

Non-technical summary
Neonatal diabetes is a rare genetic form of diabetes that develops within the first 6 months of life. It is often caused by genetic defects (mutations) in a specialised membrane protein known as the KATP channel. This protein acts as a tiny pore in the membrane of the insulin-secreting cells in the pancreas and its opening and closing is regulated by blood glucose levels. Low blood glucose holds the pore open and prevents insulin secretion whereas high blood sugar leads to channel closure and insulin secretion. We identified a novel mutation (Kir6.2-W68R) that prevents channel closure and insulin secretion, and so results in neonatal diabetes. Detailed molecular studies showed that channel opening and closing is disrupted, suggesting Kir6.2-W68 may act as a channel ‘gatekeeper’. Our results also indicated that the mutant channel could be shut by sulphonylurea drugs, which enabled the patient to transfer from insulin injections to tablet therapy.
Abstract
Abstract
We identified a novel heterozygous mutation, W68R, in the Kir6.2 subunit of the ATP-sensitive potassium (KATP) channel, in a patient with transient neonatal diabetes. This tryptophan is absolutely conserved in mammalian Kir channels. The functional effects of mutations at residue 68 of Kir6.2 were studied by heterologous expression in Xenopus oocytes, and by homology modelling. We found the Kir6.2-W68R mutation causes a small reduction in ATP inhibition in the heterozygous state and an increase in the whole-cell KATP current. This can explain the clinical phenotype of the patient. The effect of the mutation was not charge or size dependent, the order of potency for ATP inhibition being W<M∼L<R∼E∼K∼A<C∼F<Y. Replacement with tyrosine (Y) rendered the KATP channel almost completely insensitive to ATP block, dramatically increased the channel open probability, and affected the interaction of Kir6.2 with SUR1. In different Kir crystal structures the residue corresponding to W68 adopts two distinct positions. In one state, the tryptophan lies in a position that would impede movement of transmembrane domain 2 (TM2) and opening of the gate. In the other state, it is flipped out, enabling movement of TM2. We therefore hypothesise that W68 may act as a molecular ‘gatekeeper’ for Kir channels.
Introduction
Gain-of-function mutations in the Kir6.2 or SUR1 subunits of the ATP-sensitive potassium (KATP) channel are a common cause of neonatal diabetes, a rare inherited disorder characterised by the development of diabetes within the first 6 months of life (Gloyn et al. 2004; Proks et al. 2004; Hattersley & Ashcroft, 2005; Babenko et al. 2006; Ashcroft, 2007). The diabetes may be permanent or follow a remitting–relapsing time course (Flanagan et al. 2007). A very few patients (<3%) experience neurological problems, such as motor and mental developmental delay, muscle hypotonia, and epilepsy, in addition to neonatal diabetes (DEND syndrome) (Hattersley & Ashcroft, 2005; McTaggart et al. 2010). Rather more (∼20%) manifest an intermediate condition consisting of developmental delay, muscle hypotonia and neonatal diabetes (iDEND syndrome).
It is now clear that this spectrum of symptoms arises because the KATP channel couples cell metabolism to electrical activity in both endocrine and neuronal cells (Miki & Seino, 2005; Nichols, 2006; McTaggart et al. 2010). Metabolically induced changes in channel activity are produced by changes in the intracellular concentration of adenine nucleotides, with ATP inhibiting and MgADP (and MgATP) stimulating channel activity (Nichols et al. 1996; Tucker et al. 1997; Gribble et al. 1997b; Shyng et al. 1997). In the pancreatic beta-cell, the KATP channel plays a critical role in insulin secretion (Ashcroft et al. 1984). When blood glucose levels are low, the KATP channel is open holding the beta-cell hyperpolarised and preventing insulin exocytosis. However, when blood glucose levels rise, glucose uptake and metabolism by the beta-cells increase, elevating the intracellular concentration of ATP and reducing that of MgADP. This leads to KATP channel closure, membrane depolarization, activation of voltage-sensitive calcium channels, calcium entry and calcium-mediated insulin secretion. KATP channels also influence the electrical excitability of many different types of neurons and recent evidence argues that not only the developmental problems, but also the muscle hypotonia are a consequence of expression of mutant channels in neuronal tissue (Clark et al. 2010). Sulphonylurea drugs, which close the open KATP channels (Gribble & Reimann, 2003), stimulate insulin secretion in patients with neonatal diabetes and have now replaced insulin as the therapy of choice for this condition (Pearson et al. 2006; Ashcroft, 2010). The drugs also ameliorate (to differing extents) the hypotonia, developmental problems and epilepsy in some patients (Slingerland et al. 2006; Mlynarski et al. 2007; Koster et al. 2008; Slingerland et al. 2008).
The KATP channel is an octameric complex of four Kir6.2 subunits and four SUR1 subunits (Clement et al. 1997; Mikhailov et al. 2005). Kir6.2 is a member of the inwardly rectifying family of potassium channels, with two transmembrane domains (TMs), linked by a pore loop, and cytosolic N- and C-termini. It forms a tetrameric pore, each subunit of which possesses a site to which ATP binds in an Mg-independent manner to close the channel. SUR1 is an ATP-binding cassette protein that endows the channel with sensitivity to Mg-nucleotides, K-channel openers and sulphonylureas (Aguilar-Bryan et al. 1995). Binding and/or hydrolysis of MgATP and MgADP at the nucleotide-binding domains of SUR1 stimulate channel activity (Shyng et al. 1997; Gribble et al. 1998; de Wet et al. 2007).
Gain-of-function mutations in Kir6.2 that cause neonatal diabetes all act by reducing the ability of MgATP to block the channel (Ashcroft, 2007; McTaggart et al. 2010). They may do so directly, by reducing ATP binding, or indirectly, by affecting the transitions between the open and closed states (gating) of the channel leading to an increase in the channel open probability in both the presence and absence of ATP. Such ‘gating’ mutations are often in the regions of the channel thought to move during gating, such as in the helix–bundle crossing region at the cytosolic end of TM2, within the slide helix (an amphipathic helix that lies in the plane of the membrane and is believed to move during gating), or in the cytosolic gating loops of the channel (see Fig. 1).
Figure 1. W68 is highly conserved.
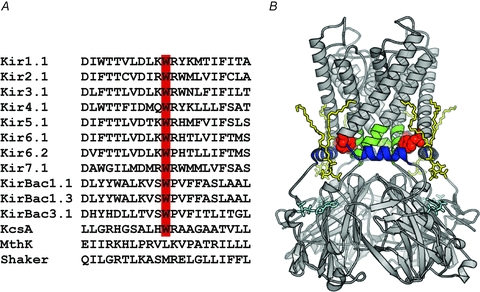
A, sequence alignment for Kir channels showing the conservation of tryptophan at the residue equivalent to W68 in Kir6.2. The tryptophan residue is not conserved in the majority of other K+ channel species. B, model of Kir6.2 showing the location of W68 (red). The slide helix is coloured blue. The transmembrane gate is formed by the C-terminal end of TM2 and is shown in green. The putative bound conformations for ATP (cyan) (Antcliff et al. 2005) and PIP2 (yellow) (Stansfeld et al. 2009) are shown as sticks.
In this paper we identify and characterise a novel Kir6.2 mutation causing a remitting–relapsing form of neonatal diabetes. This mutation changes a tryptophan residue at the cytosolic end of TM1, which is absolutely conserved across the Kir family, to an arginine (W68R). The mutation causes a reduction in the ATP sensitivity of the channel, explaining the diabetes of the patient. We show that mutation of W68 to other amino acids also affects the channel ATP sensitivity, suggesting that the tryptophan has an essential role in channel function, perhaps by acting as a molecular gatekeeper controlling the opening and closing of the channel.
Methods
Mutation detection
All subjects (or their parents) provided informed consent for genetic testing. The procedures were approved by the local ethics committee and the studies conformed to the standards set by the Declaration of Helsinki. Genomic DNA was extracted from peripheral leukocytes using standard procedures. The single-coding exon of KCNJ11 was amplified by the polymerase chain reaction (PCR; primer sequences are available on request). Unidirectional sequencing was performed using universal M13 primers and a Big Dye Terminator Cycler Sequencing Kit v3.1 (Applied Biosystems, Warrington, UK) according to manufacturer's instructions. Reactions were analysed on an ABI 3730 Capillary sequencer (Applied Biosystems, Warrington, UK) and sequences compared to the reference sequence NM000525 using Mutation Surveyor v2.61 (SoftGenetics, PA, USA).
Molecular biology and oocyte preparation
Human Kir6.2 (Genbank NM000525; E23 and I337) and rat SUR1 (Genbank L40624) were used. Site-directed mutagenesis, synthesis of capped mRNA and preparation of Xenopus laevis oocytes were performed as previously reported (Gribble et al. 1997a; Proks et al. 2006). The oocytes were co-injected with ∼4 ng of SUR1 mRNA and ∼0.8 ng wild-type or mutant Kir6.2 mRNA. To simulate the heterozygous state, SUR1 was co-expressed with a 1:1 mixture of wild-type and mutant Kir6.2. In some experiments oocytes were injected with wild-type or mutant Kir6.2 possessing a C-terminal 36 amino acid truncation (Kir6.2ΔC) (Tucker et al. 1997). For each batch of oocytes, all mutations were injected to enable direct comparison of their effects. Oocytes were incubated in Barth's solution and studied 1–4 days after injection.
Electrophysiology
Whole-cell currents were recorded using a two-electrode voltage clamp in response to voltage steps of ±20 mV from a holding potential of –10 mV, in a solution containing (mm): 90 KCl, 1 MgCl2, 1.8 CaCl2 and 5 Hepes (pH 7.4 with KOH). Metabolic inhibition was induced by 3 mm sodium azide and 0.5 mm tolbutamide was used to block KATP channels, as indicated.
Patch-clamp recordings were performed in inside-out patches using an EPC7 amplifier (List Electronik) at a constant holding potential of −60 mV. The pipette solution contained (mm): 140 KCl, 1.2 MgCl2, 2.6 CaCl2 and 10 Hepes (pH 7.4 with KOH). The Mg-free intracellular solution contained (mm): 107 KCl, 1 K2SO4, 10 EGTA, 10 Hepes (pH 7.2 with KOH) and K2ATP, as indicated. The Mg-containing intracellular solution contained (mm): 107 KCl, 11 EGTA, 2 MgCl2, 1 CaCl2, 10 Hepes (pH 7.2 with KOH) and MgATP rather than K2ATP. For experiments with neomycin the intracellular solution contained (mm): 100 KCl, 10 Hepes and 2 EGTA (pH 7.4 with KOH). To control for possible rundown or activation by MgATP, Ic was taken as the mean of the current in control solution before and after ATP application. Concentration–response curves were fitted with a modified Hill equation:
| (1) |
where [X] is the concentration of the test substance, IC50 is the concentration at which inhibition is half maximal, h is the slope factor (Hill coefficient) and a represents the fraction of unblocked current at saturating [ATP] (a = 0 except where specified).
Single-channel currents were recorded at −60 mV, filtered at 5 kHz, sampled at 20–50 kHz, and analysed using a combination of Clampfit (Axon Instruments) and Origin (OriginLab Corporation). The open probability in the absence of ATP (Po(0)) was determined from single-channel patches as the fraction of time spent in the open state for recordings of 30 s duration. First, channel activity (NPo(0)) was measured as the mean current (I) divided by the single-channel amplitude. Then Po(0) was calculated from NPo(0)/N, where N is number of channels in the patch estimated from the maximum number of superimposed events. Data are given as mean ± SEM. Significance was evaluated using Student's t test.
Homology modelling
Homology models of the Kir6.2 transmembrane domain were created using Modeller 9v8 (Sali & Blundell, 1993). Closed state models were created with W68 either in the ‘flipped-in’ state, based on KirChim (2QKS), or in the flipped-out conformation, based on Kir2.2 (3JYC). An open state model of Kir6.2 was created using KvAP (1ORQ) to model TM2, with the remainder of the channel based on Kir2.2. Side-chain conformations for the W68 mutant channels were predicted using the Hunter algorithm (Cohen et al. 2009). All structures were visualized using Pymol (Schrödinger LLC).
Results
Patient characteristics and genetics
The proband was born at 37 weeks gestation with a birth weight of 1.8 kg. She was diagnosed with diabetes at 8 years of age and treated with insulin. Her mother had transient neonatal diabetes diagnosed at 16 weeks and was treated with insulin for 10 weeks. Her diabetes relapsed at 7 years of age and insulin therapy was resumed. There were no obvious neurological complications.
Sequence analysis identified a heterozygous KCNJ11 mutation, W68R (c.202T>C, p.Trp68Arg), in both the proband and her mother. Testing of the maternal grandparents showed that the W68R mutation had arisen de novo in the proband's mother. Following identification of the KCNJ11 mutation, both mother and daughter successfully transferred from insulin to oral sulphonylureas.
Effects on whole-cell KATP currents
The tryptophan at the position corresponding to W68 in Kir6.2 is absolutely conserved within the family of inward rectifying potassium channels (Fig. 1), suggesting it has a critical role. We analysed the effects of the Kir6.2-W68R mutation on the metabolic regulation of the KATP channel by measuring whole-cell currents. When wild-type (WT) KATP channels are expressed in Xenopus oocytes they are normally closed, due to the high intracellular ATP concentration ([ATP]i), but they can be opened by lowering ATP using a metabolic inhibitor (3 mm sodium azide; Fig. 2) (Gribble et al. 1997a). Mutations that reduce the channel ATP sensitivity normally increase the whole-cell current in the absence of metabolic inhibition, reflecting the fact that they are less blocked by resting concentrations of [ATP]i (Ashcroft, 2007). Resting currents in oocytes expressing homomeric Kir6.2-W68R/SUR1 channels (homW68R channels) were significantly larger than wild-type (WT) currents. They were also increased by metabolic inhibition, suggesting that homW68R channels are not fully open at resting [ATP]i.
Figure 2. Effect of the W68R mutation on whole-cell KATP currents.
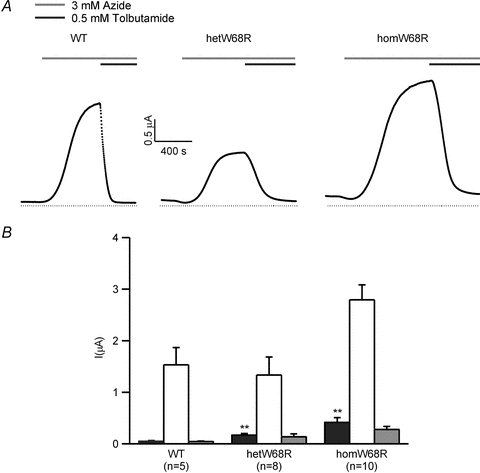
A, representative whole-cell currents evoked by repeated voltage steps from −10 to −30 mV for wild-type (WT), hetKir6.2-W68R/SUR1 (hetW68R) and Kir6.2-W68R/SUR1 (homW68R) channels. The bars indicate the duration of application of 3 mm azide (black) and 0.5 mm tolbutamide (grey). The dotted line indicates the zero current level. B, mean steady-state whole-cell currents evoked by a voltage step from −10 to −30 mV before (control, black bars), and after application of 3 mm azide (white bars), and in the presence of 3 mm azide plus 0.5 mm tolbutamide (grey bars) for wild-type (WT, n = 5), and heterozygous (n = 8) and homomeric (n = 10) Kir6.2-W68R/SUR1 channels, as indicated. **P < 0.01.
To simulate the heterozygous state of the patients, we co-injected a 1:1 mixture of mutant and wild-type Kir6.2, together with SUR1. The resulting population of channels will contain a variable number of mutant subunits (between 0 and 4) in the Kir6.2 tetramer. We refer to this mixed channel population as heterozygous (het) channels. Resting hetW68R currents were significantly larger than wild-type but not as large as homW68R currents. They were also activated by azide to a larger extent.
The sulphonylurea tolbutamide (500 μm) blocked azide-activated homW68R currents by 90 ± 2% (n = 10) and hetW68R channels by 90 ± 1% (n = 7), compared with 97 ± 1% (n = 10) for wild-type channels.
Effects on KATP channel ATP sensitivity
The increase in resting whole-cell KATP currents found for mutant channels suggests that the W68R mutation may reduce the channel ATP sensitivity. We tested this idea directly by measuring the sensitivity of wild-type and mutant channels to inhibition by ATP in inside-out patches. We first did so in the absence of Mg2+ (Fig. 3A), because this allows the effects of the mutation on ATP inhibition at Kir6.2 to be isolated from the stimulatory effects mediated via SUR1 (as ATP does not interact with SUR1 in the absence of Mg2+; Gribble et al. 1998). Both hetW68R and homW68R currents showed significantly impaired ATP sensitivity: half-maximal block (IC50) was produced by 74 μm ATP for homW68R channels and 28 μm ATP for hetW68R channels, compared with 10 μm ATP for wild-type channels (Fig. 3A, Table 1).
Figure 3. Effect of the W68R mutation on ATP sensitivity in the absence and presence of Mg2+.
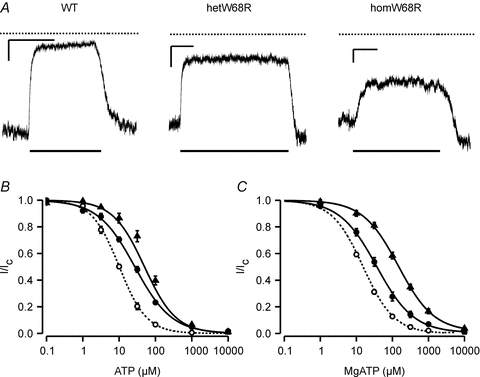
A, representative currents recorded at −60 mV from inside-out patches expressing wild-type (Kir6.2/SUR1), hetW68R or homW68R channels. Patches were exposed to 100 μm ATP as indicated by the lower bar. The dotted line indicates the zero current level. The x-axis scale bar is 10 s, and the y-axis scale bar is 500 pA (Kir6.2/SUR1), 500 pA (hetW68R) and 100 pA (homW68R). B and C, mean relationship between [ATP] and KATP current (I), expressed relative to the current in the absence of nucleotide (Ic) for Kir6.2/SUR1 (open circles), heterozygous (filled circles) and homomeric (filled triangles) Kir6.2-W68R/SUR1 channels. Experiments were carried out in the absence (B) or presence (C) of 2 mm Mg2+. The smooth curves are the best fit of eqn (1) to the data. Mg-free: WT IC50 = 10 μm, h = 1.15 (n = 9); hetW68R IC50 = 27 μm, h = 0.87 (n = 9); homW68R IC50 = 50 μm, h = 1.0 (n = 7). 2 mm Mg2+: WT IC50 = 15 μm, h = 1.01 (n = 26); hetW68R IC50 = 37 μm, h = 0.85, a = 0.005 (n = 12); homW68R IC50 = 146 μm, h = 0.88, a = 0.014 (n = 13).
Table 1.
ATP inhibition of wild-type and mutant channels
| IC50, Mg-free ATP (μm) | n | IC50, MgATP (μm) | % unblocked current, 3 mm MgATP | n | |
|---|---|---|---|---|---|
| WT | 10.1 ± 0.6 | 9 | 15.7 ± 0.9 | 0.6 ± 0.1 | 26 |
| hetW68R | 27.6 ± 2.3 | 9 | 40.4 ± 6.5 | 3.1 ± 0.6 | 12 |
| homW68R | 73.7 ± 8.0 | 7 | 142 ± 12 | 7.9 ± 0.8 | 13 |
| homW68E | 153 ± 18 | 5.8 ± 1.1 | 7 | ||
| homW68K | 256 ± 29 | 10.0 ± 1.2 | 10 | ||
| homW68A | 228 ± 39 | 8.7 ± 1.5 | 6 | ||
| homW68C | 837 ± 338 | 29 ± 4 | 6 | ||
| homW68L | 107 ± 17 | 5.8 ± 0.5 | 7 | ||
| homW68M | 77 ± 13 | 5.9 ± 1.0 | 5 | ||
| homW68F | 1027 ± 490 | 30 ± 6 | 11 | ||
| homW68Y | >10 mm | 4 | >10 mm | 87 ± 1 | 8 |
| Kir6.2ΔC | 175 ± 25 | 5.4 ± 0.6 | 5 | ||
| Kir6.2ΔC-W68Y | 1.8 ± 0.4 mm | 34 ± 6 | 5 |
Data are the mean of the fits of the Hill equation (eqn 1) to the individual dose–response curves. IC50, half-maximal inhibitory concentration of ATP. n, number of patches.
We next examined the ATP sensitivity of wild-type and mutant channels in the presence of 2 mm Mg2+, to more closely approximate the physiological condition. ATP inhibition was further reduced in the presence of Mg2+, the IC50 being 142 μm for homW68R, 40 μm for hetW68R and 14 μm for wild-type channels. (Fig. 3B, Table 1). At 3 mm MgATP, a concentration within the physiological range, the unblocked current was estimated at 3.1% for hetW68R channels, compared with 0.6% for wild-type channels.
Effect of additional mutations at W68 on ATP sensitivity
The W68R mutation replaces the hydrophobic tryptophan with a positively charged arginine. To examine the role of the introduced charge on the channel ATP sensitivity, we tested the effect of substituting a negative charge. Interestingly, introduction of a glutamate residue produced an identical effect to that of arginine: the IC50 for MgATP block of the homW68E channels was 153 μm, compared with 142 μm for homW68R (Fig. 4A, Table 1). Substitution of a smaller positively charged lysine at W68 had a similar effect (IC50 = 256 μm). These data suggest that reduced ATP sensitivity of W68R is not due to specific electrostatic interactions.
Figure 4. Effect of other mutations at position W68 on MgATP sensitivity.
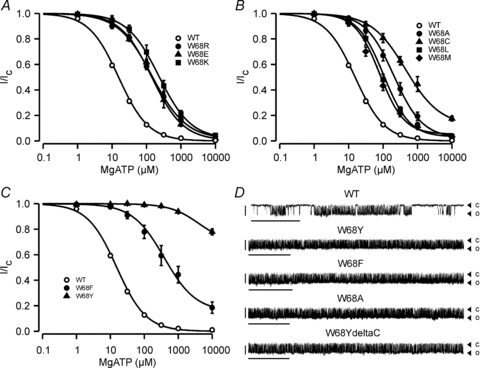
A, mean relationship between [MgATP] and KATP channel current (I), expressed relative to the current in the absence of nucleotide (Ic) for WT (open circles, n = 26), homW68R (filled circles, n = 12), homW68E (filled triangles, n = 7) and homW68K (filled squares, n = 10) channels. The smooth curves are the best fit of eqn (1) to the data: WT (IC50 = 15 μm, h = 1.01), homW68R (IC50 = 146 μm, h = 0.88, a = 0.014), homW68E (IC50 = 142 μm, h = 0.95, a = 0.004), homW68K (IC50 = 229 μm, h = 0.93, a = 0.016). B, mean relationship between [MgATP] and KATP channel current (I), expressed relative to the current in the absence of nucleotide (Ic) for WT (open circles, n = 26), homW68A (filled circles, n = 6), homW68C (filled triangles, n = 6), homW68L (filled squares, n = 7) and homW68M mutants (filled diamonds, n = 5). The smooth curves are the best fit of eqn (1) to the data: WT (IC50 = 15 μm, h = 1.01), homW68A (IC50 = 206 μm, h = 1.05, a = 0.024), homW68C (IC50 = 454 μm, h = 0.87, a = 0.121), homW68L (IC50 = 104 μm, h = 1.16, a = 0.031), homW68M (IC50 = 80 μm, h = 1.22, a = 0.053). C, mean relationship between [MgATP] and KATP channel current (I), expressed relative to the current in the absence of nucleotide (Ic) for WT (open circles, n = 26), homW68F (filled circles, n = 11), and homW68Y (filled triangles, n = 8) channels. The smooth curves are the best fit of eqn (1) to the data: WT (IC50 = 15 μm, h = 1.01), homW68F (IC50 = 388 μm, h = 0.93, a = 0.153): that through the homW68Y data is drawn by eye. D, single-channel currents recorded at −60 mV from an inside-out patch expressing WT and homW68 mutant channels (as indicated). Closed (c) and open (o) current levels are indicated by triangles. Scale bars are 2 s (x-axis) and 5 pA (y-axis).
Tryptophan has a large hydrophobic surface area with a nitrogen that introduces polarity. Its amphipathic nature is well suited for life at the membrane–water interface, where it is enriched. The other polar amino acid to have an aromatic hydrophobic ring is tyrosine, which is also enriched at the membrane–water interface. Remarkably, the presence of a tyrosine at position 68 caused the most dramatic reduction in channel ATP sensitivity, with 10 mm MgATP blocking the homW68Y channel by less than 25%. This suggests that it is the specific molecular properties of the tryptophan, not just its amphipathic nature, which is required for proper KATP channel function. The smallest shift in ATP sensitivity was produced by methionine (IC50 = 77 μm) and the order of potency was W<M∼L<R∼E∼K∼A<C∼F<Y (Fig. 4B and C; Table 1). Thus, hydrophobic residues at residue 68 can cause either greater or lesser effects on ATP sensitivity than charged residues, depending on the specific mutation. Introduction of a larger hydrophobic side-chain, such as methionine or leucine, produces a lesser shift in ATP sensitivity than the smaller alanine or cysteine. However, the size of the residue does not appear to be the sole determinant of its effect on ATP sensitivity.
Mutations at W68 affect channel gating
Many neonatal diabetes mutations reduce the ATP sensitivity of the KATP channel by impairing ATP binding and/or transduction, whereas others influence ATP sensitivity indirectly by increasing the intrinsic (ligand-independent) open probability (Po(0)) of the channel (Ashcroft, 2007). We therefore examined the effect of W68 mutations on Po(0) by recording single-channel activity in the absence of nucleotides. This was 0.86 ± 0.01 (n = 11) for W68Y and 0.4 ± 0.04 (n = 8) for WT channels (Fig. 4D). Similarly, the Po(0) was increased to 0.77 ± 0.06 (n = 6) when W68 was mutated to alanine (W68A) and 0.83 ± 0.01 (n = 5) when mutated to phenylalanine (W68F) (Fig. 4D). Thus, mutations at W68 appear to decrease the channel ATP sensitivity indirectly, by altering Po(0). The Po(0) of W68R could not be measured accurately due to the rapid rundown (see Supplementary Fig. S1).
We next explored if the shift in ATP sensitivity was intrinsic to Kir6.2, or secondary to impaired modulation by SUR1, by examining the effects of the W68Y mutation on a truncated form of Kir6.2 (Kir6.2ΔC) that expresses in the absence of SUR1 (Tucker et al. 1997). The W68Y mutation caused a dramatic increase in Po(0) from 0.01 for Kir6.2ΔC (Drain et al. 1998) to 0.78 ± 0.02 (n = 9) for Kir6.2ΔCW68Y, and a marked reduction in the ATP sensitivity of the Kir6.2ΔC channel (Fig. 5, Table 1). This indicates that the effect of the W68Y mutation on the channel open probability is intrinsic to Kir6.2 and independent of SUR1. It is likely that the reduced ATP sensitivity is secondary to the increased Po(0).
Figure 5. Kir6.2ΔC-W68Y currents have reduced ATP sensitivity.
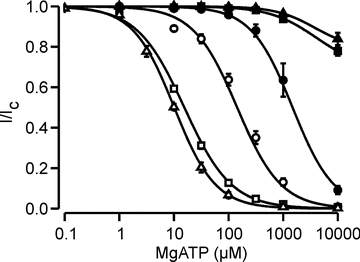
Mean relationship between [ATP] and KATP current (I), expressed relative to the current in the absence of nucleotide (Ic) for Kir6.2/SUR1 (open triangles, Mg-free, n = 9), Kir6.2/SUR1 (open squares, with Mg2+, n = 26), Kir6.2-W68Y/SUR1 (filled triangles, Mg-free, n = 4), Kir6.2-W68Y/SUR1 (filled squares, with Mg2+, n = 8), Kir6.2ΔC (open circles, with Mg2+, n = 5) and Kir6.2ΔC-W68Y/SUR1 channels (filled circles, with Mg2+, n = 5). The smooth curves are the best fit of eqn (1) to the data: Kir6.2/SUR1 (Mg-free; IC50 = 10 μm, h = 1.15), Kir6.2/SUR1 (with Mg2+, IC50 = 15 μm, h = 1.01), Kir6.2-W68Y/SUR1 (Mg-free, drawn by eye), Kir6.2-W68Y/SUR1 (with Mg2+, drawn by eye), Kir6.2ΔC (with Mg2+, IC50 = 144 μm, h = 1.08) and Kir6.2ΔC-W68Y/SUR1 channels (with Mg2+, IC50 = 1.5 mm, h = 1.14).
As previously reported, Kir6.2ΔC is about 10-fold less ATP sensitive when expressed in the absence of SUR1 (Fig. 5; Table 1) (Tucker et al. 1997). Thus, despite increasing the channel open probability (Drain et al. 1998), SUR1 enhances the ATP sensitivity of the channel. The mechanism of this effect is not known. In marked contrast, Kir6.2ΔC-W68Y was considerably more ATP sensitive than Kir6.2-W68Y/SUR1, suggesting that, in contrast to wild-type Kir6.2, SUR1 is unable to enhance the ATP sensitivity of Kir6.2-W68Y. Furthermore, there was no difference in ATP sensitivity between Kir6.2-W68Y/SUR1 in the presence and absence of Mg2+. To our knowledge, the W68Y mutation is the first to be identified that affects the ability of SUR1 to enhance the ATP sensitivity of Kir6.2. This result and suggests that mutation of W68 alters the interactions of Kir6.2 with SUR1.
Mutations at W68 affect PIP2 sensitivity
The tryptophan at residue 68 lies in close proximity to the putative PIP2 binding site of the channel, which is thought to include residue K67 (Haider et al. 2007), and the rapid rundown of W68R channels is typical for mutations that affect PIP2 binding to the KATP channel (Shyng et al. 2000). PIP2 is known to enhance the Po(0) of KATP channels and reduce their sensitivity to ATP inhibition, both of which are also seen for mutation at W68. We therefore explored the possibility that introduction of a charged residue at W68 might affect the PIP2 affinity of the channel, by assessing the effect of neomycin on channel behaviour. Neomycin is a polyanion that reduces the functional effects of PIP2 (Schulze et al. 2003). It is presumed to do so by screening the negative charges on the PIP2 headgroups and decreasing the effective availability of membrane PIP2. Thus, it may be expected that if PIP2 binds to the KATP channel with high affinity, a higher concentration of neomycin will be required to reduce the KATP current, whereas if PIP2 binding is weak, a lower concentration of neomycin will be sufficient to inhibit the channel to the same extent (Schulze et al. 2003).
Neomycin inhibited WT channels with an IC50 of 129 ± 15 μm (n = 8) (Fig. 6). Introduction of either a positive or a negative charge increased neomycin block (IC50 = 11.3 ± 2.1 μm (n = 4) for homW68R and 8.2 ± 0.9 μm (n = 4) for homW68E), suggesting these substitutions decrease PIP2 affinity, while phenylalanine had no effect (IC50 = 148 ± 32 μm (n = 5)). A decreased PIP2 affinity is expected to increase the channel ATP sensitivity, and therefore cannot explain the reduced ATP block of W68R and W68E channels. Likewise, the lack of an effect of phenylalanine substitution argues against a role for PIP2 in the reduced ATP sensitivity of W68F channels. However, the marked decrease in neomycin block observed for W68Y channels (<25% block at 10 mm, Fig. 6) is consistent with an enhanced PIP2 affinity, which could contribute to both the increase in Po(0) and the reduced ATP block.
Figure 6. Effect of the W68R mutation on neomycin sensitivity.
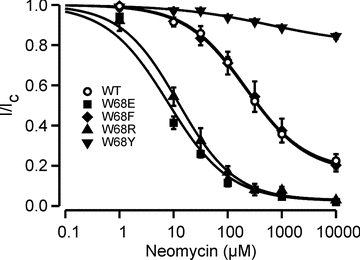
Mean relationship between [Neomycin] and KATP channel current (I), expressed relative to the current in the absence of neomycin (Ic) for Kir6.2/SUR1 (open circles, n = 8; IC50 = 216 μm, h = 0.79, a = 0.18), homW68E (filled squares, n = 4; IC50 = 9 μm, h = 0.91), homW68F (filled diamonds, n = 5; IC50 = 257 μm, h = 0.7, a = 0.14), homW68R (filled triangles, n = 4; IC50 = 13 μm, h = 0.8) and homW68Y (filled inverted triangles, n = 4; drawn by eye) channels.
Discussion
Functional implications
The results presented above provide an explanation for the patients’ diabetes and suggest that a tryptophan at residue 68 of Kir6.2 is critical for correct channel function. The small increase in the amplitude of hetW68R currents recorded in inside-out patches exposed to physiological levels of MgATP explains the increase in the resting whole-cell KATP currents. A similar increase in the beta-cell KATP current would be expected to prevent or reduce membrane depolarization, Ca2+ influx and insulin secretion evoked by glucose. This could explain the diabetic phenotype of the patients.
The amplitude of hetW68R currents recorded in the presence of MgATP is consistent with the absence of neurological symptoms in the patients. Previous studies have shown that mutations that cause transient or permanent neonatal diabetes but no neurological phenotype cause less increase in the KATP current at 3 mm MgATP than those associated with neurological symptoms (for review see Ashcroft, 2007; McTaggart et al. 2010). For mutations that cause DEND syndrome, between 26 and 40% of the heterozygous current is not blocked by 3 mm MgATP, compared to 12–20% for mutations causing iDEND, 4–10% (in general) for mutations causing neonatal diabetes alone and <1% for wild-type channels. About 3% of the maximal hetW68R current was found in 3 mm MgATP, which is at the lower end of the range that causes isolated neonatal diabetes.
The magnitude of the KATP current in 3 mm MgATP found for hetW68R currents is one of the smallest in reported to date to be associated with a Kir6.2 mutation causing neonatal diabetes (McTaggart et al. 2010). This is consistent with the remitting–relapsing diabetes in the proband's mother and the later age of onset of diabetes in the proband (8 years). Very small changes in resting whole-cell current and KATP currents at high ATP concentrations in excised patches have also been reported for other Kir6.2 mutations that cause transient diabetes (Girard et al. 2006). Because of the very high input resistance of the beta-cell when most KATP channels are closed even a small increase in current may be expected to have a large effect on the beta-cell membrane potential, and thereby on insulin release (Tarasov et al. 2006).
Molecular mechanism – W68 might act as a molecular gatekeeper
The increased whole-cell KATP current produced by the W68R mutation results from a reduced ability of ATP to close the KATP channel; this is mediated via Kir6.2 as it is present in the absence of Mg2+. This is probably accounted for by changes in the intrinsic gating of the channel rather than by a direct reduction of ATP binding to Kir6.2, as the mutation lies far from the ATP-binding site (Antcliff et al. 2005). Although the open probability of the W68R mutant could not be measured directly, due to rapid rundown of the channel in excised patches, other mutations at this position (e.g. W68Y, W68A, W68F) resulted in channels that have a markedly increased open probability. This suggests that W68 may have an important role in channel gating.
In all Kir crystal structures (Kuo et al. 2003; Nishida et al. 2007) the residue equivalent to W68 lies at the N-terminal end of TM1, close to where the protein chain undergoes a 90 degree turn to form the slide helix. In the majority of Kir channel crystal structures, including all the KirBac structures and the KirChim (Protein Data Bank (PDB) i.d.: 2QKS) structure, the residue equivalent to W68 appears to impose a constriction on the TM2 helix bundle (Fig. 7A), in a similar manner to the lock imposed on the activation gate by the S4–S5 linker in Kv channels (Long et al. 2005). Importantly, the cytosolic domains and about one third of the transmembrane domains in the KirChim are composed of Kir3.1 (the rest of the protein is KirBac1.1) (Nishida et al. 2007). Thus, the crystal structures suggest that the residue equivalent to W68 can adopt a ‘flipped-in’ position in both Kir and KirBac channels, which holds the channel closed. By contrast, in the Kir2.2 crystal structure (3JYC) the side-chain of the tryptophan is rotated away from TM2, and adopts a ‘flipped-out’ position (Fig. 7B). This is likely to release the constraint on the TM2 helices, allowing them to move and open the gate at the helix–bundle crossing (Fig. 7B, see also Supplementary videos) (Tao et al. 2009). Comparison of the two Kir crystal structures (Kir3.1 and Kir2.2) therefore suggests that W68 (or its equivalent) may act as a gatekeeper for Kir channels, by moving between ‘flipped-in’ and ‘flipped-out’ conformations.
Figure 7. Location of W68 in Kir channels.
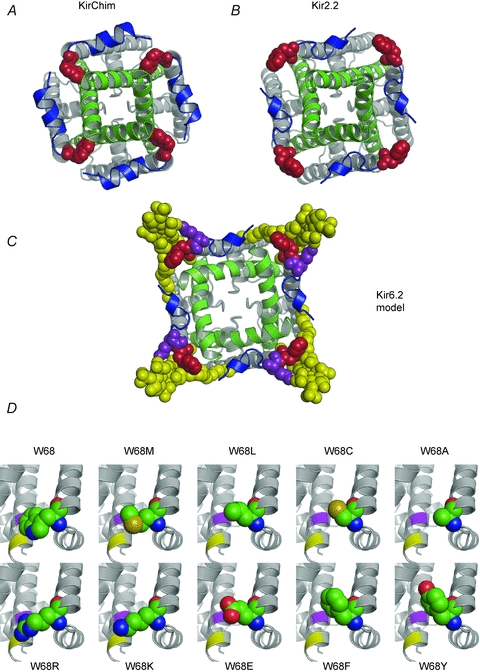
A and B, crystal structures of KirChim (PDB id: 2QKS) (A) and Kir2.2 (3JYC) (B) viewed from the intracellular side. In the majority of Kir channel X-ray structures, the residue equivalent to W68 in Kir6.2 (red) is tightly packed against the C-terminal end of TM2 (green) as seen in the crystal structure of KirChim (A). The side-chain of the residue equivalent to W68 also forms close contacts with residues in the slide helix (blue). In the Kir2.2 crystal structure (B) the tryptophan side-chain is in a different (‘flipped-out’) conformation, pointing away from the channel pore. An outward curvature of TM2 is apparent, focused on the second highly conserved glycine of TM2, although the helix–bundle crossing remains in the closed state. C, molecular model of Kir6.2 in the open state. The TM2 helices are based on KvAP; the remainder of the structure is based on Kir2.2. The ‘flipped-out’ state of W68 has allowed TM2 to undergo the opening transition. PIP2 (yellow) is shown docked into its binding site (Stansfeld et al. 2009), interacting with both W68 and with K67 (purple). D, structural models of Kir6.2 (Stansfeld et al. 2009) with the side-chain conformations of residue 68 for the mutant channels as predicted by the Hunter algorithm (Cohen et al. 2009). The locations of residues I167 (purple) and T171 (yellow) in TM2 are shown.
There is considerable sequence identity between Kir channels, and the crystal structures of Kir2.2 and the Kir3.1 domain in KirChim are highly homologous, indicating that a homology model of Kir6.2 based on the either of these structures is likely to be a good approximation to the actual structure. In a homology model of the closed ‘flipped-in’ state, the side-chain of W68 is directed towards the C-terminal end of TM2, and interacts hydrophobically with residues I167, K170 and T171. Mutations of residues K170 (K170T) and I167 (I167L) in Kir6.2 result in neonatal diabetes by reducing inhibition by ATP: at least in the case of I167L this is secondary to an increase in the channel open probability (Shimomura et al. 2007; Tarasov et al. 2007). Many different mutations at T171 also cause a large increase in Po(0) (Drain et al. 2004). This suggests that the interaction of W68 with T171 (and/or I167 and K170) favours the closed state and when this interaction is disrupted the Kir6.2 channel opens.
We also modelled Kir6.2 in the open state, using the KvAP structure (PDB i.d.: 1ORQ) as a template for TM2 and Kir2.2 as a template for the remainder of the protein (Fig. 7C). We also included PIP2, modelled in its binding site (after Stansfeld et al. 2009). In the open state model, W68 swings away from the inner TM2 helix to interact with the glycerol tail of PIP2. Thus, the presence of PIP2 may increase the probability that W68 will adopt the ‘flipped-out’ conformation, thereby eliminating the constriction placed on the helix–bundle crossing and promoting the open state. We therefore argue that the relative strength of the interactions of W68 with PIP2versus that with I167 and T171 will determine whether the channel is biased towards the open or the closed state, respectively.
This idea also suggests an explanation for why small residues (e.g. alanine, cysteine) produce large shifts in ATP sensitivity: they are too short to interact with I167 (K170/T171) so that the channel is biased towards the open state (Fig. 7D). While the charged residues (arginine, lysine or glutamate) are of suitable size to impede the motions of TM2, the addition of a charge at this region is likely to have a destabilizing effect, reducing the interactions of the residue at 68 with TM2 and destabilizing the closed state. This will indirectly reduce the channel ATP sensitivity.
Remarkably, the greatest increase in Po(0) and the largest reduction in ATP sensitivity was produced when W68 was replaced with tyrosine, the most conservative substitution. To explain why the tyrosine, and phenylanine, cause large shifts in ATP sensitivity, we postulate that they are not appropriately accommodated in the ‘binding’ pocket in which W68 lies when the channel is in the closed (flipped-in) state, as suggested by side-chain predictions by the Hunter algorithm (Fig. 7D). Therefore, the channel favours the open state. We further postulate that tyrosine may interact more strongly with PIP2 than tryptophan (or phenylanine), as there is an apparent increase in PIP2 affinity for the W68Y channel. Tyrosine is known to interact strongly with the glycerol region of lipids, and indeed serves as a marker for the headgroup position of the lipid bilayer. Whilst this is also true for tryptophan residues, we suggest that W68 is equally balanced between the ‘flipped-in’ and ‘flipped-out’ state, whereas W68Y and W68F prefer the ‘flipped-out’ state, for steric reasons.
Methionine, which shifts the ATP sensitivity the least, adopts a conformation most similar to that of tryptophan (Fig. 7D). Clearly, it is the volume and precise orientation of the tryptophan at position 68 that is critical for proper channel function, as demonstrated by the fact that this residue is conserved across all Kir channels. We therefore speculate that W68 acts as a molecular gatekeeper, a suggestion supported by the experimental observation that mutation of this residue enhances the channel open probability.
Clinical implications
Sulphonylureas are an effective therapy for most patients with neonatal diabetes caused by KATP channel mutations, and many have now successfully transferred from insulin to sulphonylureas (Pearson et al. 2006). The magnitude of tolbutamide block of mutant KATP channels in functional studies correlates well with the success of the sulfonylurea therapy (McTaggart et al. 2010). We observed that tolbutamide caused 90% block of hetW68R channels. This value lies well within the range in which patients respond to sulphonylurea therapy and explains why our patients could be successfully treated with glibenclamide.
Acknowledgments
This work was supported by Wellcome Trust (076436, 089795 and the OXION Initiative), the Royal Society, and the European Union (EuroDia-(LSHM-CT-2006–518153) and EDICT). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. F.M.A. is a Royal Society Research Professor and A.T.H. a Wellcome Trust Research Leave Fellow. A.T.H. and S.E. are members of the core staff within the NIHR-funded Peninsula Clinical Research Facility.
Glossary
Abbreviations
- KATP
ATP-sensitive K+ channel
- Kir
inward rectifier K+ channel
- SUR
sulphonylurea receptor
Author contributions
The experiments were performed in the laboratories of F.M.A and M.S at Oxford University, and in the laboratory of A.T.H/S.E. at Peninsula Medical School, Exeter. F.M.A, P.J.S, S.E. and R.M designed the experiments. R.M. and A.A. collected, analysed and interpreted the electrophysiology data, P.J.S. and M.S. the molecular modelling, and S.E. and A.T.H. the molecular genetics data. F.M.A., P.J.S. and R.M. drafted the paper. All authors contributed to the discussion, reviewed and approved the final manuscript.
Supplementary material
Supplementary Figure 1
Supplementary Video 1
Supplementary Video 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, 4th, Boyd AE, 3rd, González G, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Antcliff JF, Haider S, Proks P, Sansom MSP, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 2005;24:229–239. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM. The Walter B. Cannon physiology in perspective lecture, 2007. ATP-sensitive K-channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab. 2007;293:E880–E889. doi: 10.1152/ajpendo.00348.2007. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. New uses for old drugs: neonatal diabetes and sulphonylureas. Cell Metab. 2010;11:179–181. doi: 10.1016/j.cmet.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Harrison DE, Ashcroft SJH. Glucose induces closure of single potassium channels in isolated rat pancreatic ß-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. New Engl J Med. 2006;355:456–466. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- Clark R, Webster R, McTaggart J, Mannikko R, Iberl M, Sim X, et al. Neuronal etiology of muscle dysfunction caused by a human KATP channel mutation. Science. 2010;329:458–461. doi: 10.1126/science.1186146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, 4th, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Cohen M, Potapov V, Schreiber G. Four distances between pairs of amino acids provide a precise description of their interaction. PLoS Comput Biol. 2009;5:e1000470. doi: 10.1371/journal.pcbi.1000470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wet H, Mikhailov MV, Fotinou C, Dreger M, Craig TJ, Vénien-Bryan C, Ashcroft FM. Studies of the ATPase activity of the ABC protein SUR1. FEBS J. 2007;274:3532–3544. doi: 10.1111/j.1742-4658.2007.05879.x. [DOI] [PubMed] [Google Scholar]
- Drain P, Geng X, Li L. Concerted gating mechanism underlying KATP channel inhibition by ATP. Biophys J. 2004;86:2101–2112. doi: 10.1016/S0006-3495(04)74269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P, Li L, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci U S A. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan SE, Patch AM, Mackay DJ, Edghill EL, Gloyn AL, Robinson D, et al. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56:1930–1937. doi: 10.2337/db07-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard CA, Shimomura K, Proks P, Absalom N, Castano L, Perez de Nanclares G, Ashcroft FM. Functional analysis of six Kir6.2 (KCNJ11) mutations causing neonatal diabetes. Pflugers Arch. 2006;453:323–332. doi: 10.1007/s00424-006-0112-3. [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. New Eng J Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ammälä C, Ashcroft FM. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J Physiol. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997b;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the β cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider S, Tarasov A, Craig T, Sansom MSP, Ashcroft FM. Identification of the PIP2-binding site on Kir6.2 by molecular modelling and functional analysis. EMBO J. 2007;26:3749–3759. doi: 10.1038/sj.emboj.7601809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattersley AT, Ashcroft FM. Activating Mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights and new therapy. Diabetes. 2005;54:2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- Koster JC, Cadario F, Peruzzi C, Colombo C, Nichols CG, Barbetti F. The G53D mutation in Kir6.2 (KCNJ11) is associated with neonatal diabetes and motor dysfunction in adulthood that is improved with sulfonylurea therapy. J Clin Endocrinol Metab. 2008;93:1054–1061. doi: 10.1210/jc.2007-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, et al. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;5:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- McTaggart JS, Clark RH, Ashcroft FM. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J Physiol. 2010;588:3201–3209. doi: 10.1113/jphysiol.2010.191767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov MV, Campbell JD, de Wet H, Shimomura K, Zadek B, Collins RF, et al. 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J. 2005;24:4166–4175. doi: 10.1038/sj.emboj.7600877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Seino S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J Mol Cell Cardiol. 2005;38:917–925. doi: 10.1016/j.yjmcc.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Mlynarski W, Tarasov AI, Gach A, Girard CA, Pietrzak I, Zubcevic L, et al. Improvement of CNS function with sulphonylurea treatment in intermediate DEND syndrome caused by the novel H46L mutation in the KCNJ11 gene. Nat Clin Prac Neurol. 2007;3:640–645. doi: 10.1038/ncpneuro0640. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, 4th, Gonzalez G, et al. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, et al. Neonatal Diabetes International Collaborative Group Switching from insulin to oral sulphonylureas patients with diabetes due to Kir6.2 mutations. New Eng J Med. 2006;355:467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- Proks P, Antcliff JF, Lippiat J, Gloyn AL, Hattersley AT, Ashcroft FM. Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci U S A. 2004;101:17539–17544. doi: 10.1073/pnas.0404756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P, Arnold AL, Bruining J, Girard C, Flanagan SE, Larkin B, et al. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum Mol Genet. 2006;15:1793–1800. doi: 10.1093/hmg/ddl101. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schulze D, Krauter T, Fritzenschaft H, Soom M, Baukrowitz T. Phosphatidylinositol 4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir channels. A tale of an active and a silent PIP2 site in the N terminus. J Biol Chem. 2003;278:10500–10505. doi: 10.1074/jbc.M208413200. [DOI] [PubMed] [Google Scholar]
- Shimomura K, Hörster F, de Wet H, Flanagan SE, Ellard S, Hattersley AT, et al. A novel mutation causing DEND syndrome – a treatable channelopathy of pancreas and brain. Neurology. 2007;69:1342–1349. doi: 10.1212/01.wnl.0000268488.51776.53. [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide-binding folds of the sulfonylurea receptor. J Gen Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Cukras CA, Harwood J, Nichols CG. Structural determinants of PIP2 regulation of inward rectifier KATP channels. J Gen Physiol. 2000;116:599–608. doi: 10.1085/jgp.116.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slingerland AS, Hurkx W, Noordam K, Flanagan SE, Jukema JW, Meiners LC, et al. Sulphonylurea therapy improves cognition in a patient with the V59M KCNJ11 mutation. Diabet Med. 2008;25:277–281. doi: 10.1111/j.1464-5491.2007.02373.x. [DOI] [PubMed] [Google Scholar]
- Slingerland AS, Nuboer R, Hadders-Algra M, Hattersley AT, Bruining GJ. Improved motor development and good long-term glycaemic control with sulfonylurea treatment in a patient with the syndrome of intermediate developmental delay, early-onset generalised epilepsy and neonatal diabetes associated with the V59M mutation in the KCNJ11 gene. Diabetologia. 2006;49:2559–2563. doi: 10.1007/s00125-006-0407-0. [DOI] [PubMed] [Google Scholar]
- Stansfeld PJ, Hopkinson R, Ashcroft FM, Sansom MS. PIP2-binding site in Kir channels: definition by multiscale biomolecular simulations. Biochemistry. 2009;48:10926–10933. doi: 10.1021/bi9013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 Å resolution. Science. 2009;326:1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov AI, Girard CA, Larkin B, Tammaro P, Flanagan SE, Ellard S, Ashcroft FM. Functional analysis of two Kir6.2 (KCNJ11) mutations, K170T and E322K, causing neonatal diabetes. Diabetes Obes Metab. 2007;9:46–55. doi: 10.1111/j.1463-1326.2007.00777.x. [DOI] [PubMed] [Google Scholar]
- Tarasov AI, Welters HJ, Senkel S, Ryffel GU, Hattersley AT, Morgan NG, Ashcroft FM. A Kir6.2 mutation causing neonatal diabetes impairs electrical activity and insulin secretion from INS-1 beta cells. Diabetes. 2006;55:3075–3082. doi: 10.2337/db06-0637. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.