

Non-technical summary
The electrical activity of nerve cells is produced by the flux of ions through specialized membrane proteins called ion channels. Some ion channels can be regulated by the signalling lipid PIP2, a component of the channels’ membrane environment. Here we examine the relevance of PIP2 for the regulation of one specific channel type, termed TASK. Many chemical transmitters in the brain change neural activity by shutting off TASK channels and it has been suggested that this results from reduction of PIP2. By using novel techniques to alter the concentration of PIP2 in living cells, we find that the activity of TASK is independent of PIP2. Besides demonstrating that another signalling mechanism must control the activity of nerve cells via TASK inhibition, we delineate a general approach for clarifying the relevance of PIP2 in many cell types and organs.
Abstract
Abstract
TASK channels are background K+ channels that contribute to the resting conductance in many neurons. A key feature of TASK channels is the reversible inhibition by Gq-coupled receptors, thereby mediating the dynamic regulation of neuronal activity by modulatory transmitters. The mechanism that mediates channel inhibition is not fully understood. While it is clear that activation of Gαq is required, the immediate signal for channel closure remains controversial. Experimental evidence pointed to either phospholipase C (PLC)-mediated depletion of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) as the cause for channel closure or to a direct inhibitory interaction of active Gαq with the channel. Here, we address the role of PI(4,5)P2 for G-protein-coupled receptor (GPCR)-mediated TASK inhibition by using recently developed genetically encoded tools to alter phosphoinositide (PI) concentrations in the living cell. When expressed in CHO cells, TASK-1- and TASK-3-mediated currents were not affected by depletion of plasma membrane PI(4,5)P2 either via the voltage-activated phosphatase Ci-VSP or via chemically triggered recruitment of a PI(4,5)P2-5′-phosphatase. Depletion of both PI(4,5)P2 and PI(4)P via membrane recruitment of a novel engineered dual-specificity phosphatase also did not inhibit TASK currents. In contrast, each of these methods produced robust inhibition of the bona fide PI(4,5)P2-dependent channel KCNQ4. Efficient depletion of PI(4,5)P2 and PI(4)P was further confirmed with a fluorescent phosphoinositide sensor. Moreover, TASK channels recovered normally from inhibition by co-expressed muscarinic M1 receptors when resynthesis of PI(4,5)P2 was prevented by depletion of cellular ATP. These results demonstrate that TASK channel activity is independent of phosphoinositide concentrations within the physiological range. Consequently, Gq-mediated inhibition of TASK channels is not mediated by depletion of PI(4,5)P2.
Introduction
TWIK-related acid sensitive potassium channels (TASK-1 and TASK-3) are members of the two-pore-domain potassium channel (K2P) family (Duprat et al. 1997; Rajan et al. 2000). They are constitutively open K+-selective ‘background’ channels that dominate the resting or ‘leak’ K+ conductance in many cells, thereby setting membrane potential and basal electrical properties (reviewed in Enyedi & Czirjak, 2010). TASK channels are broadly expressed in diverse neuronal populations throughout the central nervous system (Talley et al. 2001), but also in many peripheral tissues, e.g. adrenal cortex (Czirjak et al. 2000) and heart (Putzke et al. 2007).
Both TASK-1 and TASK-3 channels are potently inhibited by receptors that signal through the Gq/11 subgroup of G-proteins, including muscarinic acetylcholine receptors, metabotropic glutamate receptors and angiotensin receptors (Enyedi & Czirjak, 2010). This inhibition is rapid and reversible. It has been observed in various native cell types and is readily reconstituted in heterologous expression systems upon co-expression of recombinant TASK with Gq-coupled receptors (e.g. Czirjak et al. 2000; Millar et al. 2000; Chemin et al. 2003; Chen et al. 2006). As TASK channels are open at resting membrane potential, their inhibition generally results in depolarization and increased excitability. A well-studied example is the cerebellar granule neuron, where TASK channels determine membrane potential and enable fast action potential firing (Millar et al. 2000; Brickley et al. 2007). Activation of Gq-coupled muscarinic m3 acetylcholine receptors and group I metabotropic glutamate receptors inhibit the TASK-mediated conductance (Boyd et al. 2000; Chemin et al. 2003), consequently changing the firing behaviour of the granule cell (Watkins & Mathie, 1996). In adrenal zona glomerulosa cells, secretion of aldosterone is promoted by the depolarization that results from inhibition of TASK-3 channels by angiotensin II via Gq-coupled AT1 receptors (Czirjak et al. 2000; Enyedi & Czirjak, 2010).
The molecular mechanism that leads to TASK channel closure remains elusive (reviewed in Mathie, 2007; Enyedi & Czirjak, 2010). While there is consensus that activation of Gαq/11 is required (Chen et al. 2006), two alternative Gq-dependent mechanisms have been proposed to mediate channel inhibition.
First, channel closure may result from a direct interaction of activated Gαq with the channel protein. This mechanism is supported, among other observations, by inhibition of TASK by active Gαq even in a cell-free (excised patch) system and by co-immunoprecipitation of Gαq with the channel protein (Chen et al. 2006). However, this direct interaction awaits confirmation by independent methods and the putative molecular interaction domains have not yet been identified.
Alternatively, TASK inhibition may result from depletion of PI(4,5)P2 by PLC activated downstream of Gαq. Evidence for this model includes an activating effect of PI(4,5)P2 applied to excised patches containing TASK channels and channel inhibition by scavengers of polyanionic lipids (Chemin et al. 2003; Lopes et al. 2005). Regulation of TASK channels by PI(4,5)P2 is an attractive model, as activity of many ion channels strictly depends on PI(4,5)P2 as a cofactor. In fact, some channel types have been shown convincingly to be controlled by PI(4,5)P2 dynamics (Suh & Hille, 2008). Specifically, Gq-mediated inhibition of KCNQ (Kv7) channels, which closely resembles inhibition of TASK by the same receptors, is directly mediated by depletion of PI(4,5)P2 (Suh & Hille, 2002; Zhang et al. 2003; Suh et al. 2006).
One pivotal experimental strategy to distinguish between both mechanisms has been the use of the PLC inhibitor U73122, because PLC acts downstream of Gαq but upstream of PI(4,5)P2 concentration changes (Horowitz et al. 2005). However, while occlusion of receptor-mediated TASK channel inhibition by U73122 was found in some studies (Czirjak et al. 2001; Chemin et al. 2003), U73122 was ineffective according to other reports (Boyd et al. 2000; Chen et al. 2006). Thus, the involvement of PLC is contentious.
In any case, if TASK inhibition results from PLC-induced depletion of PI(4,5)P2, binding of this phospholipid must be essential for maintaining channel activity and, importantly, the channel's affinity for PI(4,5)P2 must be low enough to warrant disruption of the interaction by reductions in the PI(4,5)P2 concentration that occur under physiological conditions, i.e. receptor-induced activation of PLC. Following this reasoning, we here employ an arsenal of newly developed methods to alter the concentrations of endogenous PI(4,5)P2 and related phosphoinositides in the living cell while monitoring TASK channel activity. These genetically encoded tools allowed for the acute depletion of PI(4,5)P2 without activating any of the multiple other signals generated by the Gq-mediated pathway. Neither depletion of PI(4,5)P2 and PI(4)P by various methods nor blocking resynthesis of PI(4,5)P2 after receptor-induced depletion by PLC interfered with TASK channel activity. In contrast, all of these manoeuvres reliably down-regulated the bona fide PI(4,5)P2-dependent channel, KCNQ4. Our results thus provide clear evidence that TASK channel activity does not require PI(4,5)P2. Consequently, inhibition by GPCRs is not mediated by depletion of phosphoinositides but by another signal within the complex Gq signalling cascade.
Methods
Molecular biology and heterologous expression
The following expression vectors were used: TASK-1 (NM_002246) and TASK-3 (NM_016601) both in pcDNA3.1; KCNQ4 (NM_004700.2) in pEGFP-C1 (Clontech Laboratories, Mountain View, CA, USA); PHPLCδ1-GFP (P51178; pEGFP-N1) or PHPLCδ1-YFP (pcDNA3); Ci-VSP in pRFP-C1 (NP_001028998.1); human m1R (NM_000738.2) in pSGHV0; Lyn11-FRB in pC4RHE; and CF-Inp in pCFP-N1 (Suh et al. 2006). Duplicate PH domains from Osh2p (NM_001180078, residues 256–424) were fused in tandem at XhoI/EcoRI and HindIII/KpnI sites in pEGFP-C1 (Clontech) with a short VNSKL linker, as previously described (Balla et al. 2008).
Construction of pseudojanin and its mutants were based around the mRFP-FKBP12 constructs previously reported (Varnai et al. 2006). The constructs were built using the pEGFP-C1 plasmid backbone (Clontech), with mRFP replacing the EGFP and FKBP12 fused to the 3′ end of the mRFP with a flexible linker (SGLRSRSAAAGAGGAARAALG). Next, the cytosolic catalytic fragment of S. cerevisiae Sac1p (NM_001179777, residues 2–517) was fused with a flexible linker (SAGGSAGGSAGGSAGGSAGGPRAQASRSLDA). Finally, after a third flexible linker (GGTARGAAAGAGGAGR) residues 214–644 of the human INPP5E 5-phosphatase (NM_019892) were inserted carrying a C641A mutation to remove the prenylation site. Inactivating mutations were then inserted into these phosphatase domains by site-directed mutagenesis, and the resulting mutant named for the activity they retain. Therefore, PJ-Sac carries an inactivating point mutation of the conserved DRVL motif of INPP5E (equivalent to D556A in the full length INPP5E), and thus only Sac is active. Conversely, PJ-5ptase carries an inactivating mutation in the conserved CX5R(T/S) motif of the Sac domain (equivalent to C392S in full-length Sac1p), thus only INPP5E 5-phosphatase domain is active. PJ-Dead carries both of these mutations, and so has no phosphatase activity.
Chinese hamster ovary (CHO) cells were plated onto glass-bottom dishes (WillCo Wells B. V., Amsterdam, the Netherlands) for total internal reflection fluorescence (TIRF) imaging or onto glass coverslips for electrophysiology and confocal microscopy. Cells were transfected with jetPEI transfection reagent (Polyplus Transfection, Illkirch, France) according to the manufacturer's instructions. For co-expression of ion channels with Ci-VSP-RFP, cells were selected for clear membrane localization of RFP. For co-expression experiments involving the rapamycin system (3 plasmids), cells were selected for robust cyan fluorescent protein (CFP) or RFP fluorescence, tagging the translocatable enzymes Inp54p or PJ, respectively. Only those cells were included in the analysis that showed translocation of CFP or RFP, respectively, to the plasma membrane upon application of rapamycin. This behaviour indicates successful expression of both the membrane anchor (Lyn11-FRB) and the translocatable fluorescence-tagged enzyme. The presence of PLCd1-PH-GFP and Osh2x2-PH-GFP for TIRF measurements was evidenced by the GFP fluorescence. Successful co-expression of channels was directly confirmed by the respective currents.
Electrophysiology
Whole-cell recordings were performed with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) in voltage-clamp mode. Data were sampled with an ITC-16 interface (Instrutech, HEKA, Lambrecht/Pfalz, Germany) controlled by PatchMaster software (HEKA). For experiments combining patch-clamp and fluorescence imaging, an EPC10 amplifier (HEKA) was used. Currents were low-pass filtered at 2 kHz and sampled at 5 kHz. Patch pipettes were pulled from borosilicate or quartz glass and had open pipette resistances of 1 to 3 MΩ when filled with pipette solution. Intracellular (pipette) solution contained (mm): 135 KCl, 2.41 CaCl2, 3.5 MgCl2, 5 Hepes, 5 EGTA, 2.5 Na2ATP and 0.1 Na2GTP, pH 7.3 (adjusted with KOH). Free [Ca2+] of this solution was 100 nm, as calculated with MaxChelator (maxchelator.stanford.edu). Series resistance was <10 MΩ and was not compensated. The extracellular solution was composed as follows (mm): 144 NaCl, 5.8 KCl, 0.7 NaH2PO4, 5.6 glucose, 1.3 CaCl2, 0.9 MgCl2 and 10 Hepes, pH 7.4 (adjusted with NaOH). Experiments were performed at room temperature (∼22°C).
Fluorescence imaging
TIRF imaging was performed as described elsewhere (Halaszovich et al. 2009). In brief, a BX51WI upright microscope (Olympus, Hamburg, Germany) equipped with a TIRF condenser (numerical aperture of 1.45; Olympus) and a 488 nm laser (20 mW; Picarro, Sunnyvale, CA, USA) was used. Fluorescence was imaged through a LUMPlanFI/IR 40×/0.8 NA water immersion objective (Olympus). Images were acquired with an IMAGO-QE cooled CCD camera (TILL Photonics GmbH, Gräfelfing, Germany). Wide-field fluorescence illumination was achieved with a monochromator (Polychrome IV, TILL Photonics GmbH) coupled to the BX51WI microscope through fibre optics. Green fluorescent protein (GFP) fluorescence was excited at 488 nm. The laser shutter for TIRF illumination, the monochromatic light source and image acquisition were controlled by TILLvisION software (TILL Photonics GmbH).
Confocal imaging of membrane translocation of the CF-Inp and RF–PJ constructs was performed with an upright LSM 710 Axio Examiner.Z1 microscope equipped with a W-Plan/Apochromat 20×/1.0 DIC M27 75 mm water immersion objective (Carl Zeiss GmbH, Jena, Germany). RFP was excited at 561 nm with a diode-pumped solid-state laser (Carl Zeiss), and fluorescence emission was sampled at 582–754 nm. CFP was excited at 458 nm with an argon laser (Carl Zeiss) and emission was sampled at 454–581 nm.
Chemicals
Rapamycin was purchased as a solution in Me2SO (from Merck KGaA, Darmstadt, Germany) and further diluted in extracellular solution to a concentration of 5 μm before use. 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone (XE991; Tocris, Ellisville, USA) and oxotremorine-M (Oxo-M; Tocris) were prepared as 10 mm stock solutions in H2O, and diluted with extracellular solution to their final concentration of 10 μm. Chemicals were delivered locally to the cells with a gravity-fed, custom-built application system.
Data analysis
Electrophysiological data were analysed using IgorPro (Wavemetrics, Lake Oswego, OR, USA) and imaging data were analysed with TILLvisION and IgorPro. Regions of interest (ROI) were defined to include the majority of a single cell's footprint. To avoid movement artifacts the margins of the cell were excluded from analysis. Normalized F/F0 traces were calculated from the TIRF signal intensity F, averaged from the ROI, and the initial fluorescence intensity F0 obtained before application of agonists or depolarization of the cell. Where appropriate, traces were corrected for bleaching according to a monoexponential fit to the signal decay during the pre-application interval. Fluorescence intensities were background-corrected. Time constants were obtained from monoexponential fits. All data are given as mean ± standard error of the mean (SEM), with n indicating the number of individual cells analysed. Statistical analysis was performed by ANOVA test followed by a two-tailed Dunett t test. Significance was assigned for P < 0.05.
Results
Inhibition of TASK channels by Gq-coupled muscarinic receptor
When TASK-1 or TASK-3 channels were co-expressed with the Gq-coupled muscarinic m1 acetylcholine receptor (m1R) in CHO cells, application of the muscarinic agonist Oxo-M resulted in rapid and nearly complete inhibition of TASK-mediated currents (Fig. 1A and B). After washout of Oxo-M, the current recovered to about 70% (69.1 ± 7.0% for TASK-1 and 70.3 ± 3.0% for TASK-3) within 4 min.
Figure 1. Receptor-mediated inhibition of TASK channels and concomitant depletion of PI(4,5)P2.
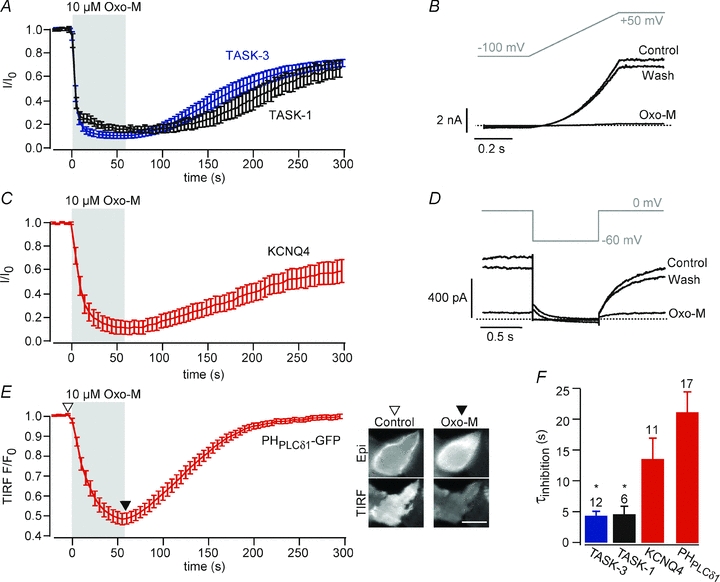
A, averaged time course of TASK currents upon activation of muscarinic m1 receptors. Data were obtained from CHO cells co-transfected with m1R and either TASK-1 (n = 6) or TASK-3 (n = 12). Application of the muscarinic agonist oxo-M (10 μm, 60 s) inhibited currents to 17.0 ± 2.4% for TASK-1 and 9.7 ± 2.4% for TASK-3. Cells were clamped at –60 mV and currents were measured in response to voltage ramps every 2 s. Traces show currents at +50 mV, normalized to current amplitude prior to application for each cell before averaging. B, representative recordings for the experiments summarized in A. Current traces in response to the voltage ramp shown were obtained from a cell co-expressing m1R and TASK-3. Dashed line indicates zero current level. C, muscarinic inhibition of KCNQ4 channels measured from CHO cells co-transfected with m1R and KCNQ4 (n = 11). Currents obtained at 0 mV using the voltage step protocol shown in D at 5 s intervals (holding potential, 0 mV) were normalized to the current amplitude prior to application of Oxo-M for each cell before averaging. D, representative recordings and voltage protocol for the experiments summarized in C. E, TIRF recordings from CHO cells expressing m1R and PHPLCδ1-GFP (n = 17 cells from 3 independent experiments). Application of 10 μm Oxo-M triggered a strong and reversible decrease in TIRF, indicative of translocation of the GFP-fused sensor from the membrane to the cytosol. Insets show epifluorescence (upper) and TIRF image (lower panel) of a representative cell before and after application of Oxo-M. Scale bar, 10 μm. F, average time constants of the onset of muscarinic current inhibition and of translocation of PHPLCδ1-GFP. Time constants were obtained from monoexponential fits to the data shown in (A–E). Asterisks indicate significantly faster time constants of TASK-1 and TASK-3 compared to KCNQ4 (P≤ 0.05).
It is well established that Gq-triggered activation of PLCß can result in a pronounced decrease of the PI(4,5)P2 concentration in the plasma membrane (e.g. Horowitz et al. 2005) Receptor-mediated PI(4,5)P2 dynamics have been implicated in the regulation of numerous ion channels (Suh & Hille, 2008). Thus, it appeared that channel deactivation due to loss of PI(4,5)P2 might be an attractive and straightforward mechanism explaining the inhibition of TASK currents by the Gq-coupled receptors, as suggested previously (Chemin et al. 2003; Lopes et al. 2005).
We tested for the putative PI(4,5)P2 depletion in CHO cells using two distinct PI(4,5)P2 biosensors, the GFP-fused pleckstrin homology (PH) domain of PLCd1 (PHPLCδ1-GFP (Stauffer et al. 1998) as an optical probe for PI(4,5)P2, and the voltage-gated potassium channel KCNQ4 (Kv7.4). PHPLCδ1-GFP specifically binds to PI(4,5)P2 and has been used widely as a genetically encoded ‘translocation biosensor’ for PI(4,5)P2. The degree of its membrane association directly reports on the concentration of PI(4,5)P2 (Varnai & Balla, 2006). Here, we used live-cell TIRF microscopy to monitor membrane association of PHPLCδ1-GFP (Halaszovich et al. 2009). In cells co-expressing m1R and PHPLCδ1-GFP, TIRF fluorescence strongly decreased during activation of m1R, indicating dissociation of the sensor from the membrane, and thus robust depletion of PI(4,5)P2 (Fig. 1E). Upon washout of the receptor agonist, the TIRF signal recovered, demonstrating reversibility of PI(4,5)P2 depletion due to resynthesis of PI(4,5)P2. It has been shown unequivocally that inhibition of KCNQ (Kv7) potassium channels by many Gq-coupled receptors results from Gq/PLC-mediated depletion of PI(4,5)P2 (Suh & Hille, 2002; Zhang et al. 2003; Suh et al. 2006; Hernandez et al. 2009). In our experimental setting, homomeric KCNQ4 channels were also strongly inhibited by activation of m1R (Fig. 1C and D), confirming depletion of PI(4,5)P2. Characteristics of inhibition resembled the inhibition observed with TASK channels: KCNQ4-mediated currents decreased by 88.0 ± 5.5% within a few seconds and recovered with a time course that matched recovery of TASK-mediated currents (Fig. 1C). However, the onset of inhibition of TASK-1 and TASK-3 occurred consistently faster than inhibition of KCNQ4 and PHPLCδ1-GFP dissociation (Fig. 1E).
TASK channel activity is resistant to depletion of PI(4,5)P2
To scrutinize the suggested role of PI(4,5)P2 concentration changes in receptor-mediated TASK channel inhibition, we used different molecular tools for the selective depletion of phosphoinositides in the intact cell.
First, we used the voltage-sensitive phosphoinositide phosphatase Ci-VSP (Murata et al. 2005; Murata & Okamura, 2007), a membrane-resident PI(4,5)P2-5-phosphatase, that is rapidly and reversibly activated by strong depolarization of the membrane potential. Upon activation, Ci-VSP efficiently removes PI(4,5)P2 from the plasma membrane by dephosphorylation to PI(4)P (Halaszovich et al. 2009; Falkenburger et al. 2010). As shown in Fig. 2A, TASK channels were completely insensitive to the activation of co-expressed Ci-VSP. After 50 s of depolarization to +80 mV, TASK-1 and TASK-3 current amplitudes were 102.0 ± 3.2% and 107.9 ± 5.8%, respectively, of current amplitude before activation of Ci-VSP. In contrast, KCNQ4 currents were strongly inhibited by the same voltage protocol when co-expressed with Ci-VSP (residual current, 32.3 ± 5.8%; Fig. 2A). This indicated efficient depletion of PI(4,5)P2 by Ci-VSP, which was further confirmed by strong translocation of the fluorescent PI(4,5)P2 probe PHPLCδ1-GFP upon depolarization (Fig. 2D).
Figure 2. TASK currents are insensitive to depletion of PI(4,5)P2 by Ci-VSP.
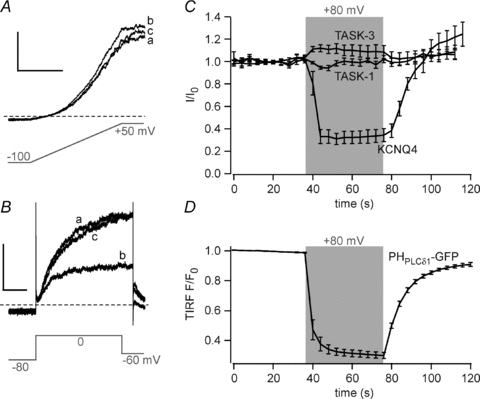
A, representative whole-cell currents from CHO cells co-transfected with TASK-3 and Ci-VSP. Currents were recorded in response to brief voltage ramps from a holding potential of –80mV (a). Holding potential was then depolarised to +80 mV for 40 s to activate Ci-VSP (b), followed by repolarisation to –80 mV (c). Scale bars, 0.5 nA and 100 ms. B, currents from cells co-transfected with KCNQ4 and Ci-VSP obtained with the same holding potential protocol as in A. Currents were evoked by brief intermittent voltage steps (–80 to 0 mV) as indicated. Scale bars, 0.1 nA and 200 ms. C, mean time course of currents obtained from experiments as in A and B from cells co-transfected with Ci-VSP and either TASK-1, TASK-3 or KCNQ4. The shaded area indicates depolarisation to +80 mV from an initial holding potential of –80 mV. Depolarisation induced rapid inhibition of KCNQ4 currents (to 34.1 ± 5.8% of initial current at the end of the depolarizing period; n = 12) but not of TASK-1 (102.0 ± 3.2%, n = 6) and TASK-3 (107.9 ± 5.8%, n = 7). Monoexponential fits to the recovery of KCNQ4 currents yielded time constants of 14.6 ± 2.2 s. D, mean TIRF intensities from voltage-clamped cells expressing Ci-VSP and PHPLCδ1-GFP obtained as in Fig. 1E (n = 12). Depolarization as in C induced fast and reversible translocation of the PI(4,5)P2 probe. Monoexponential fits to the re-association of the PH domain yielded time constants of 10.2 ± 0.8 s.
We next applied an independent approach that was developed recently to acutely modify PI(4,5)P2 concentrations (Suh et al. 2006; Varnai et al. 2006). Here, PI(4,5)P2 is depleted by chemically triggered recruitment of a yeast polyphosphoinositide-5-phosphatase, Inp54p, that specifically dephosphorylates PI(4,5)P2 at the 5′ position. This method is based on the hetero-dimerization of the FRB (FKBP/rapamycin binding) domain from mTOR and the FKBP (FK506 binding protein) in the presence of rapamycin. FRB is anchored to the plasma membrane by fusion to a membrane targeting motif from Lyn (Lyn11-FRB) and Inp54p is fused to CFP-tagged FKBP (CF-Inp) (Suh et al. 2006). Consequently dimerization induced by the addition of rapamycin results in massive and rapid translocation of Inp54p to the membrane and thus consumption of PI(4,5)P2. Recruitment can be monitored conveniently as the translocation of CFP fluorescence from the cytosol to the membrane (Fig. 3A, inset).
Figure 3. PI(4,5)P2 depletion by chemically triggered recruitment of Inp54p fails to suppress TASK currents.
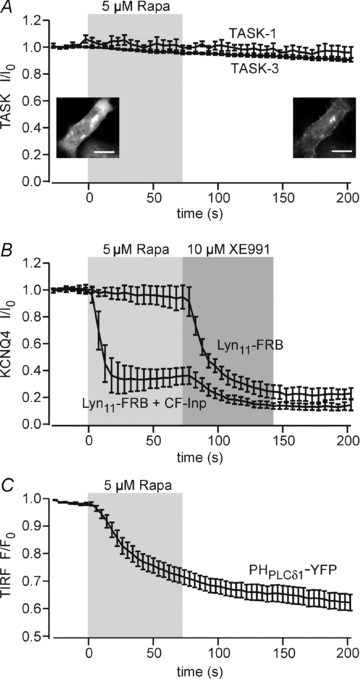
A, normalized current amplitudes measured from cells co-expressing CF-Inp, Lyn11-FRB and either TASK-1 or TASK-3. Currents were unaffected by application of rapamycin. Current amplitudes 2 min after application of rapamycin were 89.9 ± 4.1% (n = 5) and 89.6 ± 2.5% (n = 6) for TASK-1 and TASK-3, respectively. Translocation of CF-Inp to the membrane was verified for each experiment as shown in the representative confocal images before (left) and after (right) application of rapamycin. Scale bar, 10 μm. B, KCNQ4-mediated current in cells co-expressing CF-Inp and Lyn11-FRB was robustly suppressed upon application of rapamycin (n = 5). When the translocatable phosphatase was omitted (Lyn11-FRB only), currents were not affected by rapamycin, confirming current inhibition by depletion of PI(4,5)P2. Residual currents were blocked by the KCNQ channel inhibitor, XE991. Voltage protocols used were as described in Fig. 1. C, TIRF recordings from cells co-transfected with PHPLCδ1-YFP, CF-Inp and Lyn11-FRB. Application of rapamycin decreased TIRF intensity, indicating dissociation of PHPLCδ1-GFP from the membrane (n = 15 cells from 4 independent experiments).
As shown in Fig. 3A, currents mediated by TASK-1 or TASK-3 were not affected by application of rapamycin to cells co-expressing either channel with Lyn11-FRB and CF-Inp. For all recordings, efficient recruitment of Inp54p was verified by monitoring the translocation of CFP fluorescence to the membrane (insets). As before, we tested for depletion of PI(4,5)P2 by rapamycin-triggered translocation of Inp54p using the sensors KCNQ4 and PHPLCδ1-YFP. KCNQ4-mediated currents were robustly and rapidly suppressed by application of rapamycin to cells co-expressing KCNQ4, Lyn11-FRB and CF-Inp (Fig. 3B). Rapamycin applied to cells transfected with the channel alone failed to affect current amplitude, confirming that channel inhibition resulted from recruitment of Inp54p to the membrane. Additionally, dissociation of PHPLCδ1-YFP from the membrane upon addition of rapamycin (Fig. 3C) demonstrated efficient depletion of PI(4,5)P2 by recruitment of Inp54p.
Taken together, TASK channels remained fully active despite strong decrease in the concentration of membrane PI(4,5)P2 in intact cells.
Combined depletion of PI(4,5)P2 and PI(4)P does not affect TASK activity
Our experimental manipulation of PI(4,5)P2 levels differed from receptor-mediated PI(4,5)P2 depletion. The phosphatases used here stoichiometrically converted PI(4,5)P2 to PI(4)P, probably keeping the overall phosphoinositide concentration in the membrane constant. In contrast, activation of PLC depletes both PI(4,5)P2 and PI(4)P (Willars et al. 1998; Horowitz et al. 2005) without generating a structurally similar lipid. Since these two phosphoinositides constitute the majority of all phosphoinositides in the plasma membrane (Willars et al. 1998; Nasuhoglu et al. 2002), activation of Gq-coupled receptors triggers bulk depletion of phosphoinositides.
If TASK channels are activated by phosphoinositides with no or little specificity, the PI(4)P produced by Ci-VSP and Inp54p might be sufficient to sustain channel activity. We therefore sought to fully deplete both PI(4,5)P2 and PI(4)P to closely recapitulate the changes in phosphoinositide concentrations during Gq/PLC activation, but without activating the various other cellular messengers involved in Gq signalling. We therefore used a modified heterodimerization approach, where an engineered dual-specificity phosphatase, pseudojanin (PJ), replaced Inp54p. As shown schematically in Fig. 4A, pseudojanin was engineered in analogy to the native dual-specificity phosphoinositide phosphatase synaptojanin (Mani et al. 2007) and consists of two functionally distinct phosphoinositide phosphatase domains separated by a linker region. The N-terminus of PJ consists of the yeast 4′-phosphatase Sac while the C-terminus harbours a human 5-phosphatase (INPP5E). To allow for rapamycin-driven membrane recruitment, PJ was fused to FKBP and mRFP (RF–PJ).
Figure 4. TASK-3 currents are insensitive to combined depletion of PI(4,5)P2 and PI(4)P by pseudojanin.
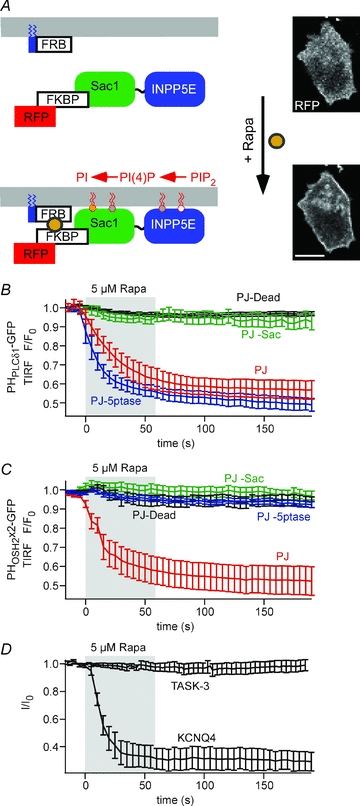
A, schematic representation of the pseudojanin membrane targeting construct (RF–PJ) and its recruitment to the plasma membrane by rapamycin. Confocal images of RF–PJ before (top) and after addition of 5 μm rapamycin (bottom) show efficient translocation of PJ. Scale bar, 10 μm. B, TIRF recordings were performed on cells co-transfected with PHPLCδ1-GFP, Lyn11-FRB, and either of the RF–PJ variants indicated. Dissociation of PHPLCδ1-GFP from the membrane upon application of rapamycin (shaded) was only observed with intact INPP5E 5-phosphatase domain. (n = 9 cells/5 independent experiments, 5/3, 10/4 and 21/8, for PJ, PJ-Sac, PJ-5ptase and PJ-Dead, respectively). C, TIRF recordings from cells co-transfected with PHOSH2×2-GFP, Lyn11-FRB and either of the RF–PJ variants indicated. Dissociation of PHOSH2×2-GFP from the membrane required functional Sac1 and INPP5E phosphatase domains (n = 14 cells /8 independent experiments, 11/5, 15/5 and 14/21, for PJ, PJ-Sac, PJ-5ptase and PJ-Dead, respectively). D, whole-cell voltage-clamp recordings from cells co-transfected with Lyn11-FRB, fully intact PJ and either TASK-3 or KCNQ4. While KCNQ currents were robustly inhibited by recruitment of PJ (residual currents, 30.6 ± 7.8%, n = 6), TASK currents were not affected (residual currents, 97.4 ± 3.9%, n = 7). Voltage protocols as shown in Fig. 1.
In cells co-transfected with RF–PJ and Lyn11-FRB, application of rapamycin triggered efficient recruitment of PJ to the plasma membrane (Fig. 4A). The resulting changes in phosphoinositide concentrations were examined by TIRF imaging of the PI(4,5)P2 sensor, PHPLCδ1-GFP, and a probe for PI(4)P, PHOSH2×2-GFP (Roy & Levine, 2004). The latter is a GFP-fused tandem construct of two identical PH domains from yeast OSH2p and has been shown to bind to the plasma membrane in a PI(4)P-dependent manner (Balla et al. 2008). PHPLCδ1-GFP dissociated from the membrane upon recruitment of PJ (Fig. 4B and C). Dissociation was also observed when the Sac1 domain was inactivated by a point mutation in its catalytic centre, but not when the INPP5E phosphatase or both phophatase domains were inactivated by mutation. Thus, recruitment of PJ leads to depletion of PI(4,5)P2 via its 5′-phoshatase domain. Similarly, recruitment of PJ resulted in dissociation of PHOSH2×2-GFP (Fig. 4C) from the membrane. In contrast to PHPLCδ1, mutational inactivation of either phosphatase domain abolished this dissociation. This indicates that PHOSH2×2 can bind to PI(4,5)P2 or PI(4)P and that the presence of only one of both phosphoinositides is sufficient for membrane localisation of this probe. However, this finding also provides clear evidence for depletion of PI(4)P by PJ recruitment. Taken together, TIRF imaging using both fluorescent probes demonstrates that rapamycin-triggered recruitment of PJ robustly and rapidly depletes both PI(4,5)P2 and PI(4)P.
We went on to examine the effects of phosphoinositide depletion by PJ on TASK channels in cells co-expressing TASK-3 with RF–PJ and Lyn11-FRB. As shown in Fig. 4D, application of rapamycin had no effect on TASK currents (remaining current, 97.4 ± 3.9%), despite successful membrane recruitment of PJ, as confirmed by association of RFP fluorescence with the plasma membrane for each recording. To independently confirm the proper activity of PJ under whole-cell patch-clamp, we also examined the effect of PJ recruitment on KCNQ4 channels. In contrast to TASK, KCNQ4-mediated currents were strongly suppressed to 30.6 ± 7.8% by PJ when subjected to the same experimental protocol (Fig. 4D). In conclusion, combined depletion of the most abundant phosphoinositides in the plasma membrane is insufficient to inhibit TASK channel activity.
Recovery from receptor-induced inhibition does not require resynthesis of PI(4,5)P2
Finally, we probed for involvement of phosphoinositides in TASK channel regulation by interfering with the resynthesis of PI(4,5)P2 after activation of Gq/PLC. If channel inhibition was due to PLC-mediated PI(4,5)P2 depletion, current recovery must depend on replenishment of PI(4,5)P2. Resynthesis occurs via sequential phosphorylation of phosphatidylinositol and PI(4)P and therefore involves ATP hydrolysis (Horowitz et al. 2005). The ATP requirement for recovery has previously been used as a criterion for defining involvement of PI(4,5)P2 in the regulation of KCNQ channels (Suh & Hille, 2002).
We examined the recovery of TASK currents from inhibition by co-expressed m1R. ATP was omitted from the pipette solution and replaced by the non-hydrolysable analogue AMP-PCP, which fully prevents resynthesis of PI(4,5)P2 as reported previously (Suh & Hille, 2002; Halaszovich et al. 2009). As shown in Fig. 5A, TASK3 currents recovered completely after withdrawal of the muscarinic agonist. The time course of recovery was not significantly different from control recordings obtained with 2.5 mm MgATP in the patch pipette (Fig. 5B). In contrast, recovery of KCNQ4 channels from m1R-mediated inhibition was completely abrogated by ATP removal as reported previously for KCNQ2/3 heteromeric channels (Suh & Hille, 2002). This finding confirmed that resynthesis was fully prevented and transient activation of PLC resulted in persistent depletion of PI(4,5)P2.
Figure 5. ATP is not required for recovery of TASK-3 currents.
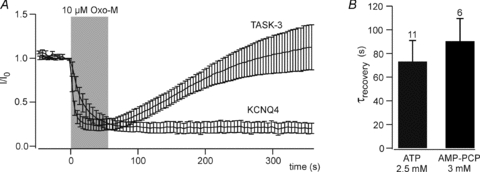
A, recovery of TASK-3 and KCNQ4 currents after removal of intracellular ATP. CHO cells co-expressing either TASK-3 or KCNQ4 with m1R were patch-clamped with a pipette solution containing no ATP and 3 mm AMP-PCP. Recordings were started after 4 min of dialysis with the pipette solution (TASK-3, n = 5; KCNQ4, n = 6). Voltage protocols as shown in Fig. 1. B, average time constants for recovery from muscarinic inhibition of TASK-3 were similar in the presence of 2.5 mm MgATP (data shown in Fig. 1C) or 3 mm AMP-PCP in the patch pipette. Time constants were derived from monoexponential fits to the current recovery for individual cells.
In conclusion, our results demonstrate that TASK channels re-open despite maximal depletion of phosphoinositides by PLC. Consequently, channel activity does not require PI(4,5)P2 or other phosphoinositides, indicating that m1R-mediated current inhibition is not mediated by reduction of the concentrations of PI(4,5)P2 or PI(4)P.
Discussion
PI(4,5)P2 is dispensable for activity of TASK channels
We used various methods to deplete the endogenous PI(4,5)P2 pool in living cells and consistently found no change in currents mediated by TASK-1 and TASK-3. These finding clearly demonstrate that TASK channel activity does not depend on the concentration of this phosphoinositide species in the plasma membrane.
Since this conclusion critically relies on the assumption that Ci-VSP and rapamycin-triggered recruitment of Inp54p efficiently degrade the PI(4,5)P2 content of the membrane, it is worth considering the degree of PI(4,5)P2 depletion achieved with these methods. We used two different sensors to follow the PI(4,5)P2 concentration in our experimental setting: a fluorescent PI(4,5)P2 probe that has been used extensively (PHPLCδ1-GFP; (Varnai & Balla, 2006) and a PI(4,5)P2-dependent ion channel, KCNQ4. Inhibition of KCNQ4 currents upon recruitment of Inp54p and PJ was faster than the dissociation of PLCδ1-PH-GFP (Figs 3 and 4) and current recovery was slower after activation of m1R or Ci-VSP (Figs 1 and 2). This difference may be explained by a lower apparent affinity of KCNQ4 for PI(4,5)P2 when compared to the fluorescent sensor. In fact, less activation of Ci-VSP, and consequently a lower degree of PI(4,5)2 depletion, is needed to fully inhibit KCNQ4 channels than to displace PLCδ1-PH-GFP from the plasma membrane (A. Rjasanow, C. R. Halaszovich and D. Oliver, unpublished results). Notwithstanding, translocation of PHPLCδ1-GFP from the membrane and inhibition of KCNQ4 channels during activation of Ci-VSP and membrane recruitment of the phosphatase quantitatively matched the responses following activation of m1R. Therefore, the degree of PI(4,5)P2 depletion achieved with the genetically encoded tools was similar to the PLC-mediated depletion during receptor stimulation.
In agreement with our observations, recent work showed that channel inhibition by Ci-VSP and rapamycin-triggered phosphatase recruitment is remarkably consistent with the sensitivity to receptor-mediated PLC activation for other PI(4,5)P2-regulated channels including voltage-gated calcium and potassium channels (Falkenburger et al. 2010; Suh et al. 2010). Moreover, even Kir2.1 inward rectifier K+ channels, which are characterized by particularly high affinity for PI(4,5)P2 (Du et al. 2004), are strongly inhibited by Ci-VSP (Murata & Okamura, 2007; A. Rjasanow and D. Oliver, unpublished results). These considerations clearly show that the genetically encoded tools used here are well suited to gauge the sensitivity of channels to PLC-mediated depletion of PI(4,5)P2.
We further note that inhibition of TASK channels by m1R activation occurred consistently faster than inhibition of KCNQ4. The latter channel has a comparatively low affinity to PI(4,5)P2, and is therefore highly sensitive to PLC activation (Hernandez et al. 2009). Thus, if depletion of PI(4,5)P2 was causing loss of TASK channel activity due to a direct PI(4,5)P2–channel interaction, the affinity of TASK channels for PI(4,5)P2 would be predicted to be even lower compared to KCNQ4 and TASK would be highly sensitive to activation of the phosphatases used in our experiments. This was not the case. We therefore conclude that TASK channels are resistant to depletion of PI(4,5)P2 within a physiological range of concentrations.
It was previously shown that application of PI(4,5)P2 to excised membrane patches can enhance TASK-mediated currents (Chemin et al. 2003; Lopes et al. 2005). This is seemingly in contradiction to our results and has been taken to suggest a role of PI(4,5)P2 in the regulation of these channels. However, it should be noted that our results do not necessarily exclude that a reduction of PI(4,5)P2 to levels below those reached under physiological conditions (by PLC or phosphatases) may lead to channel closure. In other words, it remains possible that TASK channels bind PI(4,5)P2 with a very high affinity, whereby PI(4,5)P2 would be a permissive cofactor but not a physiological regulator of TASK (Gamper & Shapiro, 2007). Such a role of PI(4,5)P2 may be consistent with the partial block of TASK-mediated currents by PI(4,5)P2-scavenging polycations such as poly-lysine and neomycin observed previously (Chemin et al. 2003; Lopes et al. 2005). Alternatively, artificially high levels of PI(4,5)P2, as may be reached by application to excised patches, may enhance TASK currents. In any case, our experiments exclude that inhibition of TASK channels by Gq-coupled receptors results from PLC-mediated depletion of PI(4,5)P2.
A role for other phosphoinositides?
Although resistance to Ci-VSP and Inp54p unequivocally show that PI(4,5)P2 depletion is not sufficient for channel down-regulation, it remained possible that other phosphoinositides might be involved in receptor-mediated inhibition of TASK. Specifically, PI(4)P might be relevant since it is present at similar concentrations as PI(4,5)P2 in the resting plasma membrane and is also depleted during receptor-induced activation of PLC (Willars et al. 1998; Horowitz et al. 2005). If TASK channels had a non-selective dependence on phosphoinositides, similar to some inward rectifier K+ (Kir) channels (Rohacs et al. 1999; Tucker & Baukrowitz, 2008), then activation of PI(4,5)P2-5-phosphatases might not suffice for channel deactivation despite strong depletion of PI(4,5)P2. In other words, the differential sensitivity of TASK to PLC versus phosphatases might result from the different changes in the overall concentration of phosphoinositides according to the different enzymatic activities.
To examine such a possible role of combined depletion of PI(4,5)P2 and PI(4)P in receptor-induced TASK inhibition, we used PJ, a novel engineered dual-specificity phosphatase for dephosphorylation of PI(4,5)P2 and PI(4)P. Recruitment of PJ to the membrane efficiently depleted both phosphoinositides as indicated by specific fluorescent sensors. However, the complete insensitivity of TASK to this manoeuvre indicated that neither PI(4,5)P2 nor PI(4)P concentrations contribute to regulation of TASK channels by PLC. This conclusion is further strongly supported by the full reactivation of TASK channels from receptor-induced inhibition observed after substitution of ATP by the non-hydrolysable analogue AMP-PCP. Under this condition, resynthesis of PI(4)P and PI(4,5)P2 is abrogated, resulting in persistent depletion of both phosphoinositides after activation of Gq-coupled receptors.
A TASK-associated pool of PI(4,5)P2 inaccessible to exogenous phosphatases?
It has been suggested that the plasma membrane may harbour functionally distinct pools of PI(4,5)P2 (Wang et al. 2004; Vasudevan et al. 2009). Such pools may correspond to PI(4,5)P2 localized to spatially separated membrane domains, such as lipid rafts or caveolae versus non-raft membranes (Johnson et al. 2008; Cui et al. 2010), although such local domains are difficult to reconcile with the fast diffusion of phosphoinositides as determined in native plasma membranes (Hilgemann, 2007). Experimental findings suggested that localized PI(4,5)P2 pools resulting from restricted diffusion may determine regulation of ion channels by PI(4,5)P2 in cardiomyocytes (Cho et al. 2005). The efficient inhibition of KCNQ2/3 heteromeric channels by Gq-coupled muscarinic receptors in HEK cells required co-localization of channels and receptors to detergent-resistant membrane fractions (Oldfield et al. 2009). In principle, the observed differential action of receptor-activated PLC and recombinant phosphatases could be compatible with PI(4,5)P2-mediated regulation of TASK channels, if the channels interacted with a distinct ‘private’ pool of PI(4,5)P2 that is depleted by endogenous PLC but is inaccessible to the phosphatase constructs used in this study. However, any membrane pool of PI(4,5)P2 must remain exhausted after depletion by PLC when replenishment of PI(4,5)P2 is blocked. Therefore, such a possibility is clearly ruled out by our finding of full current recovery in the absence of intracellular ATP.
In summary, our results obtained upon depletion of PI(4,5)P2 and PI(4)P by exogenous phosphatases and inhibition of their resynthesis after depletion by endogenous PLC rule out a requirement of these phosphoinositides for TASK channel activity. Specifically, we conclude that the inhibition of TASK channels by Gq-coupled receptors is not mediated by depletion of phosphoinositides.
Possible mechanisms for Gq/PCR-mediated TASK channel inhibition
Since depletion of PI(4,5)P2 can be ruled out as the signal that mediates channel closure downstream of Gq/PCR activation, which other mechanisms of channel inhibition remain? In principle, several other intermediates within the Gq-mediated signalling cascade may interact with TASK channels to induce channel closure.
The currently most complete evidence suggests that a direct interaction of activated Gαq protein (Gαq*) can deactivate TASK channels (Chen et al. 2006). This scenario is supported by elegant experiments including the demonstration of channel activation by purified Gαq* in excised patches and co-immunoprecipitation of Gαq* with TASK channels (Chen et al. 2006). However, it should be noted that previous studies demonstrated the suppression of GqPCR-mediated TASK channel inhibition by the PLC inhibitor U73122 (Czirjak et al. 2001; Chemin et al. 2003), suggesting at least a contribution of signals downstream of PLC activation. Among these signals, channel phosphorylation by PKC has been ruled out, since elimination of candidate phosphorylation sites did not impede Gq/PCR-dependent channel inhibition (Talley & Bayliss, 2002; Veale et al. 2007). This conclusion is supported by our current results showing that inhibition is unaffected by removal of the intracellular ATP required for phosphorylation.
Given the high physiological relevance of Gq/PCR-mediated inhibition of TASK-1 and TASK-3, further work is needed to unequivocally identify the mechanism of channel regulation.
Methods for probing the phosphoinositide regulation of ion channels
The combined methods used here provide a toolbox for probing the physiological relevance of phosphoinositides and their concentration dynamics for the regulation of ion channels. Many ion channels have been shown to be affected by PI(4,5)P2 (Suh & Hille, 2008). However, in many cases the proposal of a functional role for PI(4,5)P2 is solely based on the application of exogenous phosphoinositides to excised membrane patches. In addition to TASK channels, examples for the alteration of channel behaviour by exogenous PI(4,5)P2 include TREK channels as additional members within the K2P channel family (Chemin et al. 2005), voltage-gated K+ (Kv) channels (Oliver et al. 2004), various TRP channels (Rohacs & Nilius, 2007) and HCN channels (Zolles et al. 2006), among others. As shown here, such sensitivity to applied phosphoinositides may not always provide strong evidence for a role in the regulation of channels under physiological conditions. Furthermore, pharmacological tools for alterations of phosphoinositide concentrations are poorly developed, and a control for the effectiveness of the intended changes is usually not performed. Thus, genetically encoded tools for the acute manipulation of endogenous phosphoinositide pools combined with fluorescence-based sensors for control of these manipulations should help substantially in obtaining a clearer picture of the genuine roles of PI(4,5)P2 in ion channel regulation. Moreover, these methods are not limited to the analysis of ion channels but may be used to address the role of phosphoinositides in other cellular processes as well.
Acknowledgments
We thank Drs Jürgen Daut and Thomas Baukrowitz for insightful discussions. cDNA constructs used in this study were kindly provided by J. Daut (TASK-1/3), T. Jentsch (KCNQ4), T. Meyer (PHPLCδ1-GFP, rapamycin recruitment system) and Y. Okamura (Ci-VSP). We are indebted to S. Petzold, O. Haeckel and G. Fischer for excellent technical assistance and to D. Schreiber for advice on molecular biological techniques. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 593, TP A12; D.O.)
Glossary
Abbreviations
- CHO
Chinese hamster ovary
- Ci-VSP
Ciona intestinalis voltage-sensitive phosphatase
- GPCR
G-protein-coupled receptor
- m1R
muscarinic m1 acetylcholine receptor
- Oxo-M
oxotremorine-M
- PH
pleckstrin homology
- PI
phosphoinositide
- PI(4)P
phosphatidylinositol-4-phosphate
- PI(4,5)P2
phosphatidyl-inositol-4,5-bisphosphate
- PLC
phospholipase C
- TIRF
total internal reflection fluorescence
Author contributions
M.L. designed and performed most experiments, analysed data and wrote parts of the manuscript. M.G.L. and C.R.H. performed experiments and contributed to the design of the experiments. G.R.V.H. developed pseudojanin, advised on and designed experiments, and revised the manuscript. D.O. conceived the project, designed experiments, analysed data and wrote the manuscript. All authors approved the final version. Experiments were performed at the Philipps University Marburg.
References
- Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, Balla T. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIα. Mol Biol Cell. 2008;19:711–721. doi: 10.1091/mbc.E07-07-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd DF, Millar JA, Watkins CS, Mathie A. The role of Ca2+ stores in the muscarinic inhibition of the K+ current IK(SO) in neonatal rat cerebellar granule cells. J Physiol. 2000;529:321–331. doi: 10.1111/j.1469-7793.2000.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Aller MI, Sandu C, Veale EL, Alder FG, Sambi H, Mathie A, Wisden W. TASK-3 two-pore domain potassium channels enable sustained high-frequency firing in cerebellar granule neurons. J Neurosci. 2007;27:9329–9340. doi: 10.1523/JNEUROSCI.1427-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Girard C, Duprat F, Lesage F, Romey G, Lazdunski M. Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J. 2003;22:5403–5411. doi: 10.1093/emboj/cdg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honore E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 2005;24:44–53. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B, Viana F, Garrison JC, Bayliss DA. Inhibition of a background potassium channel by Gq protein α-subunits. Proc Natl Acad Sci U S A. 2006;103:3422–3427. doi: 10.1073/pnas.0507710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kim YA, Yoon J-Y, Lee D, Kim JH, Lee SH, Ho W-K. Low mobility of phosphatidylinositol 4,5-bisphosphate underlies receptor specificity of Gq-mediated ion channel regulation in atrial myocytes. Proc Natl Acad Sci U S A. 2005;102:15241–15246. doi: 10.1073/pnas.0408851102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Ho WK, Kim ST, Cho H. Agonist-induced localization of Gq-coupled receptors and G protein-gated inwardly rectifying K+ (GIRK) channels to caveolae determines receptor specificity of phosphatidylinositol 4,5-bisphosphate signaling. J Biol Chem. 2010;285:41732–41739. doi: 10.1074/jbc.M110.153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirjak G, Fischer T, Spat A, Lesage F, Enyedi P. TASK (TWIK-related acid-sensitive K+ channel) is expressed in glomerulosa cells of rat adrenal cortex and inhibited by angiotensin II. Mol Endocrinol. 2000;14:863–874. doi: 10.1210/mend.14.6.0466. [DOI] [PubMed] [Google Scholar]
- Czirjak G, Petheo GL, Spat A, Enyedi P. Inhibition of TASK-1 potassium channel by phospholipase C. Am J Physiol Cell Physiol. 2001;281:C700–C708. doi: 10.1152/ajpcell.2001.281.2.C700. [DOI] [PubMed] [Google Scholar]
- Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of Kir channels by diverse modulators. J Biol Chem. 2004;279:37271–37281. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997;16:5464–5471. doi: 10.1093/emboj/16.17.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- Falkenburger BH, Jensen JB, Hille B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol. 2010;135:99–114. doi: 10.1085/jgp.200910345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8:921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- Halaszovich CR, Schreiber DN, Oliver D. Ci-VSP is a depolarization-activated phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate 5′-phosphatase. J Biol Chem. 2009;284:2106–2113. doi: 10.1074/jbc.M803543200. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Falkenburger B, Shapiro MS. Affinity for phosphatidylinositol 4,5-bisphosphate determines muscarinic agonist sensitivity of Kv7 K+ channels. J Gen Physiol. 2009;134:437–448. doi: 10.1085/jgp.200910313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D. Local PIP2 signals: when, where, and how? Pflugers Archiv. 2007;455:55–67. doi: 10.1007/s00424-007-0280-9. [DOI] [PubMed] [Google Scholar]
- Horowitz LF, Hirdes W, Suh B-C, Hilgemann DW, Mackie K, Hille B. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, Chichili GR, Rodgers W. Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J Biol Chem. 2008;283:29920–29928. doi: 10.1074/jbc.M805921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CMB, Rohacs T, Czirjak G, Balla T, Enyedi P, Logothetis DE. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol. 2005;564:117–129. doi: 10.1113/jphysiol.2004.081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, Ryan TA. The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron. 2007;56:1004–1018. doi: 10.1016/j.neuron.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A. Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J Physiol. 2007;578:377–385. doi: 10.1113/jphysiol.2006.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JA, Barratt L, Southan AP, Page KM, Fyffe RE, Robertson B, Mathie A. A functional role for the two-pore domain potassium channel TASK-1 in cerebellar granule neurons. Proc Natl Acad Sci USA. 2000;97:3614–3618. doi: 10.1073/pnas.050012597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- Murata Y, Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J Physiol. 2007;583:875–889. doi: 10.1113/jphysiol.2007.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasuhoglu C, Feng S, Mao J, Yamamoto M, Yin HL, Earnest S, Barylko B, Albanesi JP, Hilgemann DW. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal Biochem. 2002;301:243–254. doi: 10.1006/abio.2001.5489. [DOI] [PubMed] [Google Scholar]
- Oldfield S, Hancock J, Mason A, Hobson SA, Wynick D, Kelly E, Randall AD, Marrion NV. Receptor-mediated suppression of potassium currents requires colocalization within lipid rafts. Mol Pharmacol. 2009;76:1279–1289. doi: 10.1124/mol.109.058008. [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- Putzke C, Wemhoner K, Sachse FB, Rinne S, Schlichthorl G, Li XT, Jae L, Eckhardt I, Wischmeyer E, Wulf H, Preisig-Muller R, Daut J, Decher N. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res. 2007;75:59–68. doi: 10.1016/j.cardiores.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Xin Liu G, Preisig-Muller R, Daut J, Karschin A, Derst C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J Biol Chem. 2000;275:16650–16657. doi: 10.1074/jbc.M000030200. [DOI] [PubMed] [Google Scholar]
- Rohacs T, Chen J, Prestwich GD, Logothetis DE. Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J Biol Chem. 1999;274:36065–36072. doi: 10.1074/jbc.274.51.36065. [DOI] [PubMed] [Google Scholar]
- Rohacs T, Nilius B. Regulation of transient receptor potential (TRP) channels by phosphoinositides. Pflugers Archiv. 2007;455:157–168. doi: 10.1007/s00424-007-0275-6. [DOI] [PubMed] [Google Scholar]
- Roy A, Levine TP. Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. J Biol Chem. 2004;279:44683–44689. doi: 10.1074/jbc.M401583200. [DOI] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B-C, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Leal K, Hille B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–238. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Bayliss DA. Modulation of TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels: volatile anesthetics and neurotransmitters share a molecular site of action. J Biol Chem. 2002;277:17733–17742. doi: 10.1074/jbc.M200502200. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21:7491–7505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Baukrowitz T. How highly charged anionic lipids bind and regulate ion channels. J Gen Physiol. 2008;131:431–438. doi: 10.1085/jgp.200709936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Balla T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta. 2006;1761:957–967. doi: 10.1016/j.bbalip.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Varnai P, Thyagarajan B, Rohacs T, Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol. 2006;175:377–382. doi: 10.1083/jcb.200607116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan L, Jeromin A, Volpicelli-Daley L, De Camilli P, Holowka D, Baird B. The β- and γ-isoforms of type I PIP5K regulate distinct stages of Ca2+ signaling in mast cells. J Cell Sci. 2009;122:2567–2574. doi: 10.1242/jcs.048124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veale EL, Kennard LE, Sutton GL, MacKenzie G, Sandu C, Mathie A. Gαq-mediated regulation of TASK3 two-pore domain potassium channels: the role of protein kinase C. Mol Pharmacol. 2007;71:1666–1675. doi: 10.1124/mol.106.033241. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Li WH, Wang J, Xu K, Dong P, Luo X, Yin HL. Critical role of PIP5KIγ87 in InsP3-mediated Ca2+ signaling. J Cell Biol. 2004;167:1005–1010. doi: 10.1083/jcb.200408008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins CS, Mathie A. A non-inactivating K+ current sensitive to muscarinic receptor activation in rat cultured cerebellar granule neurons. J Physiol. 1996;491:401–412. doi: 10.1113/jphysiol.1996.sp021224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willars GB, Nahorski SR, Challiss RA. Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J Biol Chem. 1998;273:5037–5046. doi: 10.1074/jbc.273.9.5037. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- Zolles G, Klocker N, Wenzel D, Weisser-Thomas J, Fleischmann BK, Roeper J, Fakler B. Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron. 2006;52:1027–1036. doi: 10.1016/j.neuron.2006.12.005. [DOI] [PubMed] [Google Scholar]