

Abstract
To study the protein structure–function relationship, we propose a method to efficiently create three-dimensional maps of structure space using a very large dataset of > 30,000 Structural Classification of Proteins (SCOP) domains. In our maps, each domain is represented by a point, and the distance between any two points approximates the structural distance between their corresponding domains. We use these maps to study the spatial distributions of properties of proteins, and in particular those of local vicinities in structure space such as structural density and functional diversity. These maps provide a unique broad view of protein space and thus reveal previously undescribed fundamental properties thereof. At the same time, the maps are consistent with previous knowledge (e.g., domains cluster by their SCOP class) and organize in a unified, coherent representation previous observation concerning specific protein folds. To investigate the function–structure relationship, we measure the functional diversity (using the Gene Ontology controlled vocabulary) in local structural vicinities. Our most striking finding is that functional diversity varies considerably across structure space: The space has a highly diverse region, and diversity abates when moving away from it. Interestingly, the domains in this region are mostly alpha/beta structures, which are known to be the most ancient proteins. We believe that our unique perspective of structure space will open previously undescribed ways of studying proteins, their evolution, and the relationship between their structure and function.
Keywords: global map of protein universe, protein function prediction, protein structure universe
Investigating protein structure space and its relationship to function space is a fundamental scientific challenge. Characterizing this relationship may also carry practical implications to protein function prediction, whereby one wishes to infer the biological role of a protein from its structure [as is the case with many of the structures solved in the high-throughput pipeline of the Structural Genomics projects (1, 2)]. One way to approach this challenge is to represent protein structure space by three-dimensional maps. Maps of structure space were first introduced by Holm and Sander (3) and were later used by Kim and colleagues (4–6). To calculate their maps, they first calculate the structural similarity between all pairs of protein structures. Then, they use multidimensional scaling (MDS) to find a collection of points in three dimensions, each of which corresponds to a protein, and where the distance between any two points depends on the structural similarity of the proteins they represent. Such a representation provides a comprehensive visual view of structure space, which is not constrained by a hierarchical system such as the Structural Classification of Proteins (SCOP) (7).
We propose an efficient way to calculate maps of protein structure space, using the recently introduced FragBag model (8). Using FragBag, we represent each structure as a point in a high-dimensional space and project these points to three dimensions. It was recently shown that the similarity between the FragBag vectors, or the points in the high-dimensional space, can identify near structural neighbors as accurately as the state-of-the-art structural aligners STRUCTAL and CE, for several definitions of near structural neighbors (8). Because FragBag models structures as fixed-size vectors, we can replace MDS with a more efficient procedure, Principal Component Analysis (PCA) (9). Thus, we can map a very large set of > 30,000 protein structures. Rather than studying single structures, we study properties such as structural density and functional diversity, which are defined at each point of structure space through a whole collection of structures in the vicinity of that point. By coloring the maps according to the values of these properties, we are able to visualize their distribution across structure space. This way we discover that structure space has a region of high functional diversity and that this region consists mainly of alpha/beta structures, which are known to be the most ancient proteins (10). We believe that studying such maps holds great promise to revealing important properties of protein structure space, its relation to function, and perhaps even to sequence.
Results
Constructing Functional Diversity Maps of Protein Structure Space.
To study protein structure space we analyze a set of 31,155 SCOP v1.71 (7) domains. We initially represent each such domain by a 400-long FragBag vector, which may be thought of as a point in 400-dimensional space. In the FragBag model, a protein structure is represented by a count vector of backbone fragments taken from a library of 400 commonly occurring 12-residue fragments. For each contiguous (and overlapping) 12-residue segment along the protein backbone, we identify the library fragment that fits it best in terms of RMSD after optimal superposition. The ith entry in the FragBag vector is the number of times the library’s ith fragment was found to be the best fit. The FragBag distance between two domains is the distance between their FragBag vectors. We have recently shown that this distance is a good approximation of the structural distance, as quantified by structural alignment (8). Using principal component analysis (PCA) (11), we then project the points to three-dimensional space. The eigenvalues of the resulting data covariance matrix (Fig. S1) drop sharply and the fourth largest eigenvalue (0.0106) is 8% of the first largest eigenvalue (0.1326); this indicates that three dimensions can adequately represent the essential features of protein structure space. Fig. 1 B–D shows a three-dimensional map of protein structure space, in which each domain is colored by its SCOP class (7); we show three views of the map from three angles, to get a better sense of it. As expected, the domains cluster by their SCOP class.
Fig. 1.

Maps of protein structure space. Each point represents a SCOP domain, and the distance between any two points approximates the structural distance between their corresponding domains. B–D show the map of the SCOP classes: As expected, the points are clustered. F–H show the structural density map, where the color of each point indicates the number of domains that lie in its vicinity of fixed distance (denoted Vfd). We see that the highest density is within the regions of the all-alpha domains, followed by a region in the alpha/beta domain and in the all-beta domain. Fig. S2 shows a similar density map when considering sequence nonredundant samples of the protein world.
The density of protein structure space is uneven—i.e., certain regions have more domains per “unit volume” than others. This can be seen in Fig. 1 F–H, which shows again the three views of the map, now colored according to the density score of each domain—the number of domains that are within a 0.005 distance from it. Certain proteins are more studied than others, and as a result, more variants thereof are included in our dataset. To rule out this bias as the source of the observed uneven density, we prepared similar density maps, based on 40% and 95% sequence nonredundant subsets of the original data (containing 2,517 and 4,238 domains, respectively). The results, shown in Fig. S2, are qualitatively identical to the original density map, and the correlation between the original density scores and those based on the restricted sets are very high (r = 0.945 and r = 0.960 for the 40% and the 95% sets, respectively). In the remainder of this study, we use the full dataset.
Upon inspecting Fig. 1, one can see that there is a relation between the SCOP class and the density score of a domain. Fig. 2A, which is a histogram of the density scores of the domains, color-coded by their SCOP class, shows this more clearly: The alpha + beta (yellow) and the all-beta (red) domains tend to reside in low-density regions, whereas the all-alpha (blue) domains constitute the vast majority in the very high-density regions.
Fig. 2.

Structural density and functional diversity by SCOP class. We calculate the separate histograms of structural density (A) and functional diversity (B) of each of the SCOP classes and stack them one on top of the other. We see that the densest regions are populated by all-alpha domains, and the most functionally diverse regions by the alpha/beta domains. See Table S2 (listing the exact proportions of each of the SCOP classes, among the top 10%/20% most dense/functionally diverse domains) and Fig. S12 for supporting evidence.
Next, we investigate how functional diversity varies across structure space; for this, we quantify the functional diversity in the vicinity of each domain in our dataset. We consider three definitions for the vicinity of a domain d: (i) Vfn is a fixed number (100) of the nearest structural neighbors of d, (ii) Vsamp is a sample of fixed size (100) from the domains that lie within a fixed structural distance (0.005) from d, and (iii) Vfd is the collection of all domains that are within some fixed structural distance (0.005) from d. Although Vfd is perhaps the most natural definition, it makes the vicinities of domains in denser regions contain far more members, which may bias the results.
Our measure for the functional diversity in a vicinity of a protein, however vicinity is defined, is the number of distinct functions that the domains within this vicinity possess. To determine function, we use the functional annotations of the proteins from the Gene Ontology molecular function (GO-MF) controlled vocabulary (12), and the mapping of terms to SCOP domains calculated by Lopez and Pazos (13). When a single domain is annotated as having more than one function, we include all its functions toward the count.
Structure Space Has a Core of High Functional Diversity.
Fig. 3 B–D shows a functional diversity map of protein structure space. The domains in the map are color-coded according to the functional diversity of their vicinities (red for the most diverse ones; blue for the least diverse), and vicinity is defined to be Vsamp (when there were fewer than 100 domains within this distance, all were included). This map shows a striking pattern: Protein space has a highly diverse core, and diversity drops gradually toward its periphery (we denote the high diversity region “core,” because of its location in our maps). Figs. S3 and S4 show the maps constructed using the two alternative definitions of a vicinity, Vfn and Vfd; the results are very similar.
Fig. 3.

Functional-diversity map of protein structure space. The color of a point indicates the degree of functional diversity measured by the number of distinct GO-MF terms annotating the domains in its vicinity. Here, we use the Vsamp definition for a vicinity of a protein: a sample of fixed size from all domains that fall within a fixed distance from it. A–D show the functional diversity for the true data; E–H show the functional diversity of a random world, in which the proteins have the same structures, yet their functions are assigned at random. We see that when using the true functional annotations, there is a core of high functional diversity, and that functional diversity drops toward the periphery. Alternatively, when the functions are assigned at random, there is no such core, and function diversity is uniformly high. The figures in SI Appendix, and Table S1, show that the results are qualitatively similar when using alternative datasets, scoring functions, and the more uniform (coarser) annotation graph GO slim.
As a control for the validity of our finding, we re-created the diversity map (using Vsamp again) after randomly permuting the functional annotations across all domains (i.e., the set of functional annotations originally associated with each domain was associated with a different, randomly chosen domain). If our finding were merely an artifact of the projection to three dimensions, or of some feature of protein structure space (say, the uneven density), the resulting diversity map would show again a highly diverse core. Fig. 3 F–H shows that this is not the case: Under the random permutation, the diversity score of almost all domains is very high (colored in orange and red), and the map has no prominent diverse core; the relatively few domains with low diversity scores (colored in blue) are mostly isolated domains, having fewer than 100 neighbors within a 0.005 distance, and thus necessarily less diverse vicinities (there are 5,356 such domains). The existence of the diverse core is indeed a statistically significant finding (p < 0.005; see Methods for details). When using Vfn, the results are very similar (Fig. S3); as expected, when using Vfd, diversity is highly correlated (r = 0.953) with density, because domains in denser regions now have more members in their vicinities, and thus more functional annotations (Fig. S4).
We can reliably predict the functional diversity of structures in a randomly chosen test set, using the mapping calculated for a training set. Our test set consists of 250 randomly chosen structures from the sequence nonredundant set (using a 40% sequence identity threshold); it has 52, 40, 92, 52, and 14 domains of the SCOP classes all-alpha, all-beta, alpha/beta, alpha + beta, and others, respectively. The training set has the 29,014 domains that share no sequence similarity with the test set proteins (BLAST E-value threshold of 10-3 and sequence identity of 40%). Using PCA of the training set FragBag data, we calculate the projection Ptrain to R3. For each test set proteins p, we calculate Ptrain(p) and identify the structures in p’s training-set vicinity. The predicted functional diversity score is the number of unique GO-MF terms within this vicinity. Fig. S5 plots the predicted functional diversity scores vs. the ones calculated using the complete dataset for the three definitions of vicinity, Vsamp, Vfn, and Vfd, and shows that these scores are highly correlated (r > 0.96).
A potential explanation for the high functional diversity in the core is that the core contains a high proportion of multiple-function domains, compared to the periphery (recall that multiple-function domains contribute all their functions toward the diversity). This is not the case: Fig. S6 shows a functional multiplicity map of structure space, i.e., a map in which each point is colored according to the number of GO-MF annotations of the domain it represents. The high functional diversity core seen in Fig. 3 and Fig. S3 does not overlap with a region of high functional multiplicity. Further, we see the highly diverse core even after reconstructing the functional diversity maps using only domains annotated by only one function (61% of the data); see Fig. S7.
Another potential, yet invalid, explanation for the high functional diversity in the core is related to the uneven degree of detail in the GO-MF vocabulary. The GO is implemented as a hierarchical directed graph, in which the terms are placed at the nodes and the edges direct from the general to the specific. The level of detail in the GO-MF graph is uneven: Some areas are better studied and correspond to subgraphs of the GO-MF graph that have more levels and, ultimately, more functional annotations. In addition, proteins of the same function sometimes have annotations at different levels (14). One could argue that perhaps the proteins that lie in the core happen to have functions that are described in finer detail, and the apparent high diversity of this core is merely an artifact of the uneven level of detail in the GO-MF graph. To demonstrate that this, again, is not the case, we create functional diversity maps based on Watson et al.’s GO-slim controlled vocabulary (14). GO slim is a trimmed variant of GO-MF in which function is defined more broadly, by only 190 terms (out of > 7,800); in particular, GO slim targets a level of detail in which neighboring proteins in structure space have similar functions. Fig. S8 shows a map that was constructed similarly to the one in Fig. 3 and Fig. S3, except that the function annotations are replaced by their more general terms in the GO-slim graph. Once again we see the same phenomenon: a diverse core and more homogeneous periphery. Indeed, these alternative scores are highly correlated with the original diversity score (r > 0.895); see Table S1.
We also consider three alternatives to the functional diversity score used above. Two of these alternatives are based on a weighted count of distinct GO-MF terms within a vicinity, rather than on a simple count. In the first, commonly occurring terms have a lower weight, and in the second, more specific terms (i.e., ones that are farther from the root in the GO-MF graph) have a lower weight. In the third alternative, the score is based on the coherence measure proposed in refs. 15 and 16, which quantifies the contribution of a functional annotation term to a vicinity based on statistical tests. When using vicinity definitions Vsamp, and Vfn, these alternative scores are correlated with the original diversity score (r > 0.79); see Table S1. Indeed, the functional diversity maps under each of the three alternative scores, shown in Figs. S9–S11, look similar to the one in Fig. 3.
Characterizing the Core’s Structures.
A comparison of the functional diversity maps (Fig. 3 B–D) and the SCOP-class maps (Fig. 1 B–D) reveals that the core of high functional diversity consists mainly of alpha/beta domains (colored in green). Fig. 2B and Fig. S13 A and B show this finding in another way, via histograms detailing the contribution of each SCOP class to the functional diversity scores. Table S2 lists the exact proportions of the various SCOP classes among the domains with the top 10% and 20% functional diversity scores; in all cases (including when considering the diversity scores only within the sequence nonredundant sets), the majority of the high functional diversity domains are alpha/beta proteins.
Fig. 4 highlights several SCOP folds that lie in the diverse core of structure space. The full dataset is shown in Fig. 4A with a black outline; Fig. 4 B–F show specific SCOP folds within this outline, alongside the histograms of their functional diversity scores. The most obvious candidate for the SCOP fold whose structures lie in the core is the TIM barrels (c.1), which are well known to accommodate many functions (2). Indeed, these lie in the core, and their functional diversity scores are clearly higher compared with the full dataset (Fig. 4B). We see, however, that the TIM barrels are only a part of the picture, as the core contains also many other domains. Fig. 4C shows SCOP fold adenine nucleotide alpha hydrolase-like (c.26) that was also noted as accommodating many functions (1) and also lies within the core.
Fig. 4.

SCOP folds that lie in the functionally diverse core. We highlight the location in structure space of specific SCOP folds and show histograms of the diversity of the domains of these folds; for comparison, A shows the full dataset (a copy of Fig. 3 A and B) outlined in black. B and C show two SCOP folds that are known to be functionally diverse, the TIM barrel fold (c.1) and the adenine nucleotide alpha hydrosase-like fold (c.26). Indeed, the domains of these two folds are located in the highly diverse core of structure space. There are, however, many other domains in the core. D–F show three more examples of SCOP folds that lie in the highly diverse core: phosphorylase/hydrolase-like (c.56), alpha/beta-Hydrolases (c.69), and protein kinase-like (PK-like, d.144), respectively. Table S3 lists the mean and average functional diversity scores for several SCOP folds that lie in the core.
To identify more SCOP folds in the core, we search for folds with (more than 25) domains that lie in functionally diverse vicinities. We quantify the diversity of a SCOP fold by the average and the median of the diversity scores of its domains, using the diversity scores based on the three definitions of vicinity. Table S3 lists the 20 most diverse folds under these measures: Each of the resulting six measures identifies different SCOP folds as the most diverse. To identify SCOP folds that are truly diverse, we consider folds that are among the 20 most diverse folds under all six measures. Nine folds satisfy this condition: 7-stranded beta/alpha barrel (c.6), ClpP/crotonase (c.14), methylglyoxal synthase-like (c.24), arginase/deacetylase (c.42), phosphorylase/hydrolase-like (c.56), alpha/beta-hydrolases (c.69), AraD-like aldolase/epimerase (c.74), amidase signature enzymes (c.117), protein kinase-like (PK-like) (d.144). As expected, the domains in these folds are indeed located in the core; Fig. 4 D–F shows three examples.
Better Predicting of Function from Structure in Regions of Low Functional Diversity.
We use the set of 90 proteins* studied by Watson et al. (14) to assess if one can indeed better predict function for proteins in regions of structure space having low functional diversity. Watson et al. predicted function using global structural similarity [as detected by secondary-structure matching (SSM) (17)] and evaluated the correctness of their predictions. Fig. S13 maps the protein structures used in their experiment: on the right these structures within our dataset, and on the left, the same structures with markers indicating if the prediction was correct. We see that Watson et al. better succeed in predicting the function of proteins that lie in regions of low functional diversity.
We quantify this difference by separating the proteins to two sets, according to their functional diversity, and comparing the success rate in these sets. The first set consists of 35 proteins having high diversity (≥45) vicinities, and the second consists of 55 proteins having low diversity (< 45) vicinities. Among the high diversity proteins, only 43% of the predictions were correct, significantly lower than the 67% of the correct predictions for the low diversity proteins (p = 0.021 in a one-sided, two-sample proportion test).
Discussion
The main contributions of our work are (i) we propose a method for efficiently calculating low dimensional maps of very large sets of protein structures and (ii) we use these maps to study the spatial distribution of properties of local vicinities in protein structure space (e.g., density and functional diversity) and reveal a fundamental relationship between protein structure and function spaces. Maps of structure space offer an overall perspective that complements the more detailed view offered by hierarchical classifications such as SCOP. Although in the latter case one is typically restricted to studying sets of proteins of the same label (e.g., fold), maps display structural similarities among all proteins in a single representation. Indeed, it was previously noted that considering all structural similarities is advantageous for studying protein structure and function (18, 19). The efficiency of our method renders the calculation of maps for the full Protein Data Bank (PDB) possible, as opposed to only a sparse sample from it. This is a step forward because properties characterizing local vicinities in structure space can only be measured using such large datasets.
Although our map calculation is far more efficient, it is fundamentally similar to the maps of Kim and co-workers (4–6, 20). To calculate their maps, Kim and co-workers use MDS, whereas we use PCA. Both methods generate the same map (up to a reflection and rotation of the entire space) if the distances in the MDS matrix are the Euclidean distances between the vectors in the PCA matrix (9). The difference is in efficiency: PCA calculates the eigenvalue decomposition of an L × L matrix, where L is the length of the vector describing a protein (L = 400 in our case), whereas MDS calculates the eigenvalues decomposition of an N × N matrix, where N is the size of the dataset (N = 31,155 here). The challenge, thus, is to find a representation of proteins as vectors of fixed size, such that their Euclidean distance reflects their structural distance.
FragBag fulfills this requirement: The Euclidean distance between its normalized vectors approximates very well the structural distance between the proteins they represent (using a library of 400 fragments of length 12). We have recently demonstrated this by comparing FragBag to the state-of-the-art structural alignment methods CE and STRUCTAL and showing that they identify near structural neighbors equally well, for different definitions of near structural neighbors (8). The evaluation was carried out on a large and challenging test set (2,928 proteins), and using a very stringent gold standard: the near structural neighbors identified by a best-of-all structural aligner that uses six methods (21). Recently, in a different context of sequence-based homology detection, Melvin et al. suggested another approach for representing proteins by fixed-size vectors (of dimension 200) (22). Their algorithm ProtEmbed learns from pairs of proteins marked as similar or dissimilar a transformation from a feature vector of the size of the dataset, to a lower dimensional vector. Then, they use MDS to visualize the ranking of the near neighbors in sequence space.
The phenomena we report are not due to a particular data or parameter set, as we see them in various maps. Our SCOP-class map is generally similar to the map calculated in refs. 4 and 5; their maps were based on different datasets and used DALI to identify structural similarities. In both maps the four SCOP classes are generally separated: The all-alpha and all-beta are farthest apart and orthogonal to each other, and the alpha/beta and alpha+beta lie in between, separated from each other. The maps of structural density for the full dataset and its (40%/95%) sequence nonredundant subsets are similar. Finally, we calculated several functional diversity maps, and all of them display the same pattern.
We discovered that protein structure space has a functionally diverse core and that diversity drops toward the periphery of the space. Because this observation is made in the low dimensional projection of the data, we rule out the possibility that this is a statistical artifact, by verifying that this core is not seen in maps generated for the same set of protein structures, but with randomly assigned functions. In contrast to the maps generated for real world data, and as expected from a random assignment, functional diversity in the maps with the random association of function is uniformly high. Of course in reality, the true function of a protein is not assigned at random, but rather, depends on its structure. Further, we show that our discovery cannot be explained away by the core structures having more functional annotations. Also, it cannot be attributed to the uneven level of detail in the controlled vocabulary GO, which we use to annotate function, because even when using GO slim, a (coarser) alternative, we still observe the same phenomenon. The functional diversity of a random test set of proteins is the same as when using a mapping that was calculated for a subset of the data that shares no sequence similarity with this test set. The functional diversity of structure space has this fundamental characteristic pattern.
The highly diverse core of structure space contains mainly alpha/beta domains, which were identified by phylogenetic analysis as most ancient. Winstanley et al. estimated the ages of SCOP folds and classes through phylogenies constructed from fold occurrence data in multiple genomes and concluded that the alpha/beta SCOP class is the oldest (10). Using a different method for estimating evolutionary age, Choi and Kim reached the same conclusion (6). Winstanley et al. also calculated the relative ages of SCOP folds, and according to all of their measures, the nine folds we identified as lying in the core are among the oldest folds (relative age = 1.0). These nine core folds are all enzymes, an observation in line with Redfern et al. (1), who comment that some enzyme folds are functionally divergent because their architectures easily accommodate structural embellishment, thus allowing the exploration of different functions. Finally, Winstanley et al. also conclude that the SCOP class of small proteins is relatively young, and these proteins lie in the periphery of our structure space.
A fundamental research challenge is to extend our investigation to include also sequence information and to characterize how structure and function spaces relate to sequence space. For example, the sequence variability of local structure vicinities may vary: quantifying this variability and its spatial distribution is interesting, especially since protein structure is far more conserved than sequence (23). Alternatively, one could use the maps of protein sequence space calculated by Melvin et al. (22) to study structural and functional properties of vicinities in that space. We hope that doing so will reveal further fundamental properties of the relationship between protein sequence, structure, and function.
We also plan to investigate ways of applying our results to improve the performance of protein function prediction. A common way to predict the function of a protein is to identify other proteins of known function that have similar sequences and structures and transfer their functions to the target protein (2, 14, 24). Because it is preferable to transfer function from homologues that were identified based on sequence, the sequence variability in the vicinity of the protein is very important. However, when resorting to structure-based prediction, our study suggests that if the protein lies in the periphery of structure space, then its neighbors have only a few functions that need to be considered. If, on the other hand, the protein lies in the functionally diverse core, then its neighbors have jointly many functions to consider. This is a generalization of a call for caution recently made with respect to function prediction for TIM barrels (24). Indeed, our analysis of Watson et al.’s data (14) shows that they were more successful in predicting function from structure for proteins lying in less diverse regions of structure space. Thus, it seems that one could use the functional diversity maps to better choose the parameters of structure-based function prediction, according to the location of the target protein in structure space, and perhaps even to assign confidence levels for the prediction.
Materials and Methods
Representing Protein Domains in 400 Dimensional Space.
For each domain in the dataset, we calculate FragBag (8) description vectors of length L = 400 based on a library of 400 12-mer fragments (http://cs.haifa.ac.il/~ibudowsk/libraries/centers400_12.txt); each entry in the vector is the number of times the corresponding library fragment was the best approximation of any of the 12-mer fragments in the backbone of the represented protein. A list of one or more GO-MF annotations is associated with each domain. Our dataset includes N = 31,155 SCOP v1.71 (7) domains for which Lopez and Pazos (13) provide a GO annotation. We have previously shown that the cosine distance between two FragBag vectors best approximates the structural alignment score (SAS) (25) between their corresponding structures (8). Notice that the Euclidean (norm 2) distance between two FragBag vectors that were normalized to length 1 is exactly twice their cosine distance. To see this, consider p1 and p2 two FragBag vectors, and let 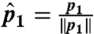 ,
, 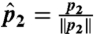 be the normalized vectors. The cosine distance between p1 and p2 is
be the normalized vectors. The cosine distance between p1 and p2 is 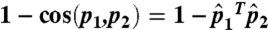 ; the Euclidean distance between the normalized vectors is
; the Euclidean distance between the normalized vectors is
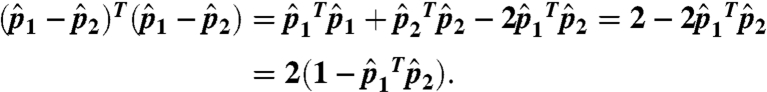 |
Thus, we normalize all FragBag vectors and consider the Euclidean (norm 2) distance; because all distances are relative, the uniform factor 2 is of no consequence.
Projecting to Three Dimensions.
We store the normalized descriptions of length L( = 400) of the N structures in our dataset in an L × N matrix and project it to three dimensions using PCA. Namely, after centering the L × N coordinates about the origin (by subtracting their mean), we calculate the L × L covariance matrix (normalized by N) and find the eigenvectors corresponding to its three largest eigenvalues. By multiplying these eigenvectors (a 3 × L matrix) by the L × N data matrix, we find the 3 × N matrix that is the projection of our data to three dimensions. There, the Euclidean (norm 2) distances between two 3D vectors is an approximation of their Euclidean (norm 2) distances in L dimensions. We emphasize that this requires only the easy computation of finding the top three eigenvalues and eigenvectors of the relatively small L × L matrix. This is in contrast to the slightly different calculation done in previous studies: Given N structures, they calculate a symmetric matrix D of size N × N of all pairwise structural distances and use MDS to find the coordinates of the points representing these N structures in three (or two) dimensions (3, 5). The technical bottleneck in the MDS calculation is finding the top three (or two) eigenvalues and eigenvectors of an N × N matrix derived from D (26); it is a challenging computation for datasets of several tens of thousands proteins. Indeed, the datasets in previous studies were smaller (e.g., less than 1,900 structures in ref. 6).
Calculating Alternative Functional Diversity Scores.
Each of the domains in the dataset has a list of its GO-MF terms; in each case, the terms are the most specific ones (rather than the term and all its parents). For each term, we calculate its weighted functional diversity in two ways: (i) (1–10* the fraction of its occurrence), where the fraction of its occurrence is the fraction of domains that are annotated by it; the scaling factor was determined to be 10, to better space the range of values in the dataset. (ii) The inverse of the depth of the term in the GO-MF annotation graph; the depth is the number of times we can replace the terms by more general ones until we reach the root. There are seven cases (out of 9,500) in which a term has two different depths, and these differ by at most three (this is a consequence of GO being a graph rather than a tree). In these cases, we use the average depth. To calculate the “coherence measure,” we check for each term and vicinity if the term is “enriched” in the vicinity, i.e., if it appears at a rate that is statistically significant. The “coherence” is the percent of the terms in a region that are enriched. Thus, the coherence measure is a value between 0–100%, and high coherence implies low diversity and vice versa; see ref. 16 for more details. Finally, the GO-slim annotation of a functional term is the most specific parent(s) of the term that is present in the GO-slim annotation graph.
Measuring the Spatial Spread of the Core in True and Random Associations of Functional Annotations to Structures.
We measure the spatial spread of the most diverse domains by their average distance from their center of mass. We consider two definitions of the most diverse proteins: (i) all domains whose diversity scores are greater than 0.8 × max_diversity, where max_diversity is the highest diversity score found in our dataset; (ii) the 20% most functionally diverse proteins. We measure the spatial spread of the most diverse domains in our dataset, and in 300 random assignments of the functional annotations to locations in structure space. The average distance in the true dataset for these two definitions is 0.0860 and 0.1131, respectively. In the random permutations, the average distances are 0.3501 ± 0.0151 and 0.3499 ± 0.0220, respectively, resulting in a p value < 0.0033.
Supplementary Material
Acknowledgments.
We thank Yuval Nov, Golan Yona, Chen Keisar, and our anonymous reviewers for their helpful comments. R.K was supported by the Marie Curie IRG Grant 224774.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1102727108/-/DCSupplemental.
*Denoted the “known-function” dataset; 1nrh, 1tea were removed because they are obsolete.
References
- 1.Redfern OC, Dessailly B, Orengo CA. Exploring the structure and function paradigm. Curr Opin Struct Biol. 2008;18:394–402. doi: 10.1016/j.sbi.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedberg I. Automated protein function prediction—the genomic challenge. Brief Bioinform. 2006;7:225–242. doi: 10.1093/bib/bbl004. [DOI] [PubMed] [Google Scholar]
- 3.Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 4.Hou J, Jun SR, Zhang C, Kim SH. Global mapping of the protein structure space and application in structure-based inference of protein function. Proc Natl Acad Sci USA. 2005;102:3651–3656. doi: 10.1073/pnas.0409772102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hou J, Sims GE, Zhang C, Kim SH. A global representation of the protein fold space. Proc Natl Acad Sci USA. 2003;100:2386–2390. doi: 10.1073/pnas.2628030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi IG, Kim SH. Evolution of protein structural classes and protein sequence families. Proc Natl Acad Sci USA. 2006;103:14056–14061. doi: 10.1073/pnas.0606239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 8.Budowski-Tal I, Nov Y, Kolodny R. FragBag, an accurate representation of protein structure, retrieves structural neighbors from the entire PDB quickly and accurately. Proc Natl Acad Sci USA. 2010;107:3481–3486. doi: 10.1073/pnas.0914097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenenbaum JB, Silva Vd, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–2323. doi: 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- 10.Winstanley HF, Abeln S, Deane CM. How old is your fold? Bioinformatics. 2005;21:i449–i458. doi: 10.1093/bioinformatics/bti1008. [DOI] [PubMed] [Google Scholar]
- 11.Jolliffe IT. Principal Component Analysis. New York: Springer; 2002. [Google Scholar]
- 12.Harris MA, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez D, Pazos F. Gene Ontology functional annotations at the structural domain level. Proteins. 2009;76:598–607. doi: 10.1002/prot.22373. [DOI] [PubMed] [Google Scholar]
- 14.Watson JD, et al. Towards fully automated structure-based function prediction in structural genomics: A case study. J Mol Biol. 2007;367:1511–1522. doi: 10.1016/j.jmb.2007.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segal E, et al. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 16.Slonim N, Atwal GS, Tkacik G, Bialek W. Information-based clustering. Proc Natl Acad Sci USA. 2005;102:18297–18302. doi: 10.1073/pnas.0507432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 18.Kolodny R, Petrey D, Honig B. Protein structure comparison: implications for the nature of ‘fold space’, and structure and function prediction. Curr Opin Struct Biol. 2006;16:393–398. doi: 10.1016/j.sbi.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Petrey D, Honig B. Is protein classification necessary? Toward alternative approaches to function annotation. Curr Opin Struct Biol. 2009;19:363–368. doi: 10.1016/j.sbi.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sims GE, Choi IG, Kim SH. Protein conformational space in higher order phi-Psi maps. Proc Natl Acad Sci USA. 2005;102:618–621. doi: 10.1073/pnas.0408746102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolodny R, Koehl P, Levitt M. Comprehensive evaluation of protein structure alignment methods: Scoring by geometric measures. J Mol Biol. 2005;346:1173–1188. doi: 10.1016/j.jmb.2004.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melvin I, Weston J, Noble WS, Leslie C. Detecting remote evolutionary relationships among proteins by large-scale semantic embedding. PLoS Comput Biol. 2011;7:e1001047. doi: 10.1371/journal.pcbi.1001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levitt M, Gerstein M. A unified statistical framework for sequence comparison and structure comparison. Proc Natl Acad Sci USA. 1998;95:5913–5920. doi: 10.1073/pnas.95.11.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loewenstein Y, et al. Protein function annotation by homology-based inference. Genome Biol. 2009;10:207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subbiah S, Laurents DV, Levitt M. Structural similarity of DNA-binding domains of bacteriophage repressors and the globin core. Current Biol. 1993;3:141–148. doi: 10.1016/0960-9822(93)90255-m. [DOI] [PubMed] [Google Scholar]
- 26.deSilva V, Tenenbaum JB. Stanford, CA: Stanford Univ; 2004. Sparse multidimensional scaling using landmark points. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.