

Non-technical summary
The α7 nicotinic acetylcholine receptors (nAChRs) are a therapeutic target for the treatment of neurological disorders such as Alzheimer's disease and schizophrenia, and drugs that potentiate α7 nAChRs through the regulation of desensitization are currently being developed. Here we show that changes to the lipid composition of the plasma membrane in rat hippocampal neurons, through either acute treatment with drugs that remove cholesterol and breakdown sphingomyelin or chronic treatment with synthesis inhibitors for cholesterol and sphingomyelin, result in significant changes in the desensitization of α7 nAChRs. These data provide evidence that the lipid composition of the plasma membrane is able to modulate the desensitization of α7 nAChRs, which will be important for the development of new therapeutic reagents.
Abstract
Abstract
The α7 nicotinic acetylcholine receptors (nAChRs) play an important role in cellular events such as neurotransmitter release, second messenger cascades, cell survival and apoptosis. In addition, they are a therapeutic target for the treatment of neurological disorders such as Alzheimer's disease and schizophrenia, and drugs that potentiate α7 nAChRs through the regulation of desensitization are currently being developed. Recently, these channels were found to be localized into lipid rafts. Here we show that the disruption of lipid rafts in rat primary hippocampal neurons, through cholesterol-scavenging drugs (methyl-β-cyclodextrin) and the enzymatic breakdown of sphingomyelin (sphingomyelinase), results in significant changes in the desensitization kinetics of native and expressed α7 nAChRs. These effects can be prevented by cotreatment with cholesterol and sphingomyelin, and can be mimicked by treatment with cholesterol and sphingomyelin synthesis inhibitors (mevastatin and myriocin, respectively), suggesting that the effects on desensitization kinetics are indeed due to changes in the levels of cholesterol and sphingomyelin in the plasma membrane. These data provide new insights into the mechanism of desensitization of α7 nAChRs by providing evidence that the lipid composition of the plasma membrane can modulate the activity of the α7 nAChRs.
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are Cys-loop ligand-gated ion channels that are widely distributed throughout the nervous system (Jones et al. 1999; Gay et al. 2008), and are pentamers formed by combinations of α2–α9 and β2–β4 subunits. Of these, only the α7–α9 subunits can form homomeric pentamers (Chen & Patrick, 1997). One important characteristic of the α7 nAChRs is their high permeability to Ca2+, allowing this nAChR subunit to play an important role in cellular events such as neurotransmitter release (Sharma & Vijayaraghavan, 2003), second messenger cascades (Sharma & Vijayaraghavan, 2001), cell survival (Shimohama & Kihara, 2001) and apoptosis (Berger et al. 1998).
Studies investigating the localization of α7 nAChRs suggest that this subunit localizes into lipid rafts (Bruses et al. 2001; Oshikawa et al. 2003; Fernandes et al. 2010), which are areas of the cell membrane enriched in cholesterol and sphingolipids. They have been proposed to form microdomains that serve a role as organizational structures in signal transduction, a concept fuelled by observations made following the disruption of lipid rafts with cholesterol-scavenging molecules such as cyclodextrins (Simons et al. 1998; Bruses et al. 2001). Pharmacological disruption of lipid rafts through cholesterol-sequestering drugs results in a change in localization and an increase in mobility of α7 nAChRs in mammalian cell cultures (Oshikawa et al. 2003) and in chick ciliary neurons (Bruses et al. 2001; Fernandes et al. 2010). Some indirect effects of lipid raft disruption on α7 nAChR function have been reported, including the loss of inhibition of the calcium inhibitable isoform adenylate cyclase 6 on PC12 cells (Oshikawa et al. 2003). However, the role that α7 nAChR localization into lipid rafts exerts on channel function remains unknown.
In this study, we investigated the direct effects that lipid rafts and their components have on α7 nAChR function. We found that removal of the main components of lipid rafts (cholesterol and sphingomyelin) by acute treatment with cholesterol-sequestering agents, alone or in combination with the enzymatic breakdown of sphingomyelin, has profound effects by slowing the kinetics of desensitization (in part by increasing the rate of recovery from desensitization) and by increasing agonist affinity. Adding back cholesterol and sphingomyelin reversed the effects on desensitization. These effects are similar to those seen by the inhibition of de novo synthesis of both cholesterol and sphingomyelin, suggesting that the disruption of lipid rafts is responsible for the changes in α7 nAChR desensitization. The mechanism of desensitization of α7 nAChRs is not completely understood; however, therapeutic drugs that potentiate α7 responses through the removal of desensitization [type II α7 positive allosteric modulators (PAMs), such as PNU-120596; Bertrand & Gopalakrishnan, 2007] are currently being developed to treat Alzheimer's disease and other neurological disorders (Young et al. 2008), highlighting the importance of understanding better the function of the α7 nAChRs.
Methods
Hippocampal neuron primary cultures
Primary cultures were prepared at embryonic day 18 using a modified version from Quitsch (2005). All procedures were approved and performed in compliance with NIEHS/NIH Humane Care and Use of Animals in Research protocols. Time pregnant Sprague Dawley rats were anestesized with isofluorane and decapitated by guillotine. Briefly, hippocampi were mechanically dissociated after treatment for 30 min at 37°C with 0.05% trypsin EDTA (Gibco, Grand Island, NY, USA). Cells were plated in neurobasal medium (Gibco) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and 1% Glutamax (Gibco) at 3500 mm−2 on coverslips coated with poly-d-lysine (Sigma, St Louis, MO, USA) and grown in a humidified atmosphere of air containing 5% CO2 at 37°C. Half the medium was replaced 24 h after plating and every 72 h thereafter with neurobasal media in which fetal bovine serum was substituted with 2% B27 (Gibco). Hippocampal neurons were transfected in Day in vitro 4 (DIV4) with α7 nAChR, Ric-3 (resistance to inhibitors of cholinesterase) and green fluorescent protein (GFP), or wild-type rat α3 and β2 nAChR subunits and GFP, using a calcium phosphate procedure described by Pottorf et al. (2006). Cells were analysed 24–48 h after transfection Day in vitro 5-6 (DIV5-6), to minimize the possible contribution of endogenous channels which express at very low levels during this period (Arnaiz-Cot et al. 2008).
Manipulation of membrane lipids
For the acute manipulation of cholesterol and sphingomyelin, hippocampal neurons were treated for 15 min at 37°C with one of the following: PBS (control); 5 mm methyl-β-cyclodextrin (MβCD; Sigma), prepared in PBS and sterile filtered; 5 mm MβCD in combination with 0.5 U ml−1 sphingomyelinase (SMase; Sigma); 5 mm MβCD in combination with 2 mm water-soluble cholesterol (Sigma); or 5 mm MβCD in combination with 0.5 U ml−1 SMase, 2 mm cholesterol and 300 μm sphingomyelin (Sigma). Chronic manipulation of cholesterol and sphingomyelin were done using mevastatin (Sigma) and myriocin (Sigma), which inhibit the synthesis of cholesterol and sphingomyelin, respectively. Hippocampal neurons were incubated for 24–48 h at 37°C with 400 μm myriocin and 2 mm mevastatin in serum-free media. After treatment, cells were immediately processed for cholesterol content or ACh-evoked current measurement.
Measurement of ACh-evoked currents
Coverslips containing transfected neurons were transferred to a chamber containing: 165 mm NaCl, 5 mm KCl, 2 mm CaCl2, 10 mm glucose, 5 mm Hepes, 1 μm atropine and 0.3 μm TTX; pH was adjusted to 7.3 with NaOH. Bath solution was perfused continuously through the chamber (1 ml volume) at 2 ml min−1 throughout the experiments. Neurons were visualized using a Nikon Eclipse TE300 microscope equipped for fluorescence detection. Borosilicate patch pipettes (3–6 MΩ) were filled with 120 mm CsCl, 2 mm MgCl2, 10 mm EGTA, 10 mm Hepes and ATP-regenerating compounds (5 mm ATP and 20 mm phosphocreatine); pH was adjusted to 7.3 with CsOH. Currents were recorded at −70 mV. Experiments were performed at room temperature (22°C). Whole-cell recordings were done using an Axopatch-200A amplifier connected to a Digidata 1322A and software (pCLAMP v. 10.1) from Axon Instruments. Currents were filtered at 1 kHz and digitized at 10 kHz with an output gain of one. Acetylcholine was applied using a synthetic quartz perfusion tube (0.7 mm i.d.) operated by a computer-controlled valve (AutoMate Scientific, Berkeley, CA, USA). The rate of solution exposure in these conditions was estimated to occur as fast as 1.5 ms. One second applications of 3 mm ACh were analysed using Clampfit 10. Data are plotted as means ± SEM and was analysed using Prism 5 (GraphPad, La Jolla, CA, USA).
Dose–response measurements were done by applying different concentrations of ACh for 1 s followed by a 60 s wash. The concentrations used were 30, 100 and 300 μm, 1 and 3 mm ACh. To construct the dose–response curve, the responses were first normalized to the response of 1 mm ACh (responses with 3 mm were indistinguishable from 1 mm) and fitted using a log (agonist) versus normalized response with a variable slope equation on GraphPad Prism 5.
Recovery from desensitization was studied by applying 3 mm ACh for 10 s to induce near-complete (>90%) desensitization, and washing out ACh for various amounts of time (1.5, 2.5, 3.5, 5 and 10 s in control conditions; 1.5, 2, 2.5, 3 and 6 s for MβCD/SMase) followed by a 2 s application of ACh. The data are represented as a percentage of the initial response and were fitted using a one-phase decay exponential equation in GraphPad Prism 5.
Single-channel currents from outside-out patches were measured using the system described above, with the difference that currents were filtered at 2 kHz and digitized at 10 kHz, with an output gain of 20. Four second applications of 50 μm ACh were analysed using Clampfit 10. Estimation of the single-channel amplitudes was done by using all-points histograms that were fitted with Gaussian distributions. Data are plotted as means ± SEM and were analysed using GraphPad Prism 5.
Quantification of cholesterol levels
Hippocampal neurons were grown in poly-d-lysine-coated six-well plates at a density of 4 × 106 cells per well. Cells were treated with the corresponding drugs and washed with PBS and scraped off the wells on Day in vitro 7 (DIV7) using 2% Triton X-100 (Sigma) in PBS. Cells were homogenized with a glass homogenizer and stored at −20°C until needed. Cholesterol levels were measured using the Amplex Red Cholesterol Assay Kit as described by the manufacturer (Molecular Probes, Eugene, OR, USA). Cells were analysed for protein concentration in parallel using the Bicinchoninic acid assay (BCA) protein assay (Pierce, Rockford, IL, USA) and then cholesterol concentration was normalized to protein concentration first, and then to cholesterol concentration in the control samples. Cholesterol data are plotted as means ± SEM and were analysed using GraphPad Prism 5.
Results
Disruption of lipid rafts affects the desensitization kinetics and acetylcholine sensitivity of neuronal α7 nAChRs
Cultured hippocampal neurons coexpressing wild-type rat α7 nAChR, Ric-3 and GFP, were exposed to a 1 s rapid application of ACh (3 mm) at a holding potential of –70 mV; this resulted in the rapid activation of an inward current that desensitized in the continued presence of agonist with a desensitization half-time of 27 ± 2 ms (Fig. 1A and D; Gay et al. 2008). These responses were completely blocked by the α7 nAChR-selective antagonist Methyllycaconitine (MLA) (10 nm, n = 5), indicating that they were due to the activation of the α7 nAChR.
Figure 1. Disruption of lipid rafts affects the desensitization kinetics and acetylcholine sensitivity of neuronal α7 nAChRs.
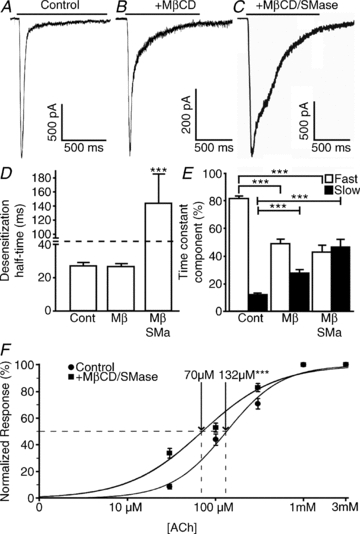
Cultured hippocampal neurons coexpressing wild-type rat α7 nAChR, Ric-3 and GFP were incubated for 15 min with PBS (control), 5 mm MβCD or 5 mm MβCD in combination with 0.5 U ml−1 SMase and subjected to a 1 s application of 3 mm ACh. A–C, response to ACh application in control conditions (A), 5 mm MβCD (B) and 5 mm MβCD and 0.5 U ml−1 SMase (C). D, MβCD treatment did not change the desensitization half-time; however, cotreatment of MβCD with SMase resulted in a fivefold increase in the desensitization half-time. E, desensitization time constants (τ) were calculated using Clampfit and fitted with a two-exponential equation. Although no difference was seen in τ values, the ratio of fast and slow desensitization was affected by both MβCD and MβCD/SMase cotreatment. F, neurons were subjected to consecutive 1 s applications of 30, 100 and 300 μm, 1 and 3 mm ACh. Responses were normalized to the response to 1 mm ACh. Dose–response curves were constructed and compared using GraphPad Prism 5. In D and E, plots show means ± SEM and were subjected to a one-way ANOVA; ***P < 0.0001. In F, log EC50 was compared using a log (agonist) versus normalized response with variable slope fit in GraphPad Prism 5; ***P < 0.0001.
The onset of desensitization, as previously described (Khiroug et al. 2002), was biphasic; the fast component (τfast = 18 ± 1 ms) comprised 81 ± 2%, while the slow component (τslow = 382 ± 41 ms) comprised12 ± 1% of the total desensitization (Fig. 1E; n = 30 cells).
As cholesterol can be either absorbed by receptor-mediated endocytosis or synthesized in the endoplasmic reticulum, we designed two approaches to disrupt lipid rafts. The first approach consisted of the acute removal of cholesterol from these microdomains through a cholesterol-sequestering agent and the enzymatic breakdown of sphingomyelin. Methyl-β-cyclodextrin has been shown to remove cholesterol from the plasma membrane of cultured cells (Klein et al. 1995). Moreover, treatment of cells with the enzyme SMase, which hydrolyses sphingomyelin into phosphocholine and ceramide, has been shown to affect lipid rafts directly by decreasing sphingomyelin (Rogasevskaia & Coorssen, 2006; Szoke et al. 2010) and indirectly through the production of ceramide, which has been shown to displace cholesterol from lipid raft membranes (Yu et al. 2005). To investigate the effects of lipid raft disruption on the function of α7 nAChR-mediated responses, cells were treated (for 15 min) with either 5 mm MβCD, or 5 mm MβCD in combination with 0.5 U ml−1 SMase. Longer treatments or higher concentrations of either MβCD or SMase resulted in decreased cell viability.
Application of MβCD alone resulted in a significant decrease in current amplitude (514 ± 77 versus 1892 ± 297 pA in control conditions; n = 13 versus n = 30, respectively), accompanied by a change in desensitization kinetics in 13 of 18 cells tested (Fig. 1B). Although desensitization half-time (26 ± 2 versus 27 ± 2 ms in control conditions; Fig. 1D) and desensitization time constants (τfast = 23 ± 2 versus 18 ± 1 ms in control conditions; τslow = 265 ± 61 versus 382 ± 41 ms in control conditions) were not significantly affected by MβCD, the ratio of the fast (49 ± 3 versus 81 ± 2% in control conditions) and slow components (27 ± 3 versus 12 ± 1% in control conditions) changed significantly (Fig. 1E).
Co-application of SMase with MβCD resulted in a larger effect on desensitization kinetics than MβCD alone (Fig. 1C; 14 of 18 cells were affected); the desensitization half-time was significantly increased to 143 ± 41 ms (versus 27 ± 2 ms in control conditions; Fig. 1D). Furthermore, although the slow time constant was not significantly different (418 ± 52 versus 382 ± 41 ms in control conditions), the fast time constant was significantly slower (65 ± 18 versus 18 ± 1 ms in control conditions). Similar to the treatment with MβCD alone, the contribution of the fast and slow components to desensitization was significantly affected with both MβCD and SMase; the fast component was 43 ± 5% (versus 81 ± 2% in control conditions) and the slow component was 46 ± 6% (versus 12 ± 1% in control conditions; Fig. 1E). Interestingly, current amplitude was not significantly different (–1072 ± 175 versus−1892 ± 297 in control conditions; n = 14 versus n = 30, respectively). Furthermore, these changes in desensitization kinetics caused by the co-application of SMase and MβCD were present when cells were dialysed with the calcium chelator BAPTA (by adding it to the recording pipette), and by replacing external Ca2+ with Ba2+ (data not shown), suggesting that Ca2+ entry through the channel is not responsible for these effects.
To test whether lipid raft disruption alters the agonist sensitivity of the α7 nAChR, we treated the neurons with MβCD and SMase and constructed ACh dose–response curves by applying different concentrations of ACh. This disruption of lipid rafts resulted in a significant increase in potency (EC50 = 70 μm, log EC50 = 1.85 ± 0.03, Hill slope = 1.02 ± 0.10; n = 7) when compared with control conditions (EC50 = 132 μm, log EC50 = 2.12 ± 0.03, Hill slope = 1.39 ± 0.13; n = 9; Fig. 1F; P < 0.0001 for log EC50 and Hill slope comparison between data sets). This suggests that changes in the lipid composition of the plasma membrane affect both the kinetics of desensitization as well as agonist potency of the α7 nAChR.
Effects of MβCD and SMase on α7 nAChR function are due to changes in the lipid composition of the plasma membrane
One possible drawback of using MβCD and sphingomyelinase to disrupt lipid rafts is that both drugs affect other cellular components. Besides sequestering cholesterol, MβCD can act as a non-specific chelator binding other phospholipids (Zidovetzki & Levitan, 2007), whereas SMase hydrolyses the membrane raft component sphingomyelin into ceramide, which can act as a second messenger altering cellular functions (Yu et al. 2005).
To determine whether the effects of MβCD and SMase on α7 nAChR desensitization kinetics were specifically caused by changes in the lipid composition of the plasma membrane, cells were treated with MβCD and SMase in the presence of water-soluble cholesterol and sphingomyelin.
Co-application of 2 mm cholesterol and 5 mm MβCD for 15 min resulted in the reversal of the effects of MβCD alone on both current amplitude (–1239 ± 162 versus−1892 ± 297 in contro conditions; n = 9 versus n = 30, respectively) and desensitization kinetics (Fig. 2A and C); in the presence of cholesterol, the fast component was 75 ± 2% (versus 49 ± 3% in MβCD and 81 ± 2% in control conditions) and the slow component was 13 ± 1% (versus 27 ± 3% in MβCD and 12 ± 1% in control conditions; Fig. 2D).
Figure 2. Cholesterol and sphingomyelin prevent the effects of lipid raft disruption through MβCD and SMase on desensitization kinetics of neuronal α7 nAChRs.
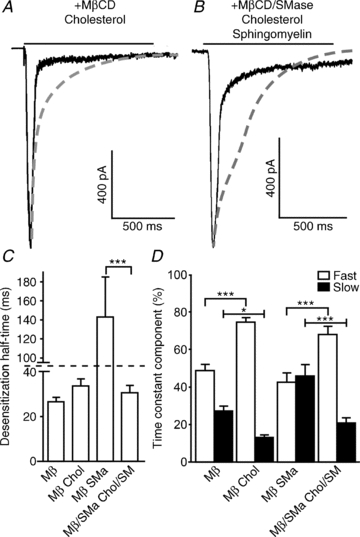
Hippocampal neurons coexpressing wild-type rat α7 nAChR, Ric-3 and GFP were incubated for 15 min with MβCD either alone or in the presence of 2 mm cholesterol; likewise, cells were subjected to MβCD/SMase alone or in the presence of both 2 mm cholesterol and 300 μm sphingomyelin; and subjected to a 1 s application of 3 mm ACh. A–B, cholesterol and sphingomyelin are able to reverse most of the effects of MβCD and SMase (dashed lines) on α7 nAChR desensitization kinetics, including the increase of desensitization half-time, caused by the cotreatment with MβCD/SMase (C), and the changes that both MβCD alone and MβCD/SMase have on the ratio of fast and slow desensitization (D). Plots show means ± SEM and were subjected to a one-way ANOVA; *P < 0.05; ***P < 0.0001.
Likewise, co-application of 2 mm cholesterol, 5 mm MβCD, 0.5 U ml−1 SMase and 300 μm sphingomyelin for 15 min resulted in the reversal of the effects of the co-application of MβCD and SMase on desensitization kinetics (n = 9; Fig. 2B–D). Desensitization half-time was significantly reduced from 143 ± 41 to 31 ± 3 ms (27 ± 2 ms in control conditions; Fig. 2C). The contributions of the fast and slow time constants to desensitization were also reversed to near control values, from 43 ± 5 to 68 ± 4% (for fast; not significantly different from control 81 ± 2%), and from 46 ± 6 to 21 ± 3% (for slow; not significantly different from control 12 ± 1%) respectively (Fig. 2D).
Effects of lipid raft disruption on neuronal α7 nAChR single-channel behaviour
Lipid raft disruption through cholesterol depletion using MβCD has been shown to affect single-channel behaviour of a variety of channels (Balse et al. 2009; Tajima et al. 2010), while it has no effect on others (Sudarikova et al. 2009). Furthermore, mutagenesis studies have shown that lipid-exposed amino acids are critical for the gating mechanism of Torpedo californica nAChRs (Lee et al. 1994; Lasalde et al. 1996; Bouzat et al. 1998; Tamamizu et al. 1999, 2000).
To study the effect of lipid raft disruption on the single-channel behaviour of the α7 nAChR, outside-out patches from neurons coexpressing wild-type rat α7 nAChR, Ric-3 and GFP (McCormack et al. 2010) were treated with either PBS or MβCD and SMase, and exposed to a 4 s rapid application of ACh (50 μm) at a holding potential of –70 mV.
For the α7 nAChR, agonist-evoked single-channel activity appears mainly as isolated brief openings with a mean open time of 0.33 ms, the frequency of which decreases with prolonged agonist application, as is expected due to desensitization (McCormack et al. 2010). The MβCD/SMase cotreated patches were characterized by significantly larger mean single-channel amplitude (–5.14 ± 0.04 versus−4.49 ± 0.05 pA in control conditions; n = 2060 and 6105 single-channel openings in control conditions and MβCD/SMase, respectively; P < 0.0001; Fig. 3A–C) and increased channel activity (Fig. 3A and B). The MβCD/SMase cotreatment had no effect on the channel mean open times (0.34 ± 0.003 versus 0.33 ± 0.006 ms in control conditions; Fig. 3D); however, this cotreatment resulted in a significant decrease in the mean closed times (2.7 ± .089 versus 7.8 ± .351 ms in control conditions; Fig. 3D). This indicates that the increase in single-channel activity is due to a large decrease in the mean closed time following MβCD/SMase cotreatment, which is consistent with our observations at the whole-cell level that lipid raft disruption reduces the rate of desensitization of neuronal α7 nAChRs (McCormack et al. 2010).
Figure 3. Effects of lipid raft disruption on neuronal α7 nAChR single-channel behaviour.
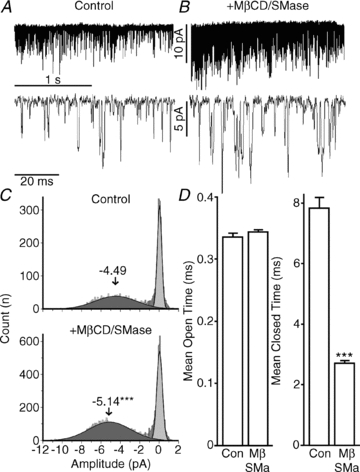
Hippocampal neurons coexpressing wild-type rat α7 nAChR, Ric-3 and GFP were incubated for 15 min with either PBS (control) or a combination of MβCD/SMase and subjected to a 4 s application of 50 μm ACh. A and B, single-channel traces using the outside-out configuration; MβCD/SMase treatment resulted in larger amplitudes and increased activity. C, all-points histogram of amplitudes of the single-channel current from six control and four MβCD/SMase-treated patches. The mean amplitudes were calculated using a single-exponential Gaussian fit and compared using GraphPad Prism 5. D, mean open and closed times were compared in control and MβCD/SMase-treated patches. Plots show means ± SEM and were subjected to Student's unpaired t test; ***P < 0.0001.
Effects of lipid raft disruption on the ACh response of native neuronal α7 nAChRs
We examined the effects of lipid raft disruption on native α7 nAChRs in hippocampal neurons that were cultured for a much longer period of time (i.e. 13–14 days), which is a period of high α7 nAChR expression in the hippocampus (Arnaiz-Cot et al. 2008). As before, neurons were exposed to a 1 s rapid application of ACh (3 mm) at a holding potential of –70 mV. This resulted in the activation of an inward current response with properties indistinguishable from those of expressed α7 nAChRs (Fig. 1), including a mean amplitude of −1066 ± 127 pA, a desensitization half-time of 37 ± 4 ms, and a biphasic onset of desensitization with a fast (τfast = 27 ± 2 ms; 74 ± 2%) and a slow component (τslow = 302 ± 52 ms; 8 ± 1%; Fig. 4; n = 14 cells). In addition, native α7 nAChRs cotreated with MβCD and SMase responded in a similar fashion to the expressed receptors; cotreatment resulted in a significant increase in desensitization half-time to 59 ± 4 ms (n = 11 cells; versus 37 ± 4 ms in control conditions; Fig. 4C). While the time constants of desensitization were not significantly different (τfast = 39 ± 4 versus 27 ± 2 ms in control conditions; τslow = 418 ± 61 ms versus 302 ± 52 ms in control conditions), the contribution of the fast and slow components was significantly affected by MβCD/SMase cotreatment; the fast component was 57 ± 2% (versus 74 ± 2% in control conditions) and the slow component was 25 ± 2% (versus 8 ± 1% in control conditions; Fig. 4D). Finally, current amplitude was not significantly different (–1066 ± 127 versus 1175 ± 182 pA in control conditions; n = 11 versus n = 14, respectively).
Figure 4. Effects of lipid raft disruption on the response of native neuronal α7 nAChRs.
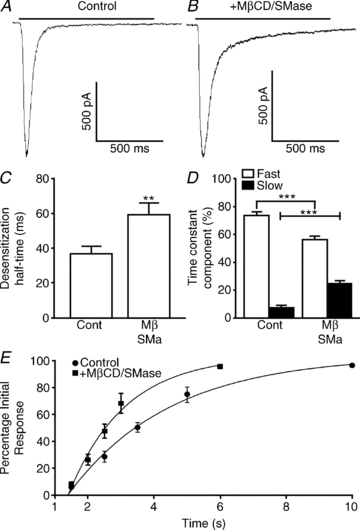
Hippocampal neurons were subjected to a 1 s application of 3 mm ACh at Day in vitro 13-14 (DIV 13-14) to record the response of native channels to a 15 min treatment with either PBS (control) or a combination of 5 mm MβCD and 0.5 U ml−1 SMase. A and B, responses to ACh application in control (A) and MβCD/ SMase-treated neurons (B). C, MβCD/SMase resulted in a significant increase in the desensitization half-time. D, desensitization time constants (τ) were calculated using Clampfit and fitted with a two-exponential equation. Although no difference was seen in τ values, the ratio of fast and slow desensitization was affected by MβCD/SMase cotreatment. E, recovery from desensitization was studied by consecutive 1 s applications of 3 mm ACh. The time of application was adjusted depending on treatment. **p < 0.001; ***p < 0.0001.
To further dissect the effects of MβCD/SMase cotreatment on α7 nAChR desensitization, we examined the rate of recovery from desensitization. This was done by continuously applying ACh (3 mm for 10 s) to induce near-complete (>90%) desensitization and, after washing out ACh for various amounts of time (1.5, 2.5, 3.5, 5 and 10 s in control conditions; 1.5, 2, 2.5, 3 and 6 s for MβCD/SMase), a brief pulse (2 s) of ACh was applied to assess the extent of recovery from the desensitized state. After MβCD/SMase cotreatment, the α7 nAChRs recovered from desensitization significantly faster; in 2.5 s, the recovery in MβCD/SMase-treated cells was 48 ± 5% (n = 7 cells) versus 29 ± 4% (n = 6 cells) in control conditions. The rate of recovery of control and treated cells was monophasic (Fig. 4E; Gay et al. 2008), with time constant values of 3.1 s (n = 6 cells) in control conditions and 1.7 s (n = 7 cells; P < 0.0001) in MβCD/SMase-treated cells, suggesting that lipid raft disruption results in a decreased probability that neuronal α7 nAChRs would become and remain in the desensitized state.
Effects of lipid raft disruption on the α3β2 nAChR
We examined the effects that lipid raft disruption has on the α3β2 nAChR subtype.
Cultured hippocampal neurons coexpressing wild-type rat α3 and β2 nAChR subunits (along with GFP) were exposed to a 1 s rapid application of ACh (3 mm) at a holding potential of –70 mV; this resulted in the rapid activation of an inward current that desensitized in the continued presence of agonist (Fig. 5A), with a desensitization half-time of 100 ± 17 ms (Fig. 5D). Similar to the α7 nAChR, the onset of desensitization was biphasic; the fast component (τfast = 95 ± 17 ms) comprised 58 ± 4%, while the slow component (τslow = 600 ± 143 ms) comprised 25 ± 2% of the total desensitization (Fig. 5E; n = 7 cells).
Figure 5. Effects of lipid raft disruption on the responses of α3β2 nAChRs.
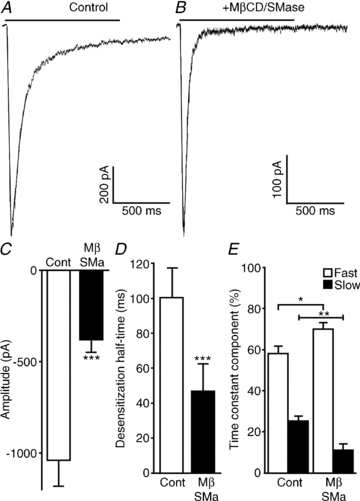
Hippocampal neurons coexpressing wild-type rat α3 and β2 nAChR subunits (along with GFP) were incubated for 15 min with either PBS (control) or a combination of 5 mm MβCD and 0.5 U ml−1 SMase and subjected to a 1 s application of 3 mm ACh. A and B, response to ACh application in control (A) and MβCD/SMase-treated neurons (B). C, MβCD/SMase resulted in a significant decrease in current amplitude. D, likewise, desensitization half-time decreased following MβCD/SMase cotreatment. E, desensitization time constants (τ) were calculated using Clampfit and fitted with a two-exponential equation. Although no difference was seen in τ values, the ratio of fast and slow desensitization was affected by MβCD/SMase cotreatment. Plots show means ± SEM, and were subjected to Student's t test; *P < 0.05; **P < 0.001; ***P < 0.0001.
Interestingly, co-application of MβCD and SMase resulted in the opposite effect on the desensitization kinetics of the α3β2 nAChR in comparison to the α7 nAChR; the desensitization half-time was significantly decreased from 100 ± 17 ms in control conditions to 47 ± 16 ms in treated cells (n = 6 cells; Fig. 5D). Although both fast and slow time constants were not significantly different, the contribution of the fast and slow components to desensitization was significantly affected; the fast component was 73 ± 3% (versus 58 ± 4% in control conditions) and the slow component was 11 ± 3% (versus 25 ± 2% in control conditions; Fig. 5E). Furthermore, the current amplitude was significantly decreased from −1040 ± 140 pA in control conditions to −380 ± 68 pA in treated cells (n = 7 versus n = 6, respectively; Fig. 5C). These results suggest that α3β2 nAChRs are also susceptible to changes in the lipid environment, although the changes in the kinetics of desensitization appear to be opposite to the effects seen with the α7 nAChRs.
Effects of cholesterol and sphingomyelin synthesis inhibitors on α7 nAChR function
So far we have demonstrated that cholesterol depletion through acute treatment with MβCD and SMase affects α7 nAChR function. These effects were reversed by cotreatment of the cells with the corresponding lipid molecules. Therefore, these treatments show the acute effects that decreasing cholesterol and sphingomyelin levels have on α7 nAChR function. To further investigate the correlation between cholesterol and sphingomyelin levels and α7 nAChR function, we inhibited the de novo synthesis of both cholesterol and sphingomyelin. We used myriocin, an inhibitor of serine palmitoyltransferase, the key enzyme in the de novo synthesis of sphingolipids used to deplete cells of sphingolipids (Szoke et al. 2010), and mevastatin, an inhibitor of cholesterol synthesis (Alberts et al. 1980); both of these have been shown to disrupt lipid rafts (Simons et al. 1998).
Cells were treated with 2 μm mevastatin and 400 nM myriocin in serum-free medium for 24–48 h. Then cells were exposed to the rapid application of ACh to activate the α7 nAChRs (3 mm for 2 s; Fig. 6A and B). Treatment with these synthesis inhibitors showed a partial effect on α7 nAChR function; alterations in desensitization kinetics were observed in nine of 21 cells. The affected cells showed no significant changes in current amplitude (–1767 ± 203 versus 1681 ± 400 pA in control conditions; n = 9 versus n = 10, respectively) or desensitization half-time (50 ± 5 versus 39 ± 3 ms control conditions), in contrast to changes seen with the MβCD/SMase cotreatment (143 ± 41 versus 27 ± 2 ms in control conditions) and desensitization time constants (τfast = 18 ± 2 versus 21 ± 4 ms in control conditions; τslow = 180 ± 56 versus 285 ± 87 ms in control conditions). However, the ratio of fast to slow components of desensitization was significantly changed to 60 ± 2% (versus 83 ± 2% in control conditions) and 26 ± 2% (versus 12 ± 1% in control conditions; Fig. 1E), respectively, indicating that inhibition of the synthesis of cholesterol and sphingomyelin had a significant effect on desensitization kinetics.
Figure 6. Inhibition of cholesterol and sphingomyelin synthesis affects the desensitization kinetics of α7 nAChRs.
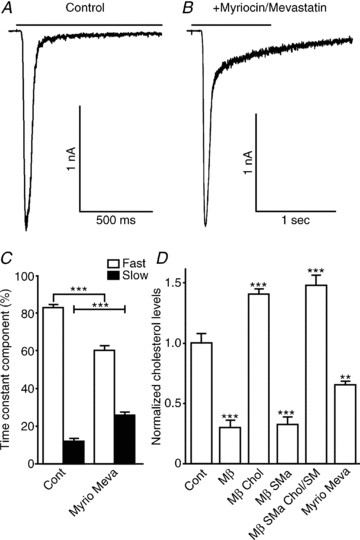
Hippocampal neurons were incubated for 24–48 h with 400 μm myriocin and 2 mm mevastatin in serum-free media and tested for their response to a 2 s ACh application. A and B, traces showing the effects of myriocin and mevastatin cotreatment in the ACh response. C, desensitization time constants (τ) were calculated using Clampfit and fitted with a two-exponential equation. Although no difference was seen in τ values, the ratio of fast and slow desensitization was affected by myriocin and mevastatin cotreatment. D, cholesterol levels measured after drug applications were normalized to protein concentration and cholesterol levels in control conditions (n = 9). Plots show means ± SEM; desensitization time constant and cholesterol measurements were analysed using Student's unpaired t test and one-way ANOVA, respectively; **P < 0.001; ***P < 0.0001.
Cholesterol levels decrease after treatment with cholesterol scavenger and synthesis inhibitors
To compare the effects of acute removal of cholesterol (MβCD) and the enzymatic breakdown of sphingomyelin (SMase) with the chronic inhibition of cholesterol synthesis (mevastatin) and sphingomyelin synthesis (myriocin) on cholesterol levels, we used the Amplex Red Cholesterol Assay Kit. Samples were tested in parallel with a BCA protein kit and first normalized to protein levels, then to cholesterol levels in control conditions. Consistent with the functional data showing partial effects of synthesis inhibitors on α7 nAChR function (Fig. 6A–C), cholesterol levels were decreased approximately 35% by cotreatment with mevastatin and myriocin. In contrast, treatment with MβCD and cotreatment with MβCD and SMase resulted in a decrease of approximately 70%, which is also consistent with the functional data (Fig. 1C–E) because these treatments resulted in larger effects on desensitization kinetics. Treatment with SMase alone showed no effects on cholesterol levels, and there was no difference in cholesterol levels between MβCD and MβCD/SMase cotreatment.
In addition, treatment with MβCD in the presence of cholesterol and MβCD/SMase in the presence of cholesterol and sphingomyelin resulted in enrichment (140 and 147% cholesterol respectively, when compared with control conditions; Borroni et al. 2007), which is consistent with the reversal these treatments had on the functional effects of MβCD and MβCD/SMase on α7 nAChR desensitization (Fig. 2B–D).
Discussion
The α7 nAChRs have previously been shown to be localized into lipid rafts (Bruses et al. 2001; Oshikawa et al. 2003; Fernandes et al. 2010); however, the role that this localization has on channel function has not been resolved. Here we have used pharmacological agents to acutely and chronically decrease the levels of cholesterol and sphingomyelin, the major lipids in lipid rafts, and show that the disruption of lipid rafts alters the functional properties of these receptors by slowing the kinetics of desensitization (in part by increasing the rate of recovery from desensitization), and by increasing agonist affinity and single-channel conductance. These data provide new insights into the mechanism of desensitization of α7 nAChRs by providing evidence that the lipid composition of the plasma membrane can modulate the activity of the α7 nAChRs.
Although the mechanism of desensitization is not completely understood, desensitization is an important characteristic of nAChRs. For example, differences in desensitization rates of nAChR subunits are thought to play a role in nicotine addiction; low doses of nicotine desensitize non-α7 nAChRs on dopaminergic and GABAergic neurons, whereas at the same time it activates α7 receptors, enhancing glutamate-mediated excitatory inputs to the dopaminergic neurons and facilitating dopamine release onto Nucleus accumbens (NAc) neurons (Mansvelder & McGehee, 2002). Moreover, abnormalities in nAChR desensitization are thought to be involved in some nAChR-related diseases, such as autosomal dominant nocturnal frontal lobe epilepsy (which is thought to be caused by mutations in α4 and β2 nAChR subunits; Bertrand et al. 2002) and congenital myasthenic syndrome (caused by mutations in endplate nAChRs; Milone et al. 1997).
Therapeutic drugs are currently being developed to treat Alzheimer's disease and other neurological disorders (Young et al. 2008) by potentiating α7 nAChRs through the removal of desensitization (e.g. type II α7 PAMs, such as PNU-120596; (Bertrand & Gopalakrishnan, 2007). However, this can in some cases (but not all) result in excessive calcium influx, resulting in cytotoxicity (Ng et al. 2007); therefore, it is critical to have a greater understanding of the mechanisms that govern the desensitization of the α7 nAChRs to aid in the development of α7 nAChR-selective PAMs that do not alter desensitization (e.g. type I PAMs).
Here we have shown that changing the levels of cholesterol and sphingomyelin in the plasma membrane using both MβCD and SMase causes a dramatic change in the desensitization kinetics of the α7 nAChRs. Furthermore, treatment with SMase alone showed no significant difference from control conditions in cholesterol levels and in ACh-evoked α7 desensitization kinetics, suggesting that significant changes in cholesterol levels are necessary for the functional effects reported here. Interestingly, the addition of SMase to MβCD was necessary for the fivefold decrease in desensitization half-time (compared with no significant change with MβCD alone), a decrease in the fast time constant of decay, and an increase in the contribution of the slow component of desensitization when compared with both control conditions and MβCD alone. Taken together, these results suggest that the cotreatment with MβCD and SMase is more effective at disrupting lipid rafts than MβCD alone. This is consistent with a previous study showing that the combination of both agents has more pronounced effects than MβCD alone on the activity of both KCC2 and NKCC1 (K+–Cl− and Na+–K+–2Cl− co-transporters, respectively), which are also associated with lipid rafts (Hartmann et al. 2009).
Here we show that disruption of lipid rafts through cotreatment with MβCD and SMase affects the functional and pharmacological properties of the α7 nAChRs, supporting the idea that lipid raft integrity is critical for proper channel function. We have also shown that the disruption of lipid rafts also affects desensitization of the α3β2 subunit-containing nAChRs (however, the effect on kinetics is opposite to the α7 receptor subtype), suggesting that lipid rafts play an important role in the modulation of more than one type of neuronal nAChRs. These modulatory effects can be mediated by changes in channel localization or by a loss of lipid raft-mediated protein–protein or protein–lipid interactions, which have been shown to result in changes to receptor conformation (Allen et al. 2007). In this study, we cannot tell whether these changes are caused by changes in receptor localization or by the loss of a physical interaction. However, cholesterol depletion with MβCD has been shown to increase the mobility (Fernandes et al. 2010) and to disperse clusters (Bruses et al. 2001) of α7 nAChRs in neurons, whereas it has been shown to cause the internalization of endplate nAChRs (Borroni et al. 2007). Although we found that cholesterol levels were not different between MβCD and MβCD/SMase treatments, current amplitude is decreased by MβCD treatment alone, suggesting that SMase breakdown of sphingomyelin into ceramide and phosphocholine is able to prevent some of the effects of MβCD on α7 nAChR function. Previous studies have shown that ceramide produced by SMase treatment displaces cholesterol from lipid rafts and forms clusters that prevent the dispersion of raft resident proteins (Yu et al. 2005). Furthermore, the generation of ceramide through pretreatment of cells with SMase has been shown to be protective against the effects of MβCD on Ca2+-triggered membrane fusion (Rogasevskaia & Coorssen, 2006). Altogether, our data suggest that differences in α7 response current amplitude between MβCD and MβCD/SMase treatments are due to changes in localization, rather than cholesterol levels.
Both cholesterol and α7 nAChRs have been shown to play a role in the development of Alzheimer's disease. Early development of Alzheimer's disease is characterized by both cholesterol dysregulation and a degeneration of cholinergic neurons (Barrantes et al. 2009). Alzheimer's disease is a neurodegenerative disorder characterized by the generation of β-amyloid (Aβ) fragments, and the α7 nAChRs have been implicated in neuroprotection against Aβ toxicity (Kihara et al. 2001). Conflicting reports have recently surfaced on the possible involvement of the α7 nAChR in Alzheimer's disease; one study using an animal model of Alzheimer's disease found that knocking out the α7 nAChR results in more severe memory (hippocampus-dependent) deficits (Hernandez et al. 2010), while another study using the same animal model found that knocking out the α7 nAChR lessens the cognitive deficiencies of Alzheimer's disease (Dziewczapolski et al. 2009). This discrepancy is explained by Hernandez et al. (2010) as a difference in the age of the animals used; the α7 nAChR activity appears to be neuroprotective in early Alzheimer's disease, whereas it may be deleterious in later stages of Alzheimer's disease (Dziewczapolski et al. 2009; Hernandez et al. 2010).
Chronic treatment of cells with a cholesterol synthesis inhibitor, followed by the acute treatment with MβCD, is able to inhibit Aβ generation in hippocampal neurons (Simons et al. 1998). Therefore, cholesterol-lowering drugs appear to be a good therapeutic approach in early Alzheimer's disease because they decrease the generation of Aβ, while at the same time they improve α7 nAChR signalling. In this study, we have shown that a decrease in cholesterol levels through synthesis inhibition is able to cause changes in desensitization kinetics without affecting the current amplitude of the α7 nAChRs, suggesting that treatment with these compounds will result in enhanced cholinergic responses in the hippocampus; this latter effect has been shown to result in improvements in cognition and memory in animal models (Hernandez et al. 2010).
Acknowledgments
This work was supported by the Intramural Research Program of the NIEHS/NIH. The authors also thank Dr Sindura Ganapathi and Dr Christian Erxleben for their helpful insights and suggestions in the manuscript preparation process; Pattie Lamb for preparing all the constructs used in this study; Dr Steve Shears for allowing use of the microplate reader; and Dr Angelika Zaremba for her technical help.
Glossary
Abbreviations
- Aβ
β-amyloid
- GFP
green fluorescent protein
- KCC2
K+–Cl− transporter
- MβCD
methyl-β-cyclodextrin
- nAChR
nicotinic acetylcholine receptor
- NKCC1
Na+–K+–2Cl− transporter
- PAM
positive allosteric modulator
- Ric
resistance to inhibitors of cholinesterase
- SMase
sphingomyelinase
Author contributions
Both authors contributed to either the conception and design or the analysis and interpretation of data. Drafting of the article was done primarily by J.O.C.-S., while both authors participated in revisions. Both authors approved the final version of the manuscript for publication.
References
- Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, Hirshfield J, Hoogsteen K, Liesch J, Springer J. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA. 1980;77:3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Arnaiz-Cot JJ, Gonzalez JC, Sobrado M, Baldelli P, Carbone E, Gandia L, Garcia AG, Hernandez-Guijo JM. Allosteric modulation of α7 nicotinic receptors selectively depolarizes hippocampal interneurons, enhancing spontaneous GABAergic transmission. Eur J Neurosci. 2008;27:1097–1110. doi: 10.1111/j.1460-9568.2008.06077.x. [DOI] [PubMed] [Google Scholar]
- Balse E, El-Haou S, Dillanian G, Dauphin A, Eldstrom J, Fedida D, Coulombe A, Hatem SN. Cholesterol modulates the recruitment of Kv1.5 channels from Rab11-associated recycling endosome in native atrial myocytes. Proc Natl Acad Sci USA. 2009;106:14681–14686. doi: 10.1073/pnas.0902809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrantes FJ, Borroni V, Vallés S. Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer's disease. FEBS Lett. 2009;584:1856–1863. doi: 10.1016/j.febslet.2009.11.036. [DOI] [PubMed] [Google Scholar]
- Berger F, Gage FH, Vijayaraghavan S. Nicotinic receptor-induced apoptotic cell death of hippocampal progenitor cells. J Neurosci. 1998;18:6871–6881. doi: 10.1523/JNEUROSCI.18-17-06871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1155–1163. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Picard F, Le Hellard S, Weiland S, Favre I, Phillips H, Bertrand S, Berkovic SF, Malafosse A, Mulley J. How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia. 2002;43(Suppl 5):112–122. doi: 10.1046/j.1528-1157.43.s.5.16.x. [DOI] [PubMed] [Google Scholar]
- Borroni V, Baier CJ, Lang T, Bonini I, White MM, Garbus I, Barrantes FJ. Cholesterol depletion activates rapid internalization of submicron-sized acetylcholine receptor domains at the cell membrane. Mol Membr Biol. 2007;24:1–15. doi: 10.1080/09687860600903387. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Roccamo AM, Garbus I, Barrantes FJ. Mutations at lipid-exposed residues of the acetylcholine receptor affect its gating kinetics. Mol Pharmacol. 1998;54:146–153. doi: 10.1124/mol.54.1.146. [DOI] [PubMed] [Google Scholar]
- Bruses JL, Chauvet N, Rutishauser U. Membrane lipid rafts are necessary for the maintenance of the α7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J Neurosci. 2001;21:504–512. doi: 10.1523/JNEUROSCI.21-02-00504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Patrick JW. The α-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the α7 subunit. J Biol Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF. Deletion of the α7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer's disease. J Neurosci. 2009;29:8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes CC, Berg DK, Gomez-Varela D. Lateral mobility of nicotinic acetylcholine receptors on neurons is determined by receptor composition, local domain, and cell type. J Neurosci. 2010;30:8841–8851. doi: 10.1523/JNEUROSCI.6236-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay EA, Giniatullin R, Skorinkin A, Yakel JL. Aromatic residues at position 55 of rat α7 nicotinic acetylcholine receptors are critical for maintaining rapid desensitization. J Physiol. 2008;586:1105–1115. doi: 10.1113/jphysiol.2007.149492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann AM, Blaesse P, Kranz T, Wenz M, Schindler J, Kaila K, Friauf E, Nothwang HG. Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J Neurochem. 2009;111:321–331. doi: 10.1111/j.1471-4159.2009.06343.x. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT. Loss of α7 nicotinic receptors enhances β-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer's disease. J Neurosci. 2010;30:2442–2453. doi: 10.1523/JNEUROSCI.5038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, Yakel JL. Rat nicotinic ACh receptor α7 and β2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol. 2002;540:425–434. doi: 10.1113/jphysiol.2001.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. α7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A β-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with β-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- Lasalde JA, Tamamizu S, Butler DH, Vibat CR, Hung B, McNamee MG. Tryptophan substitutions at the lipid-exposed transmembrane segment M4 of Torpedo californica acetylcholine receptor govern channel gating. Biochemistry. 1996;35:14139–14148. doi: 10.1021/bi961583l. [DOI] [PubMed] [Google Scholar]
- Lee YH, Li L, Lasalde J, Rojas L, McNamee M, Ortiz-Miranda SI, Pappone P. Mutations in the M4 domain of Torpedo californica acetylcholine receptor dramatically alter ion channel function. Biophys J. 1994;66:646–653. doi: 10.1016/s0006-3495(94)80838-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack TJ, Melis C, Colon J, Gay EA, Mike A, Karoly R, Lamb PW, Molteni C, Yakel JL. Rapid desensitization of the rat α7 nAChR is facilitated by the presence of a proline residue in the outer β-sheet. J Physiol. 2010;588:4415–4429. doi: 10.1113/jphysiol.2010.195495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor α subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HJ, Whittemore ER, Tran MB, Hogenkamp DJ, Broide RS, Johnstone TB, Zheng L, Stevens KE, Gee KW. Nootropic α7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc Natl Acad Sci USA. 2007;104:8059–8064. doi: 10.1073/pnas.0701321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa J, Toya Y, Fujita T, Egawa M, Kawabe J, Umemura S, Ishikawa Y. Nicotinic acetylcholine receptor α7 regulates cAMP signal within lipid rafts. AmJ Physiol Cell Physiol. 2003;285:C567–C574. doi: 10.1152/ajpcell.00422.2002. [DOI] [PubMed] [Google Scholar]
- Pottorf WJ, 2nd, Johanns TM, Derrington SM, Strehler EE, Enyedi A, Thayer SA. Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J Neurochem. 2006;98:1646–1656. doi: 10.1111/j.1471-4159.2006.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitsch A, Berhorster K, Liew CW, Richter D, Kreienkamp HJ. Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J Neurosci. 2005;25:479–487. doi: 10.1523/JNEUROSCI.2699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogasevskaia T, Coorssen JR. Sphingomyelin-enriched microdomains define the efficiency of native Ca2+-triggered membrane fusion. J Cell Sci. 2006;119:2688–2694. doi: 10.1242/jcs.03007. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci USA. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Kihara T. Nicotinic receptor-mediated protection against β-amyloid neurotoxicity. Biol Psychiatry. 2001;49:233–239. doi: 10.1016/s0006-3223(00)01100-8. [DOI] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarikova AV, Chubinskiî-Nadezhdin VI, Neguliaev IuA, Morachevskaia EA. Functional properties of sodium channels in cholesterol-depleted K562 cells. [Article in Russian.] Tsitologiia. 2009;51:676–683. [PubMed] [Google Scholar]
- Szoke E, Borzsei R, Toth DM, Lengl O, Helyes Z, Sandor Z, Szolcsanyi J. Effect of lipid raft disruption on TRPV1 receptor activation of trigeminal sensory neurons and transfected cell line. Eur J Pharmacol. 2010;628:67–74. doi: 10.1016/j.ejphar.2009.11.052. [DOI] [PubMed] [Google Scholar]
- Tajima N, Itokazu Y, Korpi ER, Somerharju P, Käkelä R. Activity of BKCa channel is modulated by membrane cholesterol content and association with Na+/K+-ATPase in human melanoma IGR39 cells. J Biol Chem. 2010;286:5624–5638. doi: 10.1074/jbc.M110.149898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamizu S, Guzman GR, Santiago J, Rojas LV, McNamee MG, Lasalde-Dominicci JA. Functional effects of periodic tryptophan substitutions in the α M4 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor. Biochemistry. 2000;39:4666–4673. doi: 10.1021/bi992835w. [DOI] [PubMed] [Google Scholar]
- Tamamizu S, Lee Y, Hung B, McNamee MG, Lasalde-Dominicci JA. Alteration in ion channel function of mouse nicotinic acetylcholine receptor by mutations in the M4 transmembrane domain. J Membr Biol. 1999;170:157–164. doi: 10.1007/s002329900545. [DOI] [PubMed] [Google Scholar]
- Young GT, Zwart R, Walker AS, Sher E, Millar NS. Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci USA. 2008;105:14686–14691. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Alterman M, Dobrowsky RT. Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J Lipid Res. 2005;46:1678–1691. doi: 10.1194/jlr.M500060-JLR200. [DOI] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]