Ded1 unwinds RNA duplexes by a mode distinct from translocating helicases. Nat Struct Mol Biol. 2006;13:981–986. doi: 10.1038/nsmb1165. [DOI] [PubMed] [Google Scholar]
- 34.Karunatilaka KS, Solem A, Pyle AM, Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature. 2010;467:935–939. doi: 10.1038/nature09422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardin JW, Hu YX, McKay DB. Structure of the RNA binding domain of a DEAD-box helicase bound to its ribosomal RNA target reveals a novel mode of recognition by an RNA recognition motif. J Mol Biol. 2010;402:412–427. doi: 10.1016/j.jmb.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karow AR, Klostermeier D. A structural model for the DEAD box helicase YxiN in solution: Localization of the RNA binding domain. J Mol Biol. 2010;402:629–637. doi: 10.1016/j.jmb.2010.07.049. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, Overgaard MT, Hu Y, McKay DB. The Bacillus subtilis RNA helicase YxiN is distended in solution. Biophys J. 2008;94:L01–03. doi: 10.1529/biophysj.107.120709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, et al. The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold. RNA. 2006;12:959–967. doi: 10.1261/rna.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajkowitsch L, et al. RNA chaperones, RNA annealers, and RNA helicases. RNA Biol. 2007;4:118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 40.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 41.Lopez-Ramirez V, Alcaraz LD, Moreno-Hagelsieb G, Olmedo-Alvarez G. Phylogenetic distribution and evolutionary history of bacterial DEAD-box proteins. J Mol Evol. 2011;72:413–431. doi: 10.1007/s00239-011-9441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: Genetics, genomics, and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kucera NJ, Hodsdon ME, Wolin SL. An intrinsically disordered C terminus allows the La protein to assist the biogenesis of diverse noncoding RNA precursors. Proc Natl Acad Sci USA. 2011;108:1308–1313. doi: 10.1073/pnas.1017085108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.