

Non-technical summary
Contraction of the muscle in the walls of blood vessels is controlled by the movement of ions into or out of the muscle cells, through proteins called ion channels. By allowing potassium (K+) to leave the cell, K+-selective ion channels keep arteries dilated. In the pulmonary artery, which carries blood to the lung for gas exchange, a type of K+ channel called TASK-1 is thought to play an important role in this regulation, but we observed that the pulmonary arteries from mice that lack the TASK-1 protein do not display any altered properties compared to wild-type (normal) mice. We found, however, that the pattern of K+ flux in muscle cells from mouse pulmonary artery is different from that of other species, including human. Our results question the use of mouse as a model for human pulmonary artery.
Abstract
Abstract
The acid-sensitive, two-pore domain K+ channel, TASK-1, contributes to the background K+ conductance and membrane potential (Em) of rat and human pulmonary artery smooth muscle cells (PASMCs), but its role in regulating tone remains elusive. This study aimed to clarify the role of TASK-1 by determining the functional properties of pulmonary artery (PA) from mice in which the TASK-1 gene was deleted (TASK-1/3 KO), in comparison with wild-type (WT) C57BL/6 controls. Small vessel wire myography was used to measure isometric tension developed by intact PA. Em and currents were recorded from freshly isolated PASMCs using the perforated patch-clamp technique. Reverse transcription–polymerase chain reaction (RT-PCR) was used to estimate K+ channel expression. We could find no difference between PA from WT and TASK-1/3 KO mice. They showed similar constrictor responses to a range of agonists and K+ concentrations, the K+ channel blockers 4-aminopyridine, tetraethylammonium ions and XE991. Treprostinil, proposed to dilate by activating TASK-1, was just as effective in TASK-1/3 KO arteries. Blocking Ca2+ influx with nifedipine (1 μm) or levcromakalim (10 μm) had no effect on resting tone in either strain. The resting Em of PASMCs and its responses to K+ channel blockers were unchanged in TASK-1/3 KO mice as were voltage-activated K+ currents, including the non-inactivating K+ current (IKN) measured at 0 mV. The Em was, however, depolarised in comparison with other species. Mouse IKN was much smaller than in rat and showed no sensitivity to pH. The results imply that TASK-1 does not form a functional channel in mouse PASMCs.
Introduction
The acid sensitive, tandem-pore domain K+ channel type-1 (TASK-1) has been proposed to play a key role in the regulation of pulmonary arterial tone. The channel is expressed in rabbit (Gurney et al. 2003), rat (Gardener et al. 2004; Manoury et al. 2009) and human (Olschewski et al. 2006) pulmonary artery smooth muscle cells (PASMCs), where it is thought to contribute to the background K+ efflux that sets the resting membrane potential (Em). Inhibition of TASK-1 channels was proposed to mediate the depolarising effects of hypoxia (Olschewski et al. 2006) and endothelin-1 (Tang et al. 2009) on PASMCs, while activation of TASK-1 may underlie the hyperpolarising effects of prostacyclin (Olschewski et al. 2006) and the β-adrenoceptor agonist isoprenaline (Bieger, 2006). The Em in turn regulates smooth muscle cell (SMC) contraction and vessel tone by controlling the open probability of voltage-gated, L-type Ca2+ channels and the associated excitation–contraction coupling.
Although microelectrode measurements on intact vessels have clearly linked Em and TASK-1 channel activity (Gardener et al. 2004), much of the data giving rise to the proposed roles for TASK-1 channels were derived from freshly isolated and cultured PASMCs. Whether TASK-1 channels regulate the tone of intact vessels remains unclear. The endocannabinoid anandamide, which inhibited 90% of recombinant TASK-1 current at 3 μm (Maingret et al. 2001) and depolarised SMCs in rat pulmonary artery by 4–6 mV at 10 μm (Gardener et al. 2004), evoked only a mild constriction of pulmonary arteries (Gardener et al. 2004; Shah, 2008). Bupivicaine, another inhibitor of TASK-1 channels (Kindler et al. 1999), was equally ineffective at constricting pulmonary artery (Gardener et al. 2004; Shah, 2008). These discrepancies may reflect poor selectivity of the drugs for TASK-1 channels. In particular, both drugs can evoke endothelium-dependent relaxation in various vascular beds (Randall et al. 1996; White et al. 2001; Choi et al. 2010), which would counteract the effect of depolarisation. On the other hand, changes in pH over the range 6–8 also had pronounced effects on the Em of PASMCs (Gurney et al. 2003; Gardener et al. 2004), but had little effect on rat pulmonary artery tone (Shah, 2008). This is surprising, because quite small changes in SMC Em can produce substantial changes in tone (Casteels et al. 1977a; Nelson et al. 1990). Thus pH changes may also have actions on vessels that mask the effect of altered Em.
The molecular nature of the K+ channels giving rise to the resting Em of PASMCs is still debated, with TASK-1 channels only one of several proposed mediators. Other K+ channels attributed a role in setting the Em include the voltage-gated delayed rectifier KV1.5 and to a lesser extent KV2.1 (Post et al. 1995; Archer et al. 2001, 2004; Remillard & Yuan, 2004; Platoshyn et al. 2006), although their biophysical and pharmacological properties do not perfectly match those of the resting K+ conductance (Gurney et al. 2002). Recent evidence suggests that PASMCs also express KCNQ-encoded Kv7 channels, which have biophysical and pharmacological properties appropriate for a role in setting the resting Em (Joshi et al. 2009). PASMCs express myriad additional voltage-gated and Ca2+-activated K+ channels that have not been implicated in Em regulation. All these K+ channels have the capacity to respond to depolarisation and/or Ca2+ influx, thereby opposing and limiting the effects of TASK inhibitors.
In order to directly address the role of TASK-1 channels in regulating the contractile properties of pulmonary artery, we made use of transgenic mice that are deficient in the TASK-1 gene. Female mice lacking a functional TASK-1 channel were found to have a mildly altered cardiovascular phenotype, consisting of elevated systolic blood pressure and bradycardia (Heitzmann et al. 2008). TASK-1 is often co-expressed with the closely related TASK-3 subunit and it has been suggested that homomeric TASK-3 channels may be able to compensate in the absence of TASK-1 expression (Aller et al. 2005). For this reason we employed TASK-1/3 double knockout (KO) mice, which had a healthy phenotype as previously described (Trapp et al. 2008a). The aims of the study were to (1) determine how the absence of TASK-1 affects the resting tone and basic contractile properties of pulmonary arteries and (2) establish the relative contribution of TASK-1 channels to the background K+ conductance and resting Em of mouse PASMCs.
Methods
Animals
All procedures were carried out with the approval of the University of Manchester Local Ethical Review Committee, under Home Office Project and Personal Licence authority. Mice used in this study were of the C57BL/6 background. Two couples of double KO mice, deficient in both knck3 (Aller et al. 2005) and kncnk9 (Brickley et al. 2007) genes (TASK-1/3 KO), described in detail previously (Trapp et al. 2008a), were kindly provided by Prof. W. Wisden, University of Aberdeen, and used to start a colony at the University of Manchester animal facility. Mice from the colony were compared to age-matched, wild type C57BL/6 mice housed in similar conditions (WT). Genotypes were confirmed by polymerase chain reaction (PCR), using genomic DNA extracted from ear biopsies as template. Male mice were used at 3–5 months old unless specified. A few experiments employed male Sprague–Dawley rats (220–300 g) as indicated. All animals were humanely killed by cervical dislocation. The heart and lungs were rapidly excised en bloc into ice-cold physiological salt solution (PSS) containing (in mm): NaCl 122, KCl 5, Hepes 10, NaH2PO4 0.5, KH2PO4 0.5, d-glucose 11, MgCl2 1, CaCl2 1.8, pH adjusted to 7.3 with NaOH. First to third order intrapulmonary arteries with diameters of 0.1–0.5 mm (mouse) and 0.2–2 mm (rat) were used. Myography experiments were performed using 2 mm segments of second order branches. For cell isolation or RNA extraction, different sized arteries were pooled.
Indices of pulmonary arterial hypertension
The ratio of right ventricle (RV) weight to left ventricle (LV) weight plus intra-ventricular septum (S) was measured to provide an index of right ventricular hypertrophy. After excising the atria, the RV was separated from the LV + S and they were separately dabbed with tissue to dry and weighed. Lungs were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS), washed in PBS and mounted in paraffin blocks. To enable microscopic assessment of vessel muscularisation, 50 μm sections were subject to Elastica Van Gieson staining by the University of Manchester core histology service. Sections were viewed at 400× magnification and vessels counted as muscularised if they displayed a double elastic lamina for more than half of their circumference, as previously described (MacLean et al. 2004). Small venules were probably included in the count as they could not easily be distinguished from arterioles. Muscularisation of vessels was assessed in sections from six WT and six TASK-1/3 KO mice.
Cell isolation
Smooth muscle cells were isolated from pulmonary arteries by enzymatic dissociation in medium (DM) of the following composition (in mm): NaCl 110, KCl 5, Hepes 10, KH2PO4 0.5, NaH2PO4 0.5, NaHCO3 10, taurine 10, EDTA 0.5, d-glucose 10, CaCl2 0.16, MgCl2 2, phenol red 0.03; pH adjusted to 7.0 with NaOH. Mouse PASMCs were usually isolated by incubating tissue with collagenase type XI (1 mg ml−1, Sigma-Aldrich, Poole, UK), protease type XXIV (0.45 mg ml−1, Sigma-Aldrich), bovine serum albumin (2 mg ml−1, Sigma-Aldrich) and trypsin inhibitor (2 mg ml−1, Sigma-Aldrich) in DM at 4°C for 8–15 min, followed by 37°C for 8 min. An alternative enzymatic protocol was used for isolating rat and occasionally mouse (where specified) PASMCs: tissue was incubated with 30 mg ml−1 papain (Fluka) for 1 h at 4°C then 6 min at 37°C after addition of 1 mg ml−1 dithiothreitol (Sigma-Aldrich), followed by incubation with 20 mg ml−1 collagenase IA (Sigma-Aldrich) for 5 min at 37°C (Manoury et al. 2009). After enzyme treatment tissues were washed in ice cold DM, transferred to a polypropylene tube containing 400–600 μl DM and gently triturated to release cells from the tissue matrix. Cells were stored at 4°C and used the same day.
Isometric tension measurement
Artery constriction was measured using a small-vessel wire myograph (Danish Myotechnology, Arhus, Denmark) as reported previously (Ward & Snetkov, 2004; Joshi et al. 2006). Data acquisition was performed using Intracept-Chart v4.6.1 software (copyright J. Dempster, University of Strathclyde, UK) with a DAQ Card-6036E and BNC2110 interface (National Instruments Corp., Newbury, UK). A basal tension of 3 mN, equivalent to a transmural pressure of around 30 mmHg (Ward & Snetkov, 2004), was applied. Throughout the experiment tissues were bathed in PSS at 37°C and continuously aerated. Vessels were allowed to equilibrate for 30–40 min, then a reference constriction to the addition of 50 mm KCl was obtained. Vessels were washed and the 50 mm KCl challenge repeated until reproducible contractions were produced. After washing and return to basal tension, a test solution was applied for 5–15 min. Contractile responses were measured as a percentage of the last response to 50 mm KCl. In experiments testing the sensitivity of PA to increasing concentrations of K+, osmolarity was maintained by replacing NaCl in the PSS with equimolar KCl. In some experiments the endothelium was disrupted by gently rubbing the vessel lumen with a rough wire or strand of hair. The absence of endothelial function was testified by the lack of response to 1 μm carbachol in vessels pre-constricted with 1 μm phenylephrine. As the physical treatment was not always sufficient to abolish the response to carbachol, NG-nitro-l-arginine methyl ester (l-NAME; 300 μm) and indomethacin (10 μm) were added to the bath to eliminate NO and prostaglandin-mediated dilator pathways. Vessels were challenged with 50 mm KCl at the end of each experiment to ensure they remained viable.
For experiments investigating hypoxic vasoconstriction, vessels were bathed in PSS containing (in mm): NaCl 118, KCl 4, NaHCO3 24, NaH2PO4 0.44, d-glucose 5.6, MgCl2 1, CaCl2 1.8; pH 7.4 after equilibrating with 5% CO2 (adapted from Ward & Snetkov, 2004). The PSS was continuously bubbled with a humidified air mixture containing 21% O2, 5% CO2 and 74% N2, which was switched to 2% O2, 5% CO2 and 93% N2 to create hypoxic conditions. The dissolved O2 level in the bath was monitored with an O2 meter (ISO2, World Precision Instruments, Sarasota, FL, USA), which was calibrated at the beginning of each experiment in 2% sodium dithionite (BDH chemicals, Hull, UK) in PSS for the zero point and PSS bubbled with 21% O2 for the baseline point. Hypoxia ( < 35 mmHg) was reached within 5 min of switching from 21% to 2% O2. During experiments the
< 35 mmHg) was reached within 5 min of switching from 21% to 2% O2. During experiments the  in the myograph chamber fell from 175 ± 2 mmHg (21% O2 gas) to 27 ± 2 mmHg (2% O2 gas) when a hypoxic challenge was presented. Vessels were pre-constricted with phenylephrine (PE) before applying hypoxia in order to facilitate hypoxic vasoconstriction.
in the myograph chamber fell from 175 ± 2 mmHg (21% O2 gas) to 27 ± 2 mmHg (2% O2 gas) when a hypoxic challenge was presented. Vessels were pre-constricted with phenylephrine (PE) before applying hypoxia in order to facilitate hypoxic vasoconstriction.
Patch clamp recordings
For electrophysiological experiments, cells were transferred to a recording chamber and superfused with PSS containing glibenclamide (10 μm). Only cells that looked clearly refringent and elongated under the microscope were studied. Unless specified, the ‘perforated patch’ mode of the patch clamp technique was used. The pipette filling solution contained (in mm): KCl 130, Hepes 10, EGTA 1, MgCl2 1; pH adjusted to 7.2 with KOH. Amphotericin B (0.3 mg ml−1, Sigma Aldrich) was freshly added to the solution each day (Rae et al. 1991). Voltage and current commands were generated with the Whole-Cell Analysis Program v3.6.6 (John Dempster, University of Strathclyde) through a BNC 2090 interface (National Instruments Corp.) and an Axopatch 200A amplifier (Molecular Devices, Sunnyvale, CA, USA). Cell capacitance was calculated from the area under the transient capacitative current elicited by a −10 mV step from a holding potential of −80 mV. External solutions were changed using a gravity-fed, multi-reservoir system connected to a common orifice positioned in the vicinity of the cell. Delayed rectifier K+ current (IKV) and the non-inactivating K+ current (IKN) were recorded from PASMCs as previously described (Osipenko et al. 1997, 1998; Manoury et al. 2009) with some modifications. IKV was elicited by applying a voltage step to 0 mV for 250 ms from a holding potential of –80 mV. The cell was then clamped at 0 mV for 4 to 10 min, allowing IKV to inactivate. The residual steady-state current was measured as the non-inactivating current, IKN. The current density versus voltage relationship for IKN was assessed by applying a 1.2 s voltage ramp from 60 mV to –100 mV. Resting Em measurements were performed using the current clamp (I = 0) mode. Membrane potentials were corrected by –3 mV to compensate for the liquid junction potential between the external and pipette solutions (Barry & Lynch, 1991). Experiments were conducted at room temperature (19–21°C) in normoxic conditions.
mRNA analysis
Reverse transcriptase PCR (RT-PCR) was used to assess the expression of genes encoding mouse K+ channel subunits. DNase-treated total RNA was extracted from isolated arteries using an RNAeasy minikit (Qiagen, Crawley, UK). cDNA was synthesised using reverse transcriptase (Superscipt III, Invitrogen, UK) with random hexameric oligonucleotides (Invitrogen, UK). For each sample, at least 200 ng of total RNA was used, and a reaction mix omitting Superscipt III was used as negative control. Primers were designed to amplify intron-spanning sequences using Primer 3 v.0.4.0 software (Whitehead Institute and Howard Hughes Medical Institute, http://primer3.sourceforge.net/): sequences are listed in Table 1. First-strand cDNA (1.5 μl), or its respective negative control, was used as the template in a PCR reaction also including forward and reverse primers (MWG Biotech, Ebersberg, Germany, 0.5 μm each) and 1.5 μl Hotstart Taq polymerase mastermix (Qiagen) in a reaction volume of 25 μl. The reaction mix was heated to 95°C for 15 min followed by 32 or 35 repetitions of the following sequence: denaturation (95°C, 30 s), annealing (60°C, 30 s) and extension (72°C, 30 s). PCR products were then analysed using 1% agarose gel electrophoresis. DNA was visualised with ethidium bromide and untraviolet illumination; images were recorded with a GelDoc2000 system (Bio-Rad, Hercules, CA, USA). The specificity of the reaction was confirmed by sequencing (DNA sequencing facility, University of Manchester) products of the expected size. For semi-quantification of gene expression, non saturating bands on gels loaded with 32 cycles of PCR product were imaged and quantified by densitometry using ImageJ 1.42q software (W. Rasband, NIH, USA). K+ channel subunit expression was normalised against expression of the hypoxanthine guanine phosphoribosyl transferase 1 (HPRT1) or β-actin housekeeping genes.
Table 1.
Primer pairs used for PCR (F, forward; R, reverse)
| Channel subunit | Gene | Accession number | Primer pairs | Amplicon size (bp) |
|---|---|---|---|---|
| TASK-1 | Kcnk3 | NM_010608.2 | F: 5′-CAGAATGTGCGCACGTTGGCTCTC-3′ R: 5′-TTACCGATGGTGGTGATGACGGTGAT-3′ | 275 |
| TASK-3 | Kcnk9 | NM_001033876.1 | F: 5′-GACGTGCTGAGGAACACCTACTT-3′ R: 5′-GTGTGCATTCCAGGAGGGA-3′ | 62 |
| TASK-2 | Kcnk5 | NM_021542.4 | F: 5′-ACAAGATCCTACAGGTGGTGTCTG-3′ R: 5′-GAAAGTCTGGTTCCCGGTGAT-3′ | 68 |
| TASK-5 | Kcnk15 | NM_001030292.1 | F: 5′-TCGCAGGCTCCTTCTACTTC-3′ R: 5′-ACACCGCACCAGTGTGTTTA-3′ | 176 |
| TWIK-2 | Kcnk6 | NM_001033525.3 | F: 5′-AGGCCTGGAGTTTTCTGGAT-3′ R: 5′-GAGGTACGCTGTGACAAGCA -3′ | 137 |
| Kv1.5 | Kcna5 | NM_145983.2 | F: 5′-TTATTCTTATGGCTGACGAGTGC-3′ R: 5′-AAGGCACCAATAGTACATCCCAG-3′ | 201 |
| Kv2.1 | Kcnb1 | NM_008420.3 | F: 5′-CACACAGCAATAGCGTTCAACTT-3′ R: 5′-AGGCGTAGACACAGTTCGGC-3′ | 229 |
| KCNQ-1 | Kcnq1 | NM_008434.2 | F: 5′-GCTGAGAAAGATGCGGTGAAC-3′ R: 5′-AGAAACAGGAGGCGATGGTCT-3′ | 151 |
| KCNQ-4 | Kcnq4 | NM_001081142.1 | F: 5′-GCTTACGGTGGATGATGTCA-3′ R: 5′-TGTGGTAGTCCGAGGTGATG-3′ | 449 |
| KCNQ-5 | Kcnq5 | NM_001160139.1 | F: 5′-GGCTTCGCACTCCTTGGCAT-3′ R: 5′-CACACTGGCATCCTTTCTCA -3′ | 593 |
| HPRT1 | Hprt1 | NM_013556.2 | F: 5′-TGGAAAGAATGTCTTGATTGTTGA-3′ R: 5′-ACTTCGAGAGGTCCTTTTCACC-3′ | 136 |
| β-Actin | Actb | NM_007393.3 | F: 5′-AGGTCATCACTATTGGCAACGA-3′ R: 5′-AGGATTCCATACCCAAGAAGGAA-3′ | 78 |
Drugs and chemicals
Nifedipine, levcromakalim, 4-aminopyridine (4-AP), phenylephrine (PE), serotonin (5-HT), endothelin-1 (ET-1), tetraethylammonium chloride (TEA), indomethacin, NG-nitro-l-arginine methyl ester (l-NAME) and glibenclamide were purchased from Sigma-Aldrich. Prostaglandin F2α (tromethamine salt, PGF2α) was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Treprostinil (Remodulin, United Therapeutics, Chertsey, UK) was a generous gift from Prof. Lucie Clapp (University College London). 10,10-bis (4-Pyridinylmethyl)-9(10H)-anthracenone (XE991) dihydrochloride, and 9α-(biphenylyl)methoxy-llβ-hydroxy-l2β-(N-piperidinyl)-ω-octanor-prost-4Z-enoic acid (GR32191) were from Tocris Bioscience (Bristol, UK).
PE, 5-HT, l-NAME and XE991 were dissolved in water to provide 10 mm stock solutions. ET-1 was prepared as a 100 μm stock solution in water. Nifedipine, levcromakalim, PGF2α, indomethacin and glibenclamide were prepared as 10 mm stock solutions in DMSO. A 10 mm stock of GR32191 was prepared in 96% ethanol. A 500 mm stock of 4-AP was prepared in PSS and pH adjusted to 7.3 with HCl. Stock solutions were aliquoted and stored at −20°C. All drugs were diluted to the working concentration in PSS so that the vehicle volume did not exceed 1% of the final bath volume.
Statistics
Data handling and statistical analysis were performed with Origin 7.5 (OriginLab Corp., Northampton, MA, USA) and GraphPad Instat (GraphPad Software Inc., La Jolla, CA, USA) software. Data in text and figures are represented as means ± SEM. For EC50 calculations, dose–response curves were fitted using the Hill equation. Normality of data distributions was assessed using the Shapiro–Wilk test and parametric or non-parametric statistical tests applied accordingly. Comparisons of two groups employed Student's t test or the Mann–Whitney U test. Where values obtained in a particular condition were compared to the baseline value in the same experiment, a paired t test or non-parametric Wilcoxon's matched-pairs signed-ranks test was performed. Multiple data sets were analysed using one-way analysis of variance (ANOVA) followed by a post hoc Dunnett's test or a Kruskal–Wallis test followed by a post hoc Dunn's multiple comparisons test when data were non-parametric. In all cases data samples were considered as significantly different if P < 0.05.
Results
TASK-1/3 KO mice as a tool to study the role of TASK-1 in PA
Tandem-pore domain K+ (K2P) channels in the vasculature, including PA of rat, rabbit and human, are well described (reviewed in Gurney & Manoury, 2009), but their presence in mouse PA has not been verified. Using RT-PCR, we detected robust expression of transcripts coding for TASK-1, TASK-2 and TWIK-2 subunits in pulmonary arteries of WT mice, but could not detect TASK-3 or TASK-5 mRNA expression (Fig. 1A and B). Consistent with their genotype, TASK-1/3 KO mice did not display any TASK-1 or TASK-3 mRNA expression in PA (Fig. 1A). Semi-quantification of RT-PCR products did not detect any significant difference between WT and TASK-1/3 KO animals in the expression pattern of the other K2P family members (Fig. 1C). Since the KCNQ gene family was recently suggested to play a role in the background K+ conductance of rat PASMCs (Joshi et al. 2006, 2009; Yeung et al. 2007), the expression of KCNQ-1, KCNQ-4 and KCNQ-5 was also investigated. All three transcripts were present in PA from WT and TASK-1/3 KO mice (Fig. 1B), although the KCNQ-5 signal was weak, but expression did not differ significantly between the two mouse strains (Fig. 1C). The mRNA expression of KV1.5 and KV2.1 channels was also similar in both strains (Fig. 1B and C).
Figure 1. Expression of K+ channel subunit mRNAs in pulmonary artery of WT and TASK-1/3 KO (KO) mice.

A, product from 35 PCR cycles using TASK-1 (left) or TASK-3 (right) primers, visualised with ethidium bromide. B, product from 32 PCR cycles using primers targeting K2P (top row), KCNQ (middle row) and delayed rectifier (bottom row) channel subunits in addition to β-actin, visualised with ethidium bromide. The presence (+) and absence (–) of reverse transcriptase is indicated above each lane. L50: DNA ladder 50bp; L100: DNA ladder 100bp. The expected product size is indicated to the right of each gel electrophoresis image. C, average densitometry values of the PCR products (n = 3). No statistical difference was detected between strains (t test).
Indices of pulmonary hypertension in WT and TASK-1/3 KO mice
A simple indicator of chronic elevated pulmonary arterial pressure is right ventricle hypertrophy, which develops in response to sustained right ventricle overload. Figure 2A shows that the RV:(LV + S) weight ratio in adult TASK-1/3 KO mice did not differ from that of WT mice. Remodelling of pulmonary arteries, characterised by muscularisation of the artery wall is another hallmark of pulmonary hypertension (Chin & Rubin, 2008). Lung sections from WT and TASK-1/3 KO mice did not appear obviously different and there was no significant difference between the strains in the percentage of vessels that appeared muscular (Fig. 2B).
Figure 2. Indices of pulmonary hypertension.

A, ratio of RV weight to the weight of the sum of the LV and septum (S), in WT and TASK-1/3 KO (KO) mice. B, percentage of vessels in lung sections showing muscularisation as assessed by the presence of a double elastic lamina for at least 50% of the circumference.
TASK-1 and the resting tone of mouse intrapulmonary artery
If, by maintaining the background K+ efflux and Em of mouse PASMCs, TASK-1 channel activity controls the resting vessel tone, then the loss of TASK-1 expression is predicted to lead to membrane depolarisation, bringing the resting potential closer to the opening threshold of L-type Ca2+ channels. Vessels from TASK-1/3 KO mice would consequently be more sensitive to the constrictor effects of depolarising agents, such as extracellular K+ and K+ channel blocking drugs. Figure 3Aa shows that the absolute force of constriction evoked by 50 mm KCl was slightly but significantly higher in WT than in TASK-1/3 KO mice. There was, however, no difference between the strains in the sensitivity to K+, as shown by the relationship between the amplitude of constriction and the extracellular K+ concentration (Fig. 3Ab). Similarly, we could not detect any difference between the mouse strains in the sensitivity to vasoconstriction by K+ channel blockers. TEA, which below 10 mm blocks large-conductance, Ca2+-activated K+ channels and some voltage-gated K+ channels (Walsh & Singer, 1980; Smirnov et al. 2002), had essentially no effect on at least four arteries from either mouse strain. Figure 3B shows concentration–constriction curves for 4-AP, which at ≤ 1 mm blocks voltage-gated K+ channels, and XE991, an inhibitor of KCNQ-encoded channels. 4-AP caused only a small constriction at >1 mm in vessels from both WT and TASK-1/3 KO mice. In contrast, XE991 was a potent constrictor of PA, inducing over 90% of the reference response to 50 mm KCl at 10 μm, but it displayed a similar sensitivity in WT (pEC50 = 6.38) and TASK-1/3 KO (pEC50 = 6.35) animals. Abolition of the XE991-induced constriction by 1 μm nifedipine confirmed that it was due to the opening of L-type Ca2+ channels (Fig. 3C), presumably activated by membrane depolarisation.
Figure 3. Responses of PA to K+ and K+ channel blockers in WT and TASK-1/3 KO (KO) mice.

Aa, mean force developed in response to 50 mm KCl (n = 45 vessels for WT and 34 vessels for KO). *P < 0.05 (Mann–Whitney test). Ab, mean constriction developed in the presence of 15, 35 and 55 mm K+, expressed relative to the response to 50 mm KCl. B, concentration–contraction relationships for cumulative addition of XE991 (n = 4–5) and 4-AP (n = 4) to arteries from WT and TASK-1/3 KO mice. C, typical recording of tension developed by a WT PA in response to cumulative addition of XE991, followed by relaxation upon addition of nifedipine (1 μm).
Another predicted consequence of membrane depolarisation due to TASK-1 deficiency is the development of a basal intrinsic tone, created by persistent Ca2+ influx through L-type Ca2+ channels (Manoury et al. 2009). If this occurred, PA might be expected to relax from baseline tone when voltage-gated Ca2+ influx is blocked, either directly by the Ca2+ antagonist nifedipine or indirectly by hyperpolarising the PASMC membrane away from the opening threshold of L-type Ca2+ channels. This hypothesis was tested by comparing the effects of nifedipine (1 μm) and the ATP-sensitive K+ (KATP) channel opener levcromakalim on the baseline tone of PA from WT and TASK-1/3 KO mice. Contrary to our prediction, neither nifedipine nor levcromakalim had any significant effect on the baseline tone of PA from either group of animals (Fig. 4). Nifedipine was similarly without effect in three vessels where endothelial function was disrupted (data not shown).
Figure 4. Pulmonary arteries from WT and TASK-1/3 KO (KO) mice lack intrinsic tone.

A, records of isometric tension from freshly isolated PA from WT or TASK-1/3 KO mice. Viability was tested by brief addition of 50 mm KCl before testing the effects of 15 mm K+, followed by 10 μm levcromakalim then 1 μm nifedipine (n = 3) with a wash period between each test. B, mean constriction to 10 μm levcromakalim and 1 μm nifedipine measured relative to the response to 50 mm KCl. A time control showing changes in the baseline are included for comparison. No significant effects were observed with either drug.
Pulmonary artery responses to vasoactive agents are not altered in TASK-1/3 KO mice
To determine if the loss of TASK-1 might have a more subtle effect on pulmonary artery function, responses to vasoconstrictor agonists were compared in vessels from WT and TASK-1/3 KO mice. Administration of PE, 5-HT, PGF2α or ET-1 evoked dose-dependent constriction, with a similar relationship observed in WT and TASK-1/3 KO vessels (Fig. 5A–D). The pEC50 values for PE were 6.7 ± 0.1 (n = 4) in WT arteries and 6.5 ± 0.2 (n = 5) in TASK-1/3 KO arteries. For 5-HT the pEC50 values were 6.5 ± 0.2 (n = 4) in WT and 6.5 ± 0.3 (n = 4) in TASK-1/3 KO vessels. ET-1 constricted WT arteries with pEC50 = 8.5 ± 0.2 (n = 4) compared with pEC50 = 8.4 ± 0.2 (n = 3) in TASK-1/3 KO vessels. As the response to the highest concentration of PGF2α tested did not clearly reach a maximum, its potency was estimated as the negative log of the concentration needed to reach half of the response to 50 mm KCl: values of 5.5 ± 0.1 (n = 5) and 5.4 ± 0.1 (n = 5) were obtained for WT and TASK-1/3 KO mice, respectively. For all four receptor agonists, the maximum observed constriction, measured relative to the response to 50 mm KCl, did not differ significantly between the two mouse strains (Fig. 5A–D). Moreover, the mean absolute force elicited by the highest concentration of each agonist tested did not differ between vessels from WT and TASK-1/3 KO mice (Fig. 5F).
Figure 5. Effects of vasoactive agents on PA from WT and TASK-1/3 KO (KO) mice.

Concentration–response curves for the constriction evoked by PE (A), 5-HT (B), PGF2α (C) and ET-1 (D) in arteries from WT and KO mice (n = 3–5). Inset in A shows original record from WT artery (upturned arrow indicates wash). E, constrictor responses of WT mouse and rat PA to hypoxia, measured as the percentage increase over the level of pre-constriction induced by PE (0.02–0.2 μm). F, maximum tension developed in response to PE (1 μm), 5-HT (10 μm), PGF2α (10 μm) and ET-1 (50 nM) by WT or KO arteries (n = 8–12 vessels except for PGF2α and ET-1 in KO vessels where n = 3). G, concentration–relaxation relationships of PA from WT or KO mice in response to cumulative addition of treprostinil, in the presence of GR32191 (1 μm). Vessels were pre-constricted with 6 μm PE and relaxation is expressed as the residual constriction. n = 6–7 animals.
As inhibition of TASK-1 channels has been implicated in hypoxic pulmonary vasoconstriction (Gardener et al. 2004; Olschewski et al. 2006) we investigated the sensitivity of isolated mouse PA to hypoxia. Responses of mouse PA to hypoxia were small and inconsistent and only observed after the vessels were pre-constricted with 0.02–0.2 μm PE, which elicited tension that amounted to 86 ± 11% (n = 13) of the response to 50 mm KCl. Even in these conditions only 6 out of 13 vessels displayed convincing constrictor responses. Challenging up to 3 times successively with hypoxia did not increase the response. In contrast, rat intra-pulmonary arteries studied under identical conditions showed a robust and reproducible constrictor response to hypoxia that developed within 9 ± 1 min (n = 25 challenges). Figure 5E shows that the mean response to hypoxia in rats was significantly larger than the mean response in the six mouse vessels where a constrictor response was apparent.
TASK-1 in human PASMCs has been shown to be modulated by the prostacyclin analogue, treprostinil (Olschewski et al. 2006), suggesting that it might be involved in the dilator response to this drug in PA. This was addressed by comparing cumulative dose–response curves for treprostinil, tested in pre-constricted PA (PE, 6 μm) from either WT or TASK-1/3 KO mice. Experiments were performed in the presence of the thromboxane receptor A2 antagonist GR32191 (1 μm) in order to reduce constrictor responses that could mask the relaxation (Fig. 5F). The maximum relaxation response to treprostinil was not different between WT (86 ± 5% of the PE-induced constriction, n = 6) and TASK-1/3 KO (80 ± 5%, n = 7) mice. The pEC50 values were also similar in WT (6.6 ± 0.1, n = 6) and TASK-1/3 KO (6.9 ± 0.2, n = 7) vessels.
Passive membrane properties of PASMCs from WT and TASK-1/3 KO mice
For all cells used in patch clamp experiments the mean input resistance was 6 ± 1 GΩ (n = 23) and 5 ± 1 GΩ (n = 15) for WT and TASK-1/3 KO mice, respectively. The mean membrane capacitance was 16 ± 1 pF in WT and 15 ± 1 pF in TASK-1/3 KO myocytes. This compares with a mean input resistance of 3 ± 1 GΩ (n = 5) and membrane capacitance of 13 ± 2 pF (n = 5) in rat PASMCs. Neither of these values was significantly different when compared across mouse groups or species.
The Em of isolated mouse PASMCs was measured using the ‘perforated’ configuration of the patch clamp technique in current clamp mode, with current clamped at zero. The mean values measured from WT and TASK-1/3 KO myocytes, compared in Fig. 6A, were not significantly different (t test). Using PASMCs from WT mice, we set out to characterise the K+ channels involved in regulating the resting Em. Figure 6B shows the effects on Em of conditions known to modulate particular types of K+ channel. Modification of extracellular pH did not have any significant effect on mouse Em (paired t test). Similarly 1 mm 4-AP had no significant effect and a high concentration of the KCNQ channel blocker XE991 (5 μm) also failed to significantly alter Em despite being applied for >10 min. Superfusing cells with 10 mm TEA depolarised 4 cells out of 14 (29%), with depolarisation reversible upon washout and ranging from 8 to 41 mV. In the remaining 10 cells TEA did not have any effect, but the mean Em values measured in the presence and absence of 10 mm TEA were significantly different (P < 0.01).
Figure 6. Em properties of PASMCs from WT and TASK-1/3 KO (KO) mice.

A, mean Em of PASMCs isolated from WT (n = 31) or TASK-1/3 KO (n = 13) mice. B, change in Em recorded in response to switching the pH of the extracellular solution to 6.3 (n = 8) or 8.3 (n = 6) or applying 10 mm TEA (n = 14), 1 mm 4-AP (n = 11) or 5 μm XE991 (n = 10). Individual values (circles) as well as the median value (dashes) are presented to show the distribution of the data. **P < 0.01 compared to baseline value (Wilcoxon's matched-pairs signed-ranks test).
The non-inactivating K+ current in mouse PASMCs is small and lacks pH sensitivity
The Em of PASMCs in several species has been proposed to be regulated by delayed rectifier channels and a non-inactivating background K+ current (IKN) (Evans et al. 1996; Osipenko et al. 1998; Olschewski et al. 2006), which in resting conditions drive the Em towards EK. Delayed rectifier currents (IKV) were described recently in mouse PA myocytes (Ko et al. 2007), but IKN has not been examined in these cells. We therefore set out to measure the non-inactivating conductance in mouse PASMCs in order to further address how the resting Em is regulated. IKN is defined as the residual current after IKV inactivation at 0 mV, in the presence of 10 μm glibenclamide and 10 mm TEA (Evans et al. 1996). When PASMCs from WT or TASK-1/3 KO mice were clamped at 0 mV for a prolonged period (≥5 min) in these conditions, there was little residual current and only a small current could be detected during a step to 60 mV and subsequent ramp to –100 mV (Fig. 7A). The mean amplitude of the residual current at 0 mV was similar in cells from both mouse strains and less than 0.5 pA pF-1 (Fig. 7B). In contrast, the current measured in exactly the same way from rat PASMCs was consistently robust, with mean amplitude (2.4 ± 1.5 pA pF-1, n = 5) significantly higher than in mouse (Fig. 7A and B).
Figure 7. Small and pH insensitive non-inactivating K+ current in mouse PASMCs.
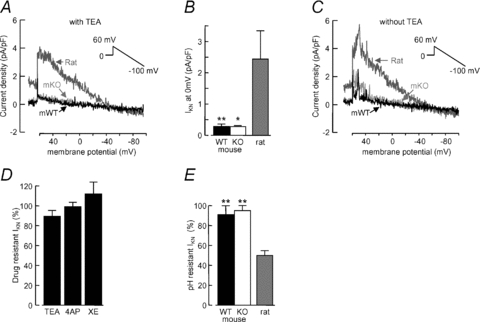
A, representative records of IKN from PASMCs bathed in PSS containing 10 μm glibenclamide and 10 mm TEA, obtained using the voltage protocol illustrated. Records shown were from a WT mouse (mWT, black trace), TASK-1/3 KO mouse (mKO, light grey trace) and rat (dark grey trace). Current amplitude is normalised to cell capacitance. B, mean IKN density at 0 mV in PASMCs isolated from WT (n = 14) or KO (n = 8) mice or from rat (n = 5). *P < 0.05,**P < 0.01 compared to rat (Kruskal–Wallis non-parametric ANOVA followed by Dunn's multiple comparisons test). C, records obtained as in A, but in the absence of TEA. D, drug-resistant IKN measured as the residual current at 0 mV following the application of 10 mm TEA (n = 6), 1 mm 4-AP (n = 6) or 5 μm XE991 (n = 5). E, pH-resistant IKN measured as the residual current at 0 mV after switching the perfusion solution to pH 6.3 PSS, in WT (n = 6) or KO (n = 6) mouse PASMCs or rat myocytes (n = 5). **P < 0.01 compared to rat (ANOVA followed by Dunnett's multiple comparisons test).
Since TEA was found to depolarise the membrane in some cells, we considered the possibility that IKN in mouse is TEA sensitive. Conversely, experiments conducted without TEA in the bath did not unmask any additional current at 0 mV, although small outward bursts were observed at very positive potentials (Fig. 7C). Indeed the mean steady-state current at 0 mV in mouse PASMCs was not significantly different in the presence or absence of TEA (Fig. 7D). Similarly, administration of 1 mm 4-AP or 5 μm XE991 did not have a significant effect on mouse IKN measured in the absence of TEA (Fig. 7D). A feature of IKN in human and rabbit PASMCs (Gurney et al. 2003; Olschewski et al. 2006) is a strong pH sensitivity, with substantial block produced by acidic pH (6.3). Consistent with this we observed a 50% reduction of IKN recorded from rat PASMCs at 0 mV in the absence of TEA (Fig. 7E). The current recorded in the same conditions from WT or TASK-1/3 KO mouse PASMCs was not, however, affected by lowering pH to 6.3 (Fig. 7E) or increasing it to 8.3 (data not shown).
Pharmacological properties of the delayed rectifier K+ current (IKV) in mouse PASMCs
The effects of K+ channel blockers were also investigated on delayed rectifier currents activated by steps to 0 mV from a holding potential of –80 mV in mouse PASMCs (Fig. 8A). The amplitude of IKV at 0 mV was similar in WT and TASK-1/3 KO myocytes (Fig. 8B) and was sensitive to inhibition by K+-channel blocking drugs. As shown in Fig. 8A and C, 10 mm TEA reduced IKV at 0 mV by 38 ± 17% (n = 9), 1 mm 4-AP caused inhibition of 33 ± 10% (n = 6) and 5 μm XE991 reduced the current by 29 ± 4% (n = 3).
Figure 8. Pharmacological properties of IKV in mouse PASMCs.

A, representative records of IKV at 0 mV from a patch-clamped WT PASMC, obtained using the voltage protocol indicated. Records are shown when cells were superfused with PSS (Control) followed by 10 mm TEA (a), 1 mm 4-AP (b) or 5 μm XE991 (c). B, mean IKV density at the end of a 250 ms depolarising step to 0 mV from a holding potential of –80 mV in WT (n = 20) or TASK-1/3 KO (KO) (n = 6) PASMCs. C, mean IK(v) density measured in WT PASMCs under control conditions (CTL) and in the presence of TEA (n = 9), 4-AP (n = 6) or XE-991 (n = 3). *P < 0.05; **P < 0.01 compared with the respective control (paired t test).
Discussion
The purpose of this study was to clarify the role of TASK-1 channels in the control of the tonic properties of pulmonary artery. Although previous studies have implicated TASK-1 in the regulation of Em in rat, rabbit and human pulmonary artery smooth muscle (Gurney et al. 2003; Gardener et al. 2004; Olschewski et al. 2006), our results do not support such a role in mouse pulmonary artery. We could not identify any difference between WT and TASK-1/3 KO mice in the physiology or pharmacology of their pulmonary arteries. The lack of effect of TASK-1 deficiency did not appear to be due to compensatory up-regulation of other K+ channels. As found in rat (Gardener et al. 2004), mouse PA expressed the K2P channel subunits TASK-2 and TWIK-2, as well as TASK-5, but they were all equally expressed in WT and TASK-1/3 KO arteries. The voltage-gated K+ channels Kv1.5 and Kv2.1, which have been implicated in the regulation of pulmonary artery function, were also expressed at similar levels in the two mouse strains. Although TASK-3 is up regulated in TASK-1 KO mice (Aller et al. 2005), mouse PA did not express this gene and use of the TASK-1/3 double knockout mouse ensured that it did not complicate our findings.
TASK-1 in the regulation of Em
Growing evidence supports a role for TASK-1 channels in regulating the Em of pulmonary artery smooth muscle. Like other K2P family members, its voltage-independent gating makes it a good candidate for carrying background conductance in cells where Em has to be kept low in order to maintain L-type Ca2+ channels closed (reviewed in Lesage & Lazdunski, 2000; Goldstein et al. 2005; Mathie, 2007; Gurney & Manoury, 2009). Our group and others established strong similarities between the biophysical and pharmacological properties of TASK channels and those of the background K+ conductance and Em of PASMCs in rabbit (TASK-1: Gurney et al. 2003), rat (TASK-2: Gonczi et al. 2006) and humans (TASK-1: Olschewski et al. 2006; Tang et al. 2009). Furthermore, as genetic validation of the importance of TASK-1, an siRNA targeted against it abrogated IKN and depolarised Em in human PASMCs (Olschewski et al. 2006; Tang et al. 2009). Similar experiments also implied a crucial role for TASK-1 and IKN in the regulation of Em in response to hypoxia (Osipenko et al. 1997; Olschewski et al. 2006), to hypertensive circulating mediators like endothelin-1 (Tang et al. 2009) and to the vasodilator treprostinil (Olschewski et al. 2006). These gene silencing experiments were, however, performed on primary cultured cells, so the importance of TASK-1 in regulating the contractile properties of the intact PA remains to be determined.
If TASK-1 contributes to the Em of PASMCs then it might be predicted that the resting potential of PASMCs from TASK-1/3 KO mice would be depolarised. In contrast, current clamp recordings confirmed that the resting potential was similar in PASMCs from WT and TASK-1/3 KO mice. Furthermore, the amplitude of IKN, proposed to be mediated by TASK-1 and to set the resting potential (Evans et al. 1996; Gurney et al. 2003; Olschewski et al. 2006), was the same in WT and TASK-1/3 KO mice, as was the amplitude of IKV. There is therefore no evidence that TASK-1 channels contribute to the background K+ conductance or resting Em in mouse pulmonary artery. The very small amplitude of IKN in mouse PASMCs was unexpected. A much larger current, with magnitude similar to previous reports (Osipenko et al. 1998; Manoury et al. 2009) was recorded from rat PASMCs under identical conditions. Moreover, a substantial non-inactivating current at 0 mV has been identified in PASMCs from all other species tested, including human (Osipenko & Gurney, 1995; Olschewski et al. 2006), rabbit (Evans et al. 1996) and pig (Evans et al. 1998). Not only was mouse IKN small, the current at 0 mV also lacked many of the distinguishing features of IKN in other species. It was unaffected by acidosis (pH 6.3), which caused substantial inhibition of rat IKN. It was also unaffected by 4-AP and XE991, both of which inhibit IKN in other species (Osipenko et al. 1997; Joshi et al. 2009).
IKN, IKV and Em
The small IKN may explain why mouse PASMCs had a resting potential close to –30 mV. A similarly depolarised resting potential was recorded from Balb/c mouse PASMCs using whole-cell recording (Ko et al. 2007), although others have reported values closer to –40 mV (Archer et al. 1999, 2001; Shimoda et al. 2001a). Comparison across these reports is complicated due to the use of diverse ionic solutions, which would have created different transmembrane gradients for K+ and Cl−, the major ions determining the Em in PASMCs (Casteels et al. 1977b). Direct comparison can, however, be made with the more polarised values of −40 to –50 mV reported in rabbit (Clapp & Gurney, 1992; Osipenko et al. 1997), pig (Evans et al. 1998) and rat (Osipenko et al. 1998; Manoury et al. 2009) PASMCs, as they were recorded under identical ionic conditions and in one case by the same experimenter. Evidence is accumulating that where the resting Em of PASMCs is depolarised, IKN is reduced or absent. For example, a depolarised Em is accompanied by inhibition or loss of IKN in response to acute (Gurney et al. 2003; Olschewski et al. 2006) or chronic (Osipenko et al. 1998) exposure to hypoxia, during organ culture of intact rat PA (Manoury et al. 2009), during the development of the piglet pulmonary circulation (Evans et al. 1998) and following siRNA targeting of TASK-1 channels in human PASMCs (Olschewski et al. 2006).
The resting potential of mouse PASMCs must be maintained by a K+ conductance, because in our experimental conditions K+ was the only ion with a negative equilibrium potential. A previous study on mouse PASMCs identified a ‘window’ current due to tonic activation of IKV, which was maximum at –31 mV and could be sufficient to generate the Em (Ko et al. 2007). Consistent with the present data, the same study found that 10 mm TEA inhibited IKV at 0 mV by about 25%. Also consistent with our data, 1 mm 4-AP was reported to inhibit mouse IKV by 25% (Archer et al. 1999), although Ko et al. (2007) required higher concentrations. We now show that 5 μm XE991 had a similar effect on IKV, yet none of these drugs consistently depolarised mouse PASMCs. Despite being statistically significant, the mean depolarisation caused by TEA was very small and the effect observed in only 4 of 14 cells. The disparity between the pharmacology of IKV and the resting potential suggests that it is not the underlying K+ current. Moreover, IKV density was comparable with PASMCs from other species, including rat (Manoury et al. 2009), rabbit (McCulloch et al. 2000), pig (Evans et al. 1998) and human (Shimoda et al. 2001b).
Although very small, IKN could still contribute to the resting potential: the high input resistance (6 GΩ) of mouse PASMCs means that 30 mV would be generated by <6 pA of current. The discrepancy between the TEA sensitivity of the Em and IKN may be reconciled by the behaviour of L-type Ca2+ channels in PASMCs, which activate with a voltage threshold between −40 and –30 mV, but are inactivated during prolonged depolarisation to 0 mV (Clapp & Gurney, 1991). Thus at the resting potential of some cells there may be sufficient Ca2+ entry through these channels to open large conductance Ca2+-activated K+ channels, which would in turn contribute to the resting potential and give rise to TEA sensitivity. In contrast, when the cell was clamped at 0 mV to record IKN, Ca2+ influx would be reduced.
Relationship between Em and artery tone
The discrepancy between the pharmacology of the Em and the vasoconstrictor effects of K+ channel blockers is less easily explained. While 4-AP was poorly effective at depolarising PASMCs and constricting arteries, TEA caused depolarisation without affecting vessel tone and XE991 caused pronounced constriction mediated by voltage-gated Ca2+ entry, but not depolarisation. One possibility is that cell dissociation or the patch-clamp recording conditions resulted in the loss of K+ channel activity, leading to PASMCs that were relatively depolarised. There was certainly no evidence that the Em of PASMCs in intact arteries was above the threshold for Ca2+ channel activation. Neither hyperpolarisation of PASMCs by levcromakalim nor direct inhibition of voltage-gated Ca2+ influx by nifedipine had any effect on the baseline tone of intact WT or TASK-1/3 KO arteries, arguing against persistent activation of L-type Ca2+ channels due to PASMC depolarisation. It is unlikely that endothelial cell activity masked a depolarisation, because there was no change in the sensitivity of vessels to nifedipine or extracellular K+ when endothelial release of nitric oxide and prostacyclin were blocked (data not shown). Although highly speculative, loss of KV7 channel activity in the isolated PASMCs could explain why XE991 failed to affect Em, but produced such a robust constriction of intact PA. KV7 channels have been proposed to contribute to IKN and the resting potential in rat PASMCs, both of which are inhibited by XE991 (Joshi et al. 2009). A potential mechanism to account for this is depletion of membrane phosphatidylinositol-4,5-bisphosphate (PIP2), which is dynamically regulated and required for KV7 channels to open (Delmas & Brown, 2005). Nevertheless, why this should occur specifically in PASMCs from mouse, but not other species is unclear. In case the differences between mouse and rat were due to different methods of cell isolation, we repeated measurements of IKN on mouse PASMCs dissociated using the rat protocol (not shown), but the amplitude of mouse IKN was similarly small in these cells.
TASK-1 is not required for normal pulmonary artery function in mouse
There was no difference between WT and TASK-1/3 KO mice in relative right ventricular weight, implying that the TASK-1/3 KO mice did not suffer from right ventricular hypertrophy. There was also no evidence of increased vessel remodelling in the TASK-1/3 KO mice. As these are well established indices of pulmonary hypertension, the results imply that the pulmonary arterial pressure of TASK-1/3 KO mice was not elevated. This agrees with the lack of intrinsic tone found in isolated arteries from both mouse strains. It also agrees with the finding that PA from WT and TASK-1/3 KO mice had the same sensitivity to the vasoconstrictors K+, PE, 5-HT, PGF2α and ET-1, despite evidence that ET-1 depolarises PASMCs by inhibiting TASK-1 channels (Tang et al. 2009). In contrast to the report that TASK-1 expression was required in human PASMCs for treprostinil to activate K+ current and cause hyperpolarisation (Olschewski et al. 2006), we also found no effect of TASK-1/3 KO on the vasodilator effect of treprostinil in intact mouse arteries. Thus these mediators are all able to modulate PA tone independently of TASK-1 channels. Iloprost, another prostacyclin analogue, has been suggested to act via KATP and large conductance Ca2+-activated K+ (BKCa) channels in rat tail arteries, probably via a cAMP-activated protein kinase-mediated mechanism (Schubert et al. 1997). BKCa and KV channels have also been implicated in the actions of ET-1 on pulmonary artery (Peng et al. 1998; Shimoda et al. 1998). Therefore it is likely that other ion channels and/or mechanisms could mediate the effects of treprostinil and ET-1 on mouse pulmonary artery.
Inhibition of TASK-1 has been proposed to contribute to hypoxic pulmonary vasoconstriction (Gurney et al. 2003; Gardener et al. 2004; Olschewski et al. 2006). Since TASK-1 KO mice were reported to show impaired carotid body function, revealed by reduced chemosensory control of breathing in response to hypoxia or hypercapnia (Trapp et al. 2008b), we hypothesised that loss of TASK-1 would similarly reduce the constrictor response of mouse PA to hypoxia. Unfortunately, the relatively small and inconsistent response of WT mouse arteries to hypoxia made such a comparison impossible. On the other hand, the lack of response is consistent with the small IKN measured in isolated cells, so perhaps it is due to the lack of TASK-1 function in mouse PA.
TASK-1 KO mice do show a cardiovascular phenotype, characterised by elevated systolic blood pressure and bradycardia, although these effects are due to hyperaldosteronism rather than a direct change in vascular function (Heitzmann et al. 2008). Interestingly, the phenotype is restricted to adult female and young mice. Although most of our study was conducted on adult males, we also compared the contractile properties of PA from females and observed no increase in intrinsic tone or sensitivity to extracellular K+ in the TASK-1/3 KO animals (data not shown).
Despite the lack of effect of TASK-1/3 KO on a range of parameters indicating PA function, TASK-1 mRNA was clearly detectable in PA from WT mice. If it is functionally expressed in mouse PASMCs, it is at a very low level and we could not find any macroscopic evidence of this. It is possible that in mouse PASMCs the message is not properly translated, or the protein is not properly trafficked to the membrane. Whatever the reason, the pulmonary arteries of mice lacking TASK-1 behave in every way like the WT vessels, indicating that TASK-1 is not an essential part of the signalling mechanisms that regulate normal pulmonary artery function.
Conclusion
We have shown that TASK-1 channels are not involved in the regulation of either the resting or the agonist-induced contractile properties of mouse PA. The responses of intact mouse PA to a range of vasoactive agents were qualitatively similar to other species, suggesting that it may not be essential in these species either. On the other hand, the data from isolated PASMCs highlight important inter-species variability in the physiology of the cells. This adds to the demonstration that mouse PA with intact endothelium did not relax to 10 μm acetylcholine, which produces pronounced vasodilatation in other species (Xu et al. 2008). Together, these findings question the use of mice as a model to investigate pulmonary vascular physiology and pathology and especially the functional roles of K+ channels.
Acknowledgments
The authors acknowledge the British Heart Foundation and the Biotechnology and Biological Sciences Research Council (BBSRC) for funding and are grateful to Prof. W. Wisden (University of Aberdeen, UK) for providing transgenic animals, and to Prof L. H. Clapp (University College London) for providing treprostinil.
Glossary
Abbreviations
- LV
left ventricle
- PA
pulmonary artery
- PASMC
pulmonary artery smooth muscle cell
- RV
right ventricle
- S
intra-ventricular septum
- SMC
smooth muscle cell
Author contributions
Experiments were performed in the laboratory of A.M.G.; B.M. and A.M.G. contributed to the concept and design of the experiments; B.M., C.L., R.O., J.R. and A.M.G. contributed to the experimental work, analysis and interpretation of data; B.M. and A.M.G. drafted and revised the article. All the authors approve the final version of the manuscript.
References
- Aller MI, Veale EL, Linden AM, Sandu C, Schwaninger M, Evans LJ, Korpi ER, Mathie A, Wisden W, Brickley SG. Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar granule neurons. J Neurosci. 2005;25:11455–11467. doi: 10.1523/JNEUROSCI.3153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, London B, Hampl V, Wu X, Nsair A, Puttagunta L, Hashimoto K, Waite RE, Michelakis ED. Impairment of hypoxic pulmonary vasoconstriction in mice lacking the voltage-gated potassium channel Kv1.5. FASEB J. 2001;15:1801–1803. doi: 10.1096/fj.00-0649fje. [DOI] [PubMed] [Google Scholar]
- Archer SL, Reeve HL, Michelakis E, Puttagunta L, Waite R, Nelson DP, Dinauer MC, Weir EK. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Natl Acad Sci U S A. 1999;96:7944–7949. doi: 10.1073/pnas.96.14.7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Wu XC, Thebaud B, Nsair A, Bonnet S, Tyrrell B, McMurtry MS, Hashimoto K, Harry G, Michelakis ED. Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells. Circ Res. 2004;95:308–318. doi: 10.1161/01.RES.0000137173.42723.fb. [DOI] [PubMed] [Google Scholar]
- Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. J Membr Biol. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- Bieger D. β-Adrenoceptor mediated responses in rat pulmonary artery: putative role of TASK-1 related K channels. Naunyn Schmiedebergs Arch Pharmacol. 2006;373:186–196. doi: 10.1007/s00210-006-0060-7. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Aller MI, Sandu C, Veale EL, Alder FG, Sambi H, Mathie A, Wisden W. TASK-3 two-pore domain potassium channels enable sustained high-frequency firing in cerebellar granule neurons. J Neurosci. 2007;27:9329–9340. doi: 10.1523/JNEUROSCI.1427-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteels R, Kitamura K, Kuriyama H, Suzuki H. Excitation–contraction coupling in the smooth muscle cells of the rabbit main pulmonary artery. J Physiol. 1977a;271:63–79. doi: 10.1113/jphysiol.1977.sp011990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteels R, Kitamura K, Kuriyama H, Suzuki H. The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol. 1977b;271:41–61. doi: 10.1113/jphysiol.1977.sp011989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:1527–1538. doi: 10.1016/j.jacc.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Choi YS, Jeong YS, Ok SH, Shin IW, Lee SH, Park JY, Hwang EM, Hah YS, Sohn JT. The direct effect of levobupivacaine in isolated rat aorta involves lipoxygenase pathway activation and endothelial nitric oxide release. Anesth Analg. 2010;110:341–349. doi: 10.1213/ANE.0b013e3181c76f52. [DOI] [PubMed] [Google Scholar]
- Clapp LH, Gurney AM. Modulation of calcium movements by nitroprusside in isolated vascular smooth muscle cells. Pflugers Arch. 1991;418:462–470. doi: 10.1007/BF00497774. [DOI] [PubMed] [Google Scholar]
- Clapp LH, Gurney AM. ATP-sensitive K+ channels regulate resting potential of pulmonary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 1992;262:H916–H920. doi: 10.1152/ajpheart.1992.262.3.H916. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Evans AM, Osipenko ON, Gurney AM. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J Physiol. 1996;496:407–420. doi: 10.1113/jphysiol.1996.sp021694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, Osipenko ON, Haworth SG, Gurney AM. Resting potentials and potassium currents during development of pulmonary artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 1998;275:H887–H899. doi: 10.1152/ajpheart.1998.275.3.H887. [DOI] [PubMed] [Google Scholar]
- Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM, Weston AH. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol. 2004;142:192–202. doi: 10.1038/sj.bjp.0705691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev. 2005;57:527–540. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- Gonczi M, Szentandrassy N, Johnson IT, Heagerty AM, Weston AH. Investigation of the role of TASK-2 channels in rat pulmonary arteries; pharmacological and functional studies following RNA interference procedures. Br J Pharmacol. 2006;147:496–505. doi: 10.1038/sj.bjp.0706649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38:305–318. doi: 10.1007/s00249-008-0326-8. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Osipenko ON, MacMillan D, Kempsill FE. Potassium channels underlying the resting potential of pulmonary artery smooth muscle cells. Clin Exp Pharmacol Physiol. 2002;29:330–333. doi: 10.1046/j.1440-1681.2002.03653.x. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, Kempsill FE. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res. 2003;93:957–964. doi: 10.1161/01.RES.0000099883.68414.61. [DOI] [PubMed] [Google Scholar]
- Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, El WA, Lalli E, Guy N, Mengual R, Reichold M, Tegtmeier I, Bendahhou S, Gomez-Sanchez CE, Aller MI, Wisden W, Weber A, Lesage F, Warth R, Barhanin J. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J. 2008;27:179–187. doi: 10.1038/sj.emboj.7601934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:31. doi: 10.1186/1465-9921-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:368–376. doi: 10.1124/jpet.108.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler CH, Yost CS, Gray AT. Local anesthetic inhibition of baseline potassium channels with two pore domains in tandem. Anesthesiology. 1999;90:1092–1102. doi: 10.1097/00000542-199904000-00024. [DOI] [PubMed] [Google Scholar]
- Ko EA, Burg ED, Platoshyn O, Msefya J, Firth AL, Yuan JX. Functional characterization of voltage-gated K+ channels in mouse pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol. 2007;293:C928–C937. doi: 10.1152/ajpcell.00101.2007. [DOI] [PubMed] [Google Scholar]
- Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279:F793−F801. doi: 10.1152/ajprenal.2000.279.5.F793. [DOI] [PubMed] [Google Scholar]
- MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, Colston J, Loughlin L, Nilsen M, Dempsie Y, Harmar A. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation. 2004;109:2150–2155. doi: 10.1161/01.CIR.0000127375.56172.92. [DOI] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lazdunski M, Honore E. The endocannabinoid anandamide is a direct and selective blocker of the background K+ channel TASK-1. EMBO J. 2001;20:47–54. doi: 10.1093/emboj/20.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoury B, Etheridge SL, Reid J, Gurney AM. Organ culture mimics the effects of hypoxia on membrane potential, K+ channels and vessel tone in pulmonary artery. Br J Pharmacol. 2009;158:848–861. doi: 10.1111/j.1476-5381.2009.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A. Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J Physiol. 2007;578:377–385. doi: 10.1113/jphysiol.2006.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch KM, Kempsill FE, Buchanan KJ, Gurney AM. Regional distribution of potassium currents in the rabbit pulmonary arterial circulation. Exp Physiol. 2000;85:487–496. [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, Kwapiszewska G, Klepetko W, Seeger W, Olschewski H. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res. 2006;98:1072–1080. doi: 10.1161/01.RES.0000219677.12988.e9. [DOI] [PubMed] [Google Scholar]
- Osipenko ON, Gurney AM. The identification of the ionic currents in human intrapulmonary artery myocytes. Br J Pharmacol. 1995;116:P406. [Google Scholar]
- Osipenko ON, Alexander D, MacLean MR, Gurney AM. Influence of chronic hypoxia on the contributions of non-inactivating and delayed rectifier K currents to the resting potential and tone of rat pulmonary artery smooth muscle. Br J Pharmacol. 1998;124:1335–1337. doi: 10.1038/sj.bjp.0702006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipenko ON, Evans AM, Gurney AM. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold, O2-sensing potassium current. Br J Pharmacol. 1997;120:1461–1470. doi: 10.1038/sj.bjp.0701075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Michael JR, Hoidal JR, Karwande SV, Farrukh IS. ET-1 modulates KCa-channel activity and arterial tension in normoxic and hypoxic human pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 1998;275:L729–L739. doi: 10.1152/ajplung.1998.275.4.L729. [DOI] [PubMed] [Google Scholar]
- Platoshyn O, Brevnova EE, Burg ED, Yu Y, Remillard CV, Yuan JX. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol. 2006;290:C907–C916. doi: 10.1152/ajpcell.00028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res. 1995;77:131–139. doi: 10.1161/01.res.77.1.131. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Randall MD, Alexander SP, Bennett T, Boyd EA, Fry JR, Gardiner SM, Kemp PA, McCulloch AI, Kendall DA. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem Biophys Res Commun. 1996;229:114–120. doi: 10.1006/bbrc.1996.1766. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Yuan JX. Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol. 2004;286:L49–L67. doi: 10.1152/ajplung.00041.2003. [DOI] [PubMed] [Google Scholar]
- Schubert R, Serebryakov VN, Mewes H, Hopp HH. Iloprost dilates rat small arteries: role of KATP- and KCa-channel activation by cAMP-dependent protein kinase. Am J Physiol Heart Circ Physiol. 1997;272:H1147–H1156. doi: 10.1152/ajpheart.1997.272.3.H1147. [DOI] [PubMed] [Google Scholar]
- Shah S. Effects of modulators of TASK potassium channels on rat pulmonary artery tone. Bioscience Horizons. 2008;1:114–121. [Google Scholar]
- Shimoda LA, Sylvester JT, Sham JS. Inhibition of voltage-gated K+ current in rat intrapulmonary arterial myocytes by endothelin-1. Am J Physiol Lung Cell Mol Physiol. 1998;274:L842–L853. doi: 10.1152/ajplung.1998.274.5.L842. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001a;281:L202–L208. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Sylvester JT, Booth GM, Shimoda TH, Meeker S, Undem BJ, Sham JS. Inhibition of voltage-gated K+ currents by endothelin-1 in human pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2001b;281:L1115–L1122. doi: 10.1152/ajplung.2001.281.5.L1115. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Beck R, Tammaro P, Ishii T, Aaronson PI. Electrophysiologically distinct smooth muscle cell subtypes in rat conduit and resistance pulmonary arteries. J Physiol. 2002;538:867–878. doi: 10.1113/jphysiol.2001.013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Li Y, Nagaraj C, Morty RE, Gabor S, Stacher E, Voswinckel R, Weissmann N, Leithner K, Olschewski H, Olschewski A. Endothelin-1 inhibits background two-pore domain channel TASK-1 in primary human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2009;41:476–483. doi: 10.1165/rcmb.2008-0412OC. [DOI] [PubMed] [Google Scholar]
- Trapp S, Aller MI, Wisden W, Gourine AV. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J Neurosci. 2008a;28:8844–8850. doi: 10.1523/JNEUROSCI.1810-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Aller MI, Wisden W, Gourine AV. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J Neurosci. 2008b;28:8844–8850. doi: 10.1523/JNEUROSCI.1810-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JV, Jr, Singer JJ. Penetration-induced hyperpolarization as evidence for Ca2+ activation of K+ conductance in isolated smooth muscle cells. Am J Physiol Cell Physiol. 1980;239:C182–C189. doi: 10.1152/ajpcell.1980.239.5.C182. [DOI] [PubMed] [Google Scholar]
- Ward JP, Snetkov VA. Determination of signaling pathways responsible for hypoxic pulmonary vasoconstriction: use of the small vessel myograph. Methods Enzymol. 2004;381:71–87. doi: 10.1016/S0076-6879(04)81004-8. [DOI] [PubMed] [Google Scholar]
- White R, Ho WS, Bottrill FE, Ford WR, Hiley CR. Mechanisms of anandamide-induced vasorelaxation in rat isolated coronary arteries. Br J Pharmacol. 2001;134:921–929. doi: 10.1038/sj.bjp.0704333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Platoshyn O, Makino A, Dillmann WH, Akassoglou K, Remillard CV, Yuan JX. Characterization of agonist-induced vasoconstriction in mouse pulmonary artery. Am J Physiol Heart Circ Physiol. 2008;294:H220–H228. doi: 10.1152/ajpheart.00968.2007. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, Greenwood IA. Molecular expression and pharmacological identification of a role for Kv7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]