

Abstract
The final interprotein electron transfer (ET) in the mammalian respiratory chain, from cytochrome c (Cyt c) to cytochrome c oxidase (CcO) is investigated by 1H-15N heteronuclear single quantum coherence spectral analysis. The chemical shift perturbation in isotope-labeled Cyt c induced by addition of unlabeled CcO indicates that the hydrophobic heme periphery and adjacent hydrophobic amino acid residues of Cyt c dominantly contribute to the complex formation, whereas charged residues near the hydrophobic core refine the orientation of Cyt c to provide well controlled ET. Upon oxidation of Cyt c, the specific line broadening of N-H signals disappeared and high field 1H chemical shifts of the N-terminal helix were observed, suggesting that the interactions of the N-terminal helix with CcO are reduced by steric constraint in oxidized Cyt c, while the chemical shift perturbations in the C-terminal helix indicate notable interactions of oxidized Cyt c with CcO. These results suggest that the overall affinity of oxidized Cyt c for CcO is significantly, but not very much weaker than that of reduced Cyt c. Thus, electron transfer is gated by dissociation of oxidized Cyt c from CcO, the rate of which is controlled by the affinity of oxidized Cyt c to CcO for providing an appropriate electron transfer rate for the most effective energy coupling. The conformational changes in Lys13 upon CcO binding to oxidized Cyt c, shown by 1H- and 1H, 15N-chemical shifts, are also expected to gate intraprotein ET by a polarity control of heme c environment.
Keywords: interaction site, ET complex, isotope edited-NMR, unidirectional ET, redox-dependent interaction
Electron transfer (ET) plays a crucial role in the process of oxidative phosphorylation in the respiratory chain. Electron flow within transmembrane complexes in the mitochondria leads to transport of protons across the inner mitochondrial membrane. This process is coupled to synthesis of ATP. The electron flow is terminated at cytochrome c oxidase (CcO), where a dioxygen molecule is reduced to two water molecules in a four-electron reduction reaction. The four electrons are consecutively donated by cytochrome c (Cyt c) a small one-electron carrier (1). To rapidly and specifically dock with CcO in the concentrated protein milieu of the intermembrane space of the mitochondria, Cyt c has been proposed to specifically interact with partner proteins to regulate the ET reaction.
One of the keys for elucidating the ET mechanism is to identify the interaction site of the protein–protein complex in the ET reaction. Based on the surface charges of Cyt c, ET from Cyt c to CcO has been considered to involve formation of one or more Cyt c-CcO ET complexes in a process guided by electrostatic interactions (1, 2). The dependence of the ET rate on ionic strength suggests that a significant contribution of electrostatic interactions leads to formation of the ET complex. The results of a docking simulation also indicate that binding of these two proteins involves electrostatic interactions between a Lys patch on the surface of Cyt c and a group of carboxylate residues on CcO (3). Extensive chemical modification studies have indicated that a group of Lys residues, Lys13, Lys72, Lys86, Lys27, and Lys87 (in decreasing order of importance), on the surface of Cyt c are responsible for the interaction of Cyt c with CcO (4). However, these chemical modification studies necessitated the addition of a bulky side chain to the modification site, which interferes with specific binding of CcO and suppresses the ET process. In fact, a docking simulation (5) has suggested that Lys72 is the only residue that forms a salt bridge with an acidic residue of CcO and that the other Lys residues would not interact with the acidic residues of CcO. Lys13, whose chemical modification induces a drastic suppression of the ET reaction, is located at the supposed interface, but the side chain of Lys13 remains rigid and buried in the interior of the protein during the docking simulation. The side chain is not in an electrostatically favorable position in terms of participating in the interaction between the two proteins (5). No experimental interpretation has been reported for the different conclusions obtained from the chemical modification analyses and the docking simulations.
Certain biological interprotein ET systems exhibit “gating” ET reactions that are controlled by dynamics of the protein (particularly at the interface of the protein) rather than intrinsic ET properties (6). The gating ET mechanism has been theoretically proposed to be a general property of biological ET systems (7) and redox-dependent protein–protein interactions are suggested to occur within the bacterial ET complex (8). In fact, the apparent ET rate (10–102 s-1) from Cyt c to CcO (9) is much slower than the rate estimated from electrochemically triggered redox reaction of a surface-modified Cyt c bound to self-assembled monolayers on a gold electrode (∼107 s-1) (10). Rapid electron injection using photoexcited metal complexes was also found to exhibit a fast ET rate (∼105 s-1) (11). The lower rate of ET from Cyt c to CcO strongly suggests that a conformational change at the protein interface is required for the interprotein ET reaction, which leads to the redox-dependent gating mechanism (11). The significant structural differences observed between reduced and oxidized Cyt c (12, 13) also support the proposal that conformational changes accompany the ET process. In contrast, no significant redox-dependent conformational changes on the surface of CcO have been reported (14, 15). Thus, it appears that redox-dependent conformational changes of Cyt c are crucial for gating the ET process which provides the reaction with unidirectional character.
One of the most powerful experimental methods for direct characterization of interaction sites of the ET complex and analysis of interactions at the atomic level is multidimensional isotope edited-NMR spectroscopy. To identify the Cyt c sites that interact with CcO in the mammalian respiratory chain, we used detergent-solubilized intact bovine CcO which includes all 13 different subunits (15) and [15N]- or [13C, 15N]-labeled Cyt c (16). By using a triple resonance technique, the NMR signals of the N-H groups in [13C, 15N]-labeled Cyt c were assigned. The chemical shift perturbations of N-H groups associated with the addition of CcO to a solution of Cyt c were monitored by 1H-15N heteronuclear single quantum coherence (HSQC) spectra, and these chemical shift perturbations were assigned to direct interactions between Cyt c and CcO. Based on this structural information of the CcO interaction site of Cyt c, we found that the hydrophobic interactions of the exposed heme group and adjacent hydrophobic amino acids drive the predominant interaction that positions the two redox centers in close proximity. The distribution of charged residues at the interaction site fine-tunes the orientation of the binding of Cyt c to CcO to regulate the ET reaction. Furthermore, these electrostatic and hydrophobic interactions which depend upon the redox states of Cyt c induce a redox-dependent conformational change at the interaction interface which is expected to facilitate unidirectional transfer of electrons from Cyt c to CcO.
Results
Chemical Shift Perturbation Mapping of Interaction Sites in Reduced Cyt c.
To identify the CcO interaction site of Cyt c and to examine the interprotein interactions which are crucial for the process of ET from Cyt c to CcO, 1H-15N HSQC spectra of [15N]-labeled reduced Cyt c were measured in the absence and presence of unlabeled fully reduced CcO. The addition of fully reduced CcO is likely to increase the rotational correlation time and nonspecific line broadening of the cross peaks induced by the large molecular mass of the complex. However, we successfully detected a specifically enhanced line broadening of the N-H peaks originating from Glu4, Ile9, Ile11, Lys13, Asn31, Lys79, Lys88, and Glu90 (Fig. S1). The line widths of these signals were found to increase over the range of the CcO titration (Fig. S2), indicating the existence of intermolecular interactions between these residues and CcO.
In addition to the specific line broadening, some of the peaks in the 1H-15N HSQC spectrum of Cyt c exhibit chemical shift changes associated with the addition of CcO (Fig. S3). These chemical shift perturbations strongly suggest that changes in local protein conformation alter the magnetic shielding and/or that the residues are in direct contact with a counterpart protein (17). Fig. 1A displays chemical shift changes in the 1H (upper box) and 1H-15N dimensions (lower box). The asterisks represent amino acid residues producing signals with specific line broadening upon addition of CcO.
Fig. 1.

Chemical shift perturbations in reduced Cyt c in the presence of CcO. (A). Overview of the combined backbone chemical shift changes (bar graph) and line broadening (asterisk) induced by binding to CcO. (B). Mapping of the affected residues on the reduced Cyt c surface. The left and right boxes are 180º rotations about the vertical axis. The residues with chemical shift changes of 0.020 > x, 0.020 ≤ x < 0.025, 0.025 ≤ x < 0.030, 0.030 ≤ x < 0.035, and 0.035 ≤ x are colored with gray, aqua, green, yellow, and orange, respectively. Residues in red show specific line broadening.
As depicted in Fig. 1A, 1H signals from Lys5, Lys7, Phe10, Met12, His18, Gly24, Ile81, Ile85, Lys86, Lys87, Glu89, Ala92, and Asp93 have significant chemical shifts (|ΔδH| > 0.02 ppm) associated with the addition of CcO. The N-H cross peaks from Lys5, Lys7, Lys8, Phe10, Met12, Cys14, Cys17, His18, Gly24, His33, Ile81, Ile85, Lys86, Lys87, Glu89, Arg91, Arg92, Arg93, and Arg95 are also found to be substantially shifted (ΔδN,H > 0.02 ppm). These amino acid residues are localized in the N-terminal helix (H1: 3–13), loop 7 (L7: 81–87), and C-terminal helix (H5: 88–101) regions (Fig. S4), suggesting that the CcO binding site includes these regions. The calculated values in Fig. 1A were used to color-code the Cyt c structure (13) in Fig. 1B. As clearly shown in Fig. 1B, perturbed residues are located in the N- and C-terminal regions on the side of the exposed hydrophobic heme periphery.
Chemical Shift Perturbation Mapping of Interaction Sites in Oxidized Cyt c.
1H-15N HSQC spectra of oxidized Cyt c were also measured in the absence and presence of fully oxidized CcO (Fig. S5) to elucidate the conformational changes occurring in the interaction sites associated with the ET process. Specific line broadening and chemical shift changes were detected in 1H-15N HSQC spectra of oxidized Cyt c in the presence of CcO as they had been for reduced Cyt c. Specific line broadening was identified for resonances originating from Asp2, Cys14, Gly29, Met80, Ile81, Val83, Lys88, and Glu90. 1H signals of Lys5, Gly6, Ile9, Phe10, Met12, Lys13, Ser15, His18, Lys79, Ile85, Lys86, Lys87, Glu89, Asp93, Ile94, and Ala96, and the N-H cross peaks from Lys5, Gly6, Ile9, Phe10, Ile11, Met12, Lys13, Ser15, Cys17, His18, Lys79, Ile85, Lys86, Lys87, Glu89, Arg91, Ala93, Ile94, and Ala96 undergo specific chemical shift changes upon addition of CcO (|ΔδH| > 0.02 ppm, ΔδN,H > 0.02 ppm) (Fig. 2A). These perturbed residues in oxidized Cyt c are also localized in the N- and C-terminal regions on the side of the exposed hydrophobic edge of the Cyt c heme moiety (Fig. 2B). A similar observation was made for reduced Cyt c (Fig. 1B).
Fig. 2.

Chemical shift perturbations in oxidized Cyt c in the presence of CcO. (A). Overview of the combined backbone chemical shift changes (bar graph) and line broadening (asterisk) induced by binding to CcO. (B). Mapping of the affected residues on the oxidized Cyt c surface. The left and right boxes are 180º rotations about the vertical axis. The residues with chemical shift changes of 0.020 > x, 0.020 ≤ x < 0.030, 0.030 ≤ x < 0.040, 0.040 ≤ x < 0.050, and 0.050 ≤ x are colored with gray, aqua, green, yellow, and orange, respectively. Residues in red show specific line broadening.
As depicted in Fig. 2A, chemical shift changes in oxidized Cyt c are enhanced to a greater extent relative to those of reduced Cyt c. Such enhanced chemical shift changes in the oxidized state originate from the paramagnetic effects of the ferric heme iron (18). Although most of the amino acid residues of oxidized Cyt c which exhibit chemical shift changes and specific line broadening correspond to those in reduced Cyt c, some of the signals are preferentially perturbed in either the reduced or the oxidized state of Cyt c. Particularly, the 1H chemical shift perturbations of amide protons of the N-terminal helix of oxidized Cyt c are significantly different from those of reduced Cyt c, while the C-terminal region in oxidized Cyt c shows a similar 1H chemical shift perturbation pattern to that in reduced Cyt c (top boxes in Figs. 1A and 2A). This chemical shift perturbation pattern suggests that certain structural perturbations associated with oxidation of the heme iron of Cyt c are induced at the interaction interface. Therefore, oxidized Cyt c interacts with CcO on the same side of the protein, but significant rearrangements in the interactions are induced when the heme iron of Cyt c is oxidized.
Discussion
Identification of the Interaction Site for CcO in Cyt c.
As shown in Figs. 1 and 2, the chemical shift perturbations were successfully detected on the specific side of Cyt c involved in formation of the distinct CcO interaction site. Most of the chemical shift changes are less than 0.05 ppm, reflecting rather small conformational changes which occur upon binding of Cyt c to CcO. These experimentally significant changes in the present NMR analyses are expected to correspond to conformational changes which are much smaller than those detectable by 3D structural analyses (at the level of 0.1 Å).
In Fig. 3, perturbed residues associated with the binding of CcO in Cyt c in both oxidation states are color-coded according to charge and hydrophobicity of the side chain. Unexpectedly negatively charged residues in addition to Lys residues are included in the interaction surface. In fact, previous studies have never focused on negatively charged residues, because it has been believed that basic residues of Cyt c predominantly control the interaction with the acidic surface of CcO. The negative charges in the positively charged patch are expected to weaken the attractive electrostatic interactions if the net positive charge is critical for the interaction between the two proteins. Thus, the electrostatic interactions between mammalian Cyt c and CcO are unlikely to represent the predominant interaction for stabilization of the complex as suggested by the previous molecular dynamics calculations for small model proteins (19). Although no direct ionic interaction was reported in the high resolution structure of the cross-linked Cyt c—CcP complex (20), the ET rate from Cyt c to CcP depends on the ionic strength (20) and thermodynamic parameters indicate significant contribution of the electrostatic interaction to the formation of the ET complex (21). Therefore, the present results strongly suggest that charged amino acid residues in the interaction site contribute to the process of adjusting the orientation of Cyt c with respect to CcO through their electrostatic interactions, in order to facilitate the strictly controlled ET reaction.
Fig. 3.

Amino acid residues affected by binding to CcO in reduced (A) and oxidized Cyt c (B). Positively charged, negatively charged, and nonpolar residues are shown in blue, red, and yellow, respectively.
It is notable here that the 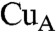 site of subunit II of CcO, which is thought to act as the site of entry of electrons into CcO, is surrounded by aromatic amino acid residues including Trp104, Tyr105, Tyr121, and Phe206, which form an exposed hydrophobic cluster (22). The hydrophobic heme edge and adjacent hydrophobic amino acid residues of Cyt c, as shown in Fig. 3, can therefore interact with the hydrophobic cluster at the
site of subunit II of CcO, which is thought to act as the site of entry of electrons into CcO, is surrounded by aromatic amino acid residues including Trp104, Tyr105, Tyr121, and Phe206, which form an exposed hydrophobic cluster (22). The hydrophobic heme edge and adjacent hydrophobic amino acid residues of Cyt c, as shown in Fig. 3, can therefore interact with the hydrophobic cluster at the 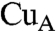 site. The involvement of the hydrophobic amino acid residues near the exposed heme edge in the interaction site indicates that the hydrophobic interaction is the dominant interaction for positioning the two redox centers in close proximity to each other.
site. The involvement of the hydrophobic amino acid residues near the exposed heme edge in the interaction site indicates that the hydrophobic interaction is the dominant interaction for positioning the two redox centers in close proximity to each other.
Although the contribution of Lys residues to the formation of the ET complex was reported in previous chemical modification studies (4, 23), some of the Lys residues that had been identified as interacting with CcO did not show a significant chemical shift perturbation. While chemical modification of Lys72 in Cyt c was found to cause severe inhibition of ET activity (4, 23) and a molecular simulation concluded that this Lys residue is the only residue that can form a hydrogen bond with an acidic amino acid residue in CcO (3, 5), the chemical shift and line width of the N-H peak from Lys72 in both the reduced and the oxidized Cyt c were found to be essentially insensitive to the addition of CcO (Figs. 1A and 2A). On the other hand, the NMR spectra clearly show that Lys5 [which has not been chemically modified because it is located quite far from the heme site (4, 23)], is involved in the interaction site of reduced and oxidized Cyt c.
The contribution of hydrophobic amino acid residues to the interaction with CcO, as suggested by the previous docking study (3), has been confirmed experimentally by the present NMR analyses. However, some of the charged residues such as Glu16 and Lys72, which surround the hydrophobic core and have been suggested to participate in interactions with CcO in the docking study, did not show specific chemical shift perturbations. As illustrated in Fig. 3, the charged residues interacting with CcO do not surround the hydrophobic core in the interaction site for CcO in contrast to the conclusion of the docking simulation (3). These findings demonstrate the unique and sensitive performance of NMR spectroscopy for identification of the amino acid residues participating in protein–protein interactions.
Comparisons of the Present Results with Other Electron Transfer Complexes Involving Cytochrome c.
The Cyt c—cytochrome c peroxidase (CcP) complex has been the most extensively analyzed ET complex at present (20, 21, 24, 25). The similarity in the complex formation mechanism in the CcO/Cyt c and CcP/Cyt c systems has been proposed, because the interaction sites for Cyt c in both CcP and CcO surfaces are surrounded by negatively charged amino acid residues. In fact, X-ray structural information of the CcP/Cyt c system was used as a model in a docking simulation of the CcO/Cyt c system (3). Furthermore, thermodynamic measurements (21) and a high-resolution X-ray analysis of the cross linked complex (20) revealed that hydrophobic interactions, and not electrostatic interactions, are the dominant interactions driving formation of the complex as demonstrated for the Cyt c-CcO complex by the present work. Detailed comparisons of the mechanisms of two ET systems which participate in completely different physiological processes, (Cyt c peroxidation vs. O2 reduction coupled with proton pumping), are expected to provide various insights into these ET mechanisms.
While some amino acid residues interacting with CcO are located far apart from the heme edge, such as Glu4, Phe10, Ile11, Lys88, and Arg91 (Fig. 1B), amino acid residues located near the heme moiety such as Glu16, Thr28, Lys72, and Phe82 in the CcP interaction site (21, 25) are not involved in interactions with CcO. The contribution of the N- and C-terminal helix regions to regulation of the binding orientation of Cyt c is more prominent in CcO than that in the interaction with CcP (Fig. S6). In the interaction site for CcO on Cyt c, it is notable that the location of heme edge of Cyt c outside the central part of the interaction area would not be appropriate for providing the maximum ET rate. However, the structure is expected to facilitate a strictly controlled ET from Cyt c to CcO, which is crucial for efficient energy transduction by CcO, because each ET to CcO is coupled with pumping of one proton equivalent (26). In fact, it has been shown that the energy coupling efficiency (H+/e- stoichiometry) is sensitively influenced by the electron flow rate and provides a maximum efficiency at a moderate flow rate (27).
In addition, the previous NMR study reported a distinct difference between reduced and oxidized Cyt c in terms of the size of the interaction site for CcP (25), but the size of the interaction site for CcO is essentially independent of the redox state (Fig. 3). These results suggest that the affinity of Cyt c for CcP is likely to be decreased significantly upon oxidation. The decreased affinity would enable CcP to readily receive the next molecule of reduced Cyt c. In contrast, in the CcO system, binding of the next molecule of reduced Cyt c is somewhat controlled and limited by the presence of oxidized Cyt c. This control may contribute to the efficiency of energy transduction mediated by CcO.
A similar NMR analysis has been reported for an ET complex consisting of fragmented cytochrome c552 (Cyt c552) with a 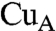 domain of Paracoccus denitrificans CcO as a model system for the ET reaction from Cyt c to CcO (18). The two hydrophobic amino acid residues Ala79 and Ala81 corresponding to Ile81 and Val83 at the interaction site of CcO in mammalian Cyt c are proposed to interact with the hydrophobic cluster at the
domain of Paracoccus denitrificans CcO as a model system for the ET reaction from Cyt c to CcO (18). The two hydrophobic amino acid residues Ala79 and Ala81 corresponding to Ile81 and Val83 at the interaction site of CcO in mammalian Cyt c are proposed to interact with the hydrophobic cluster at the 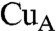 site (28, 29). However, the other hydrophobic amino acid residues at the interaction site of mammalian Cyt c, Ile9, Ile11, and Met12 are not involved in the interaction site of Cyt c552. No specific and distinct line broadening was observed for the truncated bacterial Cyt c. Thus, an enhanced hydrophobic interaction between mammalian Cyt c and CcO is expected to provide a slower exchange rate for the ET complex formation as indicated by the line broadening of the NMR signals. These results are consistent with the observation of line broadening during the binding of putidaredoxin (Pd) to putidaredoxin reductase (PdR) of Pseudomonas putida (30). The rate of this ET is much slower than that of the ET from Cyt c552 to CcO.
site (28, 29). However, the other hydrophobic amino acid residues at the interaction site of mammalian Cyt c, Ile9, Ile11, and Met12 are not involved in the interaction site of Cyt c552. No specific and distinct line broadening was observed for the truncated bacterial Cyt c. Thus, an enhanced hydrophobic interaction between mammalian Cyt c and CcO is expected to provide a slower exchange rate for the ET complex formation as indicated by the line broadening of the NMR signals. These results are consistent with the observation of line broadening during the binding of putidaredoxin (Pd) to putidaredoxin reductase (PdR) of Pseudomonas putida (30). The rate of this ET is much slower than that of the ET from Cyt c552 to CcO.
In the ET complex between cytochrome c2 and the reaction center of the bacterial photosynthetic reaction, a crucial hydrophobic amino acid residue, Phe102, is also involved in the interaction site (31). All ET complexes examined thus far indicate the existence of crucial roles for hydrophobic amino acid residues in the formation of physiologically relevant ET complexes. Hydrophobic residues are expected to play critical roles in controlling the polarity of the ET pathway.
Conformational Changes in the Interaction Site Associated with Electron Transfer: a Mechanism of the Electron Transfer Gating.
ET from Cyt c to CcO must be strictly controlled to obtain maximum efficiency in the process of energy transduction, as described above. The control mechanism remains one of the most important unresolved problems in this field. It should be noted here that, although redox-coupled conformational changes of Cyt c have been observed in 3D structural analyses, no experimental trial has been performed to elucidate the physiological relevance of the conformational changes.
As clearly shown in Fig. 4A, the chemical shift perturbation and line broadening of N-H signals originating from specific residues at the N and C termini depend upon the redox state of Cyt c. The residues which exhibit redox-dependent chemical shift perturbations are color-coded in the Cyt c structure shown in Fig. 4B. In Fig. 4A, specific line broadening was preferentially observed for the NMR signals from the N-terminal helix (Glu4, Ile9, Ile11, and Lys13) in reduced Cyt c, while chemical shift changes and line broadening of the amino acid residues in both the N- and C-terminal regions (Met12, Cys14, Ser15, His18, Met80, Ile81, Val83, Lys85, Lys86, and Asp94) were enhanced to a greater extent in oxidized Cyt c.
Fig. 4.

Comparison of the CcO binding sites between reduced and oxidized Cyt c. (A). The difference in chemical shift changes and line broadening associated with the redox state. The difference of chemical shift changes and line broadening between reduced and oxidized Cyt c (Δδoxidized-Δδreduced) is plotted against the residue number. Positive differences and line broadening in red indicate highly perturbed residues upon oxidation of Cyt c, while negative differences and line broadening in blue indicate those less perturbed. (B). Representation of the difference of the chemical shift changes and line broadening on the Cyt c surface. The left and right boxes are 90º rotations about the vertical axis. The residues at the CcO binding site are colored according to their different chemical shift changes and line broadening: positive differences (pink), line broadening in oxidized Cyt c (red), negative differences (aqua), and line broadening in reduced Cyt c (blue). Red and pink colors indicate more perturbed residues upon oxidation of Cyt c, while blue and aqua colors indicate less perturbed residues.
The specific line broadening of the NMR peak of an amide group arises from fast transverse (T2) relaxation due to the more enhanced contribution from fast relaxation of the large molecular weight complex of Cyt c and CcO. The fast relaxation suggests that the N-terminal helix in the reduced state is tightly associated with CcO. In oxidized Cyt c, no signal broadening was observed in the N-terminal helix, although specific chemical shifts associated with the addition of CcO were still observed. The N-terminal helix of oxidized Cyt c, therefore, still constitutes the interaction site for CcO, but the effect of the fast relaxation time in the Cyt c—CcO complex on the transverse relaxation time of the amide groups is reduced. The reduced effect of the fast relaxation indicates a weaker association of oxidized Cyt c with CcO after electron transfer to CcO has occurred. Redox-dependent changes in the dynamic behavior of the N-terminal helix significantly more intense than that of the C-terminals are unlikely to be induced by the intrinsic properties of the Cyt c protein, because the redox-dependence of order parameter (S2) of the N-terminal helix of free Cyt c in solution is not prominent, compared with other regions (32) (Fig. S7).
Such redox-dependent interactions of the N-terminal helix are also evident in the 1H chemical shift perturbations (top boxes of Figs. 1A and 2A). A previous study reported that the chemical shifts of amide protons correlate with the hydrogen bond length between amide proton and carbonyl oxygen in the secondary structure, and that the high field shift of the amide proton corresponds to a longer hydrogen bonding distance (33). In the reduced state, the alternating pattern of low and high field shifts was observed for both of the N- and C-terminal helices, suggesting that the binding of CcO to Cyt c induces conformational changes such as “distortion” (Fig. S8A) of the N- and C-terminal helices, where shortening and lengthening of the hydrogen bond between amide proton and the carbonyl oxygen are alternately induced.
In oxidized Cyt c, the pattern of the 1H chemical shift perturbations by the addition of CcO in the N-terminal helix is significantly different from the pattern observed for reduced Cyt c. The alternating pattern of the low and high field shifts of the C-terminal region is quite similar to the pattern observed for reduced Cyt c. In the N-terminal helix, most of the 1H chemical shifts of the amide groups showed high field shifts, corresponding to the increased distances of the hydrogen bonds between amide protons and carbonyl oxygens in the helix structure. This chemical shift perturbation pattern suggests the existence of an “expanded” helix structure (Fig. S8B). However, an alternating (or zigzag) pattern is also detectable in the high field shift pattern region which would suggest that an enhanced structural change occurs. Such a structural change would include both “expansion” and “distortion” upon CcO binding, which is a more enhanced structural change than that of reduced Cyt c upon CcO binding. The 1H chemical shifts in the N-terminal region of oxidized Cyt c suggest that the constraints to the N-terminal helix upon the CcO binding are definitely stronger in oxidized Cyt c than in reduced Cyt c.
These results together with the disappearance of line broadening in the N-terminal helix upon oxidation suggest that oxidized Cyt c has a significantly weaker affinity for CcO than reduced Cyt c. However, the 1H-, and 15N, 1H-chemical shifts in the C-terminal helix in the oxidized state are still more notable than those in the reduced state. This chemical shift perturbation pattern suggests that there are fairly strong interactions between the C-terminal helix and CcO in the oxidized state. Thus, the overall affinity of oxidized Cyt c to CcO seems to be significantly lower than that of reduced Cyt c, but the affinity difference is unlikely to be very large. The finding that the size of the area of Cyt c interacting with CcO is essentially independent of the oxidation state of Cyt c, in contrast to the redox-coupled change in the size of the area of Cyt c interacting with CcP also supports a small affinity difference for CcO between the two redox states.
Previous 3D structural analysis of the reduced and oxidized states of Cyt c also indicates the redox-dependent positional shift of the N-terminal helix (12, 13) (Fig. S5). Upon oxidation of the heme iron, the hydrogen bond between Lys13 in the N-terminal helix and Glu90 in the C-terminal helix is disrupted, leading to spatial rearrangements of the two terminal helices. As clearly illustrated in Figs. 1B and 2B, Lys13 is located at the center of the interaction site for CcO and adjacent to Cys17, which forms a thioether bond to the heme moiety. In the oxidized state, the ϵ-amino group of Lys13 would not be in the protonated state, because the side chain is buried within the protein interior. Lys13 in this conformation would not form a salt bridge with an acidic amino acid in CcO, consistent with the docking simulation analyses. On the other hand, both the large 1H- and 1H, 15N-chemical shifts of Lys13 upon CcO binding in the oxidized state as given in Fig. 2A suggest that fairly significant constraints are imposed by CcO binding. The critical role of Lys13 in the enzyme function has been established by chemical modification analyses (4, 23). Namely, the above NMR shifts of Lys13 are expected to be induced by physiologically relevant conformational changes, such as a movement of the negatively polarized nitrogen atom of the ϵ-amino group of Lys13 toward the heme c iron upon CcO binding. This movement would influence the polarity of the heme c environment to decrease the electron affinity of the oxidized heme c, transiently until oxidized Cyt c bound to CcO is replaced with the next reduced Cyt c under normal enzymatic turnover. Thus, Lys13 is also expected to contribute to electron gating.
The present NMR analyses have enabled us to propose the following ET mechanism from Cyt c to CcO. Structural changes in the N-terminal helices of Cyt c, stimulating dissociation of oxidized Cyt c from the complex as well as the constraints on Lys13 induced by binding of CcO to oxidized Cyt c, and suppressing reverse electron transfer, are expected to facilitate an “electron gating mechanism” for ET from Cyt c to CcO (or facilitating unidirectional ET). On the other hand, both the strong interaction of the C-terminal region of Cyt c with CcO and the size of the interaction area as estimated by the present NMR analyses are roughly independent of the oxidation state of Cyt c. These findings suggest that the overall affinity of oxidized Cyt c for CcO is lower than but fairly close to that of reduced Cyt c. Thus, oxidized Cyt c complexed with CcO appropriately suppresses binding of reduced Cyt c to slow down (or control) the rate of ET to CcO. The overall affinity difference is expected to be a critical characteristic which enables electron donation to CcO to be effectively coupled to energy transduction mediated by CcO.
In summary, we have detected specific line broadening and chemical shift changes in the NMR signals of mammalian Cyt c associated with the formation of the ET complex with bovine CcO. Based on these chemical shift perturbations, we have nonempirically determined the interaction site for CcO on the Cyt c protein surface, and found that the interaction site consists of charged and hydrophobic amino acid residues. The hydrophobic interactions mediated by the hydrophobic heme periphery and adjacent hydrophobic amino acid residues are the dominant interactions that position the two redox centers in close proximity, while charged residues in the interaction site refine the orientation of Cyt c with respect to CcO. Furthermore, the redox-dependent changes in the Cyt c/CcO interactions shown in the present NMR analyses indicate that the ET reaction between Cyt c and CcO is gated by regulation of the affinity of Cyt c for CcO.
Materials and Methods
Construction of the Human Cyt c Expression System and Protein Preparation.
A gene encoding human Cyt c was synthesized using a modified recursive PCR strategy. Both genes of human Cyt c and yeast Cyt c heme lyase, which form the thioether bonds necessary for the maturation of Cyt c in the host cell (32), were cloned into the pET-21 (+) vector using SacI and HindIII restriction sites. The protein expression and purification were performed according to previously published procedures (32). Purified protein with an R-value (= A409/A280) over 4.5 was collected to dissolve into 50 mM sodium phosphate buffer at pH 7.0 by centrifugal ultrafiltration.
CcO was purified from bovine heart as described previously (22) and dissolved into 50 mM sodium phosphate buffer at pH 7.4 containing 0.2% n-decyl-β-D-maltoside (DM) (15). After freezing CcO in liquid nitrogen, CcO was stored at -80 °C until use.
NMR Experiments.
NMR samples for the backbone assignments contained 8 mM Cyt c in 50 mM sodium phosphate buffer at pH 5.8 (16). The reduced form of Cyt c was obtained by anaerobically adding the sodium dithionite solution to the oxidized Cyt c solution. All NMR experiments were performed at 298 K on a Bruker DRX600 spectrometer equipped with a cryoprobe (32). All spectra were processed by NMR pipe (34) and the NMR data were analyzed with SPARKY (Goddard TD and Kneller DG, SPARKY 3, University of California, San Francisco http://www.cgl.ucsf.edu/home/sparky/).
In the 1H-15N HSQC experiments, each sample contained 0.4 mM Cyt c, 5% D2O, and 0.2% n-decyl-β-D-maltoside (DM) in 50 mM sodium phosphate buffer, and the pH was adjusted to 6.8. In the presence of CcO, the molar ratios of reduced and oxidized Cyt c to CcO were set at 1∶0.8 and 1∶0.6, respectively. The reduced Cyt c-CcO complex was formed by anaerobically adding the sodium dithionite solution. The 1H-15N HSQC experiments were performed using the published pulse sequence with 15N decoupling using GARP4 during the 1H acquisition period. 1H-15N HSQC spectra were collected with a spectral width of 10,000 Hz and 2,048 complex points in the 1H dimension, and with a spectral width of 2,048 and 256 complex points in the 15N dimension.
Analysis of Chemical Shift Changes.
After identifying the amide signals on the basis of the sequential assignments and peak fitting using SPARKY, the CcO-induced changes in the backbone amide proton and nitrogen chemical shifts of Cyt c were determined. According to the analysis by Mulder et al. (17), 1H and 15N chemical shifts were combined using the following equation to calculate the composite chemical shifts.
Supplementary Material
Acknowledgments.
We thank Professor A. Grant Mauk (University of British Columbia) for his helpful suggestions on the expression of Cyt c. This work is supported by Grants-in-Aid for Scientific Research on Priority Areas 20051002 (to K.I.), 23121501 (to K.I.), 16087208 (to S.Y.) and 22247012 (to S.Y.), Targeted Protein Research Program (T.U., K.S.-I., S.Y., and K.I.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology. K.S. was supported by a Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108320108/-/DCSupplemental.
References
- 1.Pettigrew GW, Moore GR. Cytochrome c—Biological Aspects. Berlin: Springer-Verlag; 1987. [Google Scholar]
- 2.Zhen Y, Hoganson CW, Babcock GT, Ferguson-Miller S. Definition of the interaction domain for Cytochrome c on Cytochrome c oxidase I. J Biol Chem. 1999;274:38032–38041. doi: 10.1074/jbc.274.53.38032. [DOI] [PubMed] [Google Scholar]
- 3.Roberts VA, Pique ME. Definition of the interaction domain for Cytochrome c on Cytochrome c oxidase. III. J Biol Chem. 1999;274:38051–38060. doi: 10.1074/jbc.274.53.38051. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson-Miller S, Brautigan DL, Margoliash E. Definition of cytochrome c binding domains by chemical modification. III. Kinetics of reaction of carboxydinitrophenyl cytochromes c with cytochrome c oxidase. J Biol Chem. 1978;253:149–159. [PubMed] [Google Scholar]
- 5.Flok D, Helms V. Protein-protein docking of electron transfer complexes: cytochrome c oxidase and cytochrome c. Proteins. 2002;47:75–85. doi: 10.1002/prot.10066.abs. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman BM, Ratner MA. Gated electron transfer: when are observed rates controlled by conformational interconversion? J Am Chem Soc. 1987;109:6237–6243. [Google Scholar]
- 7.Sumi H, Marcus RA. Dynamical effects in electron transfer reactions. J Chem Phys. 1986;84:4894–4914. [Google Scholar]
- 8.Kuznetsov VY, Poulos TL, Sevrioukova IF. Putidaredoxin-to-cytochrome P450cam electron transfer: differences between the two reductive steps required for catalysis. Biochemistry. 2006;45:11934–11944. doi: 10.1021/bi0611154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michel B, Proudfoot AEI, Wallace CJA, Bosshard HR. The cytochrome c oxidase-cytochrome c complex: spectroscopic analysis of conformational changes in the protein-protein interaction domain. Biochemistry. 1989;28:456–462. doi: 10.1021/bi00428a008. [DOI] [PubMed] [Google Scholar]
- 10.Niki K, et al. Coupling to lysine-13 promotes electron tunneling through carboxylate-terminated alkanethiol self-assembled monolayers to cytochrome c. J Phys Chem B. 2003;107:9947–9949. [Google Scholar]
- 11.Winkler JR, Malmström BG, Gray HB. Rapid electron injection into multisite metalloproteins: intramolecular electron transfer in cytochrome oxidase. Biophys Chem. 1995;54:199–209. doi: 10.1016/0301-4622(94)00156-e. [DOI] [PubMed] [Google Scholar]
- 12.Banci L, et al. Solution structure of oxidized horse heart cytochrome c. Biochemistry. 1997;36:9867–9877. doi: 10.1021/bi970724w. [DOI] [PubMed] [Google Scholar]
- 13.Banci L, Bertini I, Huber JG, Spyroulias GA, Turano P. Solution structure of reduced horse heart cytochrome c. J Biol Inorg Chem. 1999;4:21–31. doi: 10.1007/s007750050285. [DOI] [PubMed] [Google Scholar]
- 14.Muramoto K, et al. A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc Natl Acad Sci USA. 2007;104:7881–7886. doi: 10.1073/pnas.0610031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoyama H, et al. A peroxide bridge between Fe and Cu ions in the O2 reduction site of fully oxidized cytochrome c oxidase could suppress the proton pump. Proc Natl Acad Sci USA. 2009;106:2165–2169. doi: 10.1073/pnas.0806391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeng WY, Chen CY, Chang HC, Chuang WJ. Expression and characterization of recombinant human cytochrome c in E. coli. J Bioenerg Biomembr. 2002;34:423–431. doi: 10.1023/a:1022561924392. [DOI] [PubMed] [Google Scholar]
- 17.Mulder FAA, Schipper D, Bott R, Boelens R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J Mol Biol. 1999;292:111–123. doi: 10.1006/jmbi.1999.3034. [DOI] [PubMed] [Google Scholar]
- 18.Wienk H, et al. Interaction of cytochrome c with cytochrome c oxidase: an NMR study on two soluble fragments derived from Paracoccus denitrificans. Biochemistry. 2003;42:6005–6012. doi: 10.1021/bi027198f. [DOI] [PubMed] [Google Scholar]
- 19.Basdevant N, Weinstein H, Ceruso M. Thermodynamic basis for promiscuity and selectivity in protein-protein interactions: PDZ domains, a case study. J Am Chem Soc. 2006;128:12766–12777. doi: 10.1021/ja060830y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo M, Bhaskar B, Li H, Barrows TP, Poulos TL. Crystal structure and characterization of a cytochrome c peroxidase-cytochrome c site-specific cross-link. Proc Natl Acad Sci USA. 2004;101:5940–5945. doi: 10.1073/pnas.0306708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volkov AN, Bashir Q, Worrall JAR, Ubbink M. Binding hot spot in the weak protein complex of physiological redox partners yeast cytochrome c and cytochrome c peroxidase. J Mol Biol. 2009;385:1003–1013. doi: 10.1016/j.jmb.2008.10.091. [DOI] [PubMed] [Google Scholar]
- 22.Yoshikawa S, et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 23.Smith HT, Staudenmayer N, Millett F. Use of specific lysine modifications to locate the reaction site of cytochrome c with cytochrome oxidase. Biochemistry. 1977;16:4971–4974. doi: 10.1021/bi00642a005. [DOI] [PubMed] [Google Scholar]
- 24.Pelletier H, Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 25.Worrall JAR, Kolczak U, Canters GW, Ubbink M. Interaction of yeast Iso-1-cytochrome c with cytochrome c peroxidase investigated by [15N,1H] heteronuclear NMR spectroscopy. Biochemistry. 2001;40:7069–7076. doi: 10.1021/bi0025823. [DOI] [PubMed] [Google Scholar]
- 26.Bloch D, et al. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc Natl Acad Sci USA. 2004;101:529–533. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capitanio N, et al. Factors affecting the H+/e- Stoichiometry in mitochondrial cytochrome c oxidase: influence of the rate of electron flow and transmembrane. Biochemistry. 1996;35:10800–10806. doi: 10.1021/bi9606509. [DOI] [PubMed] [Google Scholar]
- 28.Witt H, Malatesta F, Nicoletti F, Brunori M, Ludwig B. Tryptophan 121 of subunit II is the electron entry site to cytochrome-c oxidase in Paracoccus denitrificans. J Biol Chem. 1998;273:5132–5136. doi: 10.1074/jbc.273.9.5132. [DOI] [PubMed] [Google Scholar]
- 29.Bertini I, Cavallaro G, Rosato A. A structural model for the adduct between cytochrome c and cytochrome c oxidase. J Biol Inorg Chem. 2005;10:613–624. doi: 10.1007/s00775-005-0011-7. [DOI] [PubMed] [Google Scholar]
- 30.Aoki M, Ishimori K, Morishima I. NMR studies of putidaredoxin: associations of putidaredoxin with NADH-putidaredoxin reductase and cytochrome P450cam. Biochim Biophys Acta. 1998;1386:168–178. doi: 10.1016/s0167-4838(98)00091-0. [DOI] [PubMed] [Google Scholar]
- 31.Axelrod HL, et al. X-ray structure determination of the cytochrome c2: reaction center electron transfer complex from Rhodobacter sphaeroides. J Mol Biol. 2002;319:501–515. doi: 10.1016/S0022-2836(02)00168-7. [DOI] [PubMed] [Google Scholar]
- 32.Sakamoto K, Kamiya M, Uchida T, Kawano K, Ishimori K. Redox-controlled backbone dynamics of human cytochrome c revealed by 15N NMR relaxation measurements. Biochem Biophys Res Commun. 2010;398:231–236. doi: 10.1016/j.bbrc.2010.06.065. [DOI] [PubMed] [Google Scholar]
- 33.Wagner G, Pardi A, Wüthrich K. Hydrogen bond length and 1H NMR chemical shifts in proteins. J Am Chem Soc. 1983;105:5948–5949. [Google Scholar]
- 34.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.