

Non-technical summary
A pressure ulcer, also known as a pressure sore, bedsore or decubitus ulcer, results from localized ulcerated tissue breakdown caused by sustained, unrelieved mechanical pressure in the body–support interface such as with a bed, wheelchair and orthoses/prostheses. Pressure ulcers are common in the wheelchair bound or bedridden frail elderly patients with neuromuscular disorder and orthoses/prostheses clients. Pressure ulcers represent a significant health problem as they impose a heavy burden on sufferers, with negative psychological, physical, social and financial consequences. Most importantly, there are currently no effective therapies for preventing and treating pressure ulcers. In the present study, our data demonstrate that pharmacological inhibition of caspase is effective in alleviating muscle damage induced by prolonged moderate compression. These findings suggest that regimens targeting caspase/apoptosis inhibition might be of use to prevent or treat pressure ulcers.
Abstract
Abstract
There are currently no effective therapies for treating pressure-induced deep tissue injury. This study tested the efficacy of pharmacological inhibition of caspase in preventing muscle damage following sustained moderate compression. Adult Sprague–Dawley rats were subjected to prolonged moderate compression. Static pressure of 100 mmHg compression was applied to an area of 1.5 cm2 in the tibialis region of the right limb of the rats for 6 h each day for two consecutive days. The left uncompressed limb served as intra-animal control. Rats were randomized to receive either vehicle (DMSO) as control treatment (n = 8) or 6 mg kg−1 of caspase inhibitor (z-VAD-fmk; n = 8) prior to the 6 h compression on the two consecutive days. Muscle tissues directly underneath the compression region of the compressed limb and the same region of control limb were harvested after the compression procedure. Histological examination and biochemical/molecular measurement of apoptosis and autophagy were performed. Caspase inhibition was effective in alleviating the compression-induced pathohistology of muscle. The increases in caspase-3 protease activity, TUNEL index, apoptotic DNA fragmentation and pro-apoptotic factors (Bax, p53 and EndoG) and the decreases in anti-apoptotic factors (XIAP and HSP70) observed in compressed muscle of DMSO-treated animals were not found in animals treated with caspase inhibitor. The mRNA content of autophagic factors (Beclin-1, Atg5 and Atg12) and the protein content of LC3, FoxO3 and phospho-FoxO3 that were down-regulated in compressed muscle of DMSO-treated animals were all maintained at their basal level in the caspase inhibitor treated animals. Our data provide evidence that caspase inhibition attenuates compression-induced muscle apoptosis and maintains the basal autophagy level. These findings demonstrate that pharmacological inhibition of caspase/apoptosis is effective in alleviating muscle damage as induced by prolonged compression.
Introduction
Pressure ulceration refers to localized damage to the soft tissues as a result of sustained pressure. The occurrence of pressure ulcers is common in people who are subjected to sustained mechanical compression resulting from diminished mobility, particularly in the wheelchair bound or bedridden frail elderly and patients with spinal cord injury. It has been reported that the prevalence of pressure ulcers is high, ranging from 10% to 23%, in hospitalized patients in westernized societies such as the United States and the Netherlands (NPUAP, 2001; Bours et al. 2002). Pressure ulcers impose a heavy burden on patients, with negative psychological, physical, social and financial consequences (Gorecki et al. 2009).
Currently existing strategies including clinical risk assessment, regular repositioning, pressure-relieving devices and wound debridement only delay the development of the ulcer lesions, and as yet no interventions have been found to be effective in treating or preventing the onset of pressure ulcers. This is partly attributed to the limited understanding of the molecular aetiology of this disorder. In general, pressure ulcers are classified into superficial and deep ulcers. Superficial ulcers are confined to the skin and can be diagnosed at an early stage whereas deep pressure ulcers originate in the underlying soft tissues in the compressed region. The development of deep ulcers includes subsequent progression of ulceration upwards until it penetrates to the skin (Gawlitta et al. 2007). Deep ulcers are of great clinical importance as the damage is already very severe by the time the ulceration becomes detectable at the skin surface. The term ‘pressure-induced deep tissue injury’ has been used by the US National Pressure Ulcer Advisory Panel (NPUAP) to emphasize that deep pressure ulcers occur as ‘a pressure-related injury to subcutaneous tissues under intact skin’ (Ankrom et al. 2005; Black et al. 2007).
Muscle tissue has been demonstrated to be particularly susceptible to external compression when compared to skin (Linder-Ganz & Gefen, 2004; Linder-Ganz et al. 2006; Stekelenburg et al. 2006). The association between compressive tissue straining and muscle cell death has been shown using an in vitro engineered muscle model (Breuls et al. 2003; Wang et al. 2005). Cell death was found to occur within 1–2 h at a gross 30% and 50% compressive muscle straining, which resulted in the deformation of individual muscle cells to an elliptical form (Breuls et al. 2003). A mouse skeletal myoblast model also showed the damaging effect of compressive strain from prolonged pressure and revealed that the proportion of apoptotic cells was increased following hours of compressive straining (Wang et al. 2005). Data from our laboratory have substantiated the in vitro observation by demonstrating that apoptosis was activated in the presence of pathohistological damage in muscle in an in vivo animal model of pressure-induced deep tissue injury (Siu et al. 2009). Significant increases in apoptotic muscle-related nuclei and DNA fragmentation, elevation of caspases mRNA content, and positive immunoreactivity of cleaved caspase-3 and Bax were exhibited in muscle following prolonged moderate compression. This suggested that activation of muscle apoptosis is associated with the pathogenesis of pressure-induced deep tissue injury (Siu et al. 2009). More recent data have demonstrated that apoptotic and autophagic changes preceded pathohistological changes in muscle in response to prolonged moderate compression (Teng et al. 2011). These results suggested that apoptosis and autophagy are early, and possibly causative, events in compression-induced muscle pathology, and led us to hypothesize that the apoptotic cell death machinery could be a potential target for exploring therapeutic intervention in compression-induced muscle disorders. Hence, this study aimed to investigate the potential beneficial effects of pharmacological inhibition of caspase, an essential effector of apoptosis, on muscle damage following sustained moderate compression.
Methods
Animals
Adult male Sprague–Dawley rats aged ∼2.5 months (body weight 260–280 g; n = 16) were used in this study. They were housed in pathogen-free conditions at an ambient temperature of ∼20 °C in The Hong Kong Polytechnic University Centralized Animal Facilities and were exposed to a light-controlled environment with a 12:12 h light–dark cycle each day. All rats were fed with standard nutrient diet and water ad libitum throughout the study period. All procedures involving animal care and handling were conducted in accordance with institutional guidelines using protocols approved by the Animal Subjects Ethics Sub-committee of The Hong Kong Polytechnic University. Our procedures are in compliance with the policies of The Journal of Physiology (Drummond, 2009) and relevant UK regulations.
Administration of caspase inhibitor and induction of pressure-induced deep tissue injury
Rats were randomized to receive either dimethyl sulphoxide (DMSO) (vehicle control; n = 8) or caspase inhibitor z-VAD-fmk (Z-Val-Ala-dl-Asp-fluoromethylketone in DMSO, Bachem; n = 8) to a cumulative dose of 6 mg kg−1. The sufficiency of the dosage of 6 mg kg−1 of z-VAD-fmk was previously reported (Petillot et al. 2007). The z-VAD-fmk or vehicle administration was carried out by two intravenous injections of 3 mg kg−1 of z-VAD-fmk (or the same volume of DMSO), each given immediately prior to the compression procedure on the two consecutive days (as described later). All rats were subjected to a pressure ulcer regimen as induced by moderate compression (Kwan et al. 2007; Siu et al. 2009; Teng et al. 2011). This experimental animal model was adopted because it has been shown to mimic the pathology of pressure-induced deep tissue injury without causing any morphological damage to the skin layer (Kwan et al. 2007). The rats were first anaesthetized by single injection of a mixture of ketamine (100 mg (kg body weight)−1, Alfasan) and xylazine (10 mg (kg body weight)−1, Alfasan). Administration of anaesthetic drugs was performed by intraperitoneal (i.p.) injection. Rats were maintained under surgical anaesthesia throughout the experimental period by ensuring the loss of the pedal withdrawal reflex. The sustainability of the anaesthetic level throughout the compression process was also determined by whisker movement. One-third of the initial dose of anaesthetic drugs was i.p. administered approximately one to two times throughout the compression procedure to maintain the level of surgical anaesthesia. Hairs of the compression site were gently shaved immediately before compression. Static pressure of 100 mmHg (13.3 kPa equivalent) was applied to an area of 1.5 cm2 over the tibialis region of the right limb of the rats (see Supplemental Data Fig. 1). The compression force was continuously monitored by a three-axial force transducer equipped in the compression indentor. The blood flow at the compression site was monitored using a laser Doppler flowmeter (DRT4, Moor Instruments, Devon, UK) as previously described (Kwan et al. 2007). The left uncompressed limb served as intra-animal control. The compression duration was 6 h on each of two consecutive days. Rats were allowed to recover from each of the two periods of anaesthesia and it was observed that ambulatory activity was restored ∼30 min after each of the compression procedure. Rats were killed 1 day after the last bout of compression by an overdose of a ketamine–xylazine mixture. Muscle tissues directly underneath the compression region, including the major hindlimb muscle groups, i.e. gastrocnemius, plantaris, soleus and tibialis anterior, in the compressed limb and the same region of control limb were harvested. Tissues were quickly frozen in liquid nitrogen-cooled isopentane and stored at −80 °C until further use.
To examine the recovery of muscle following prolonged moderate compression, three rats were exposed to the compression procedure without injection of z-VAD-fmk or DMSO. Animals were then housed in individual cages and were killed 9 days after the last bout of compression. Muscles were harvested and histological analysis was carried out (see Supplemental Data Fig. 2).
Histological analysis
Haematoxylin and eosin staining was used to demonstrate the histology of the compressed and intra-animal control muscle tissues. Ten micrometre thick frozen muscle cross-sections were cut perpendicular to the skin and in the direction of compression in a cryostat at −20 °C, then air dried and fixed with 10% formalin (HT-5011, Sigma-Aldrich) at room temperature for 10 min before counter-staining in Mayer's haematoxylin (MHS-1, Sigma-Aldrich) and 1% eosin in CaCl2 (318906, Sigma-Aldrich). The numbers of nuclei in the interstitial space and within myofibres were counted. Nuclei were counted in three randomly chosen image fields captured with a 20 × objective and the average number of nuclei was presented. The number of nuclei within myofibres was normalized to the number of myofibres in the field. Fibre area was estimated by outlining myofibres using ImageJ software. The interstitial area was approximated by subtracting the total muscle fibre area from the square field area. In addition, muscle fibre diameter was determined by measuring the major and minor axis of all the individual fibres in the fields with the reference of the scale bar.
TUNEL and dystrophin staining analysis
Nuclei with apoptotic DNA strand breaks in muscle tissues were assessed by double immunofluorescent labelling of terminal dUTP nick-end labelling (TUNEL In Situ Cell Death Detection Kit, Roche Applied Science) and dystrophin by following the previously described procedure (Siu et al. 2009). Anti-dystrophin mouse monoclonal antibody (D8168, Sigma-Aldrich) was used at 1:500 dilution. The sections were mounted with DAPI mounting medium (Vectashield Mounting Medium, Vector Laboratories, Burlingame, CA, USA) to visualize the nuclei. TUNEL- and DAPI-stained nuclei and dystrophin staining were examined under a fluorescence microscope (Biological Research Microscope 80i, Nikon) equipped with a digital camera (DXM 1200C, Nikon). Data were expressed as the TUNEL index, which was quantified by counting the number of TUNEL-positive nuclei divided by the total number of nuclei (i.e. DAPI-positive nuclei) multiplied by 100. The TUNEL index for each muscle section was estimated in four random, non-overlapping fields at an objective magnification of 20 ×.
Triple immunocytochemical analysis
Ten micrometre thick frozen muscle cross-sections were air dried at room temperature and fixed with cold acetone. Background activity was minimized by blocking the sections with 5% goat serum and 5% horse serum. After washing, sections were incubated with anti-granulocyte mouse monoclonal (554905, BD Pharmingen), anti-CD31 mouse monoclonal (MCA1334G, Serotec, Kidlington, UK), anti-CD68/ED1 mouse monoclonal (MCA341R, Serotec) or anti-CD163/ED2 mouse monoclonal (MCA342R, Serotec) antibodies at 1:500 dilutions and at 4 °C overnight. Sections were then exposed to anti-mouse IgG (H+L) fluorescein-conjugated secondary antibody (1:300 dilutions, FI-2000, Vector Laboratories) for 1 h at room temperature. Negative controls were performed by eliminating the primary or secondary antibody. Tissue sections were then labelled with laminin to visualize the basal lamina by incubating with anti-laminin rabbit polyclonal antibody (1:500 dilution, L9393, Sigma-Aldrich) at 4 °C overnight followed by an anti-rabbit IgG Cy3 conjugate F(ab′)2 fragment incubation (1:300 dilution, C2306, Sigma-Aldrich). The sections were then mounted with DAPI mounting medium (Vector Laboratories) to visualize the nuclei. The sections were examined under a fluorescence microscope (Biological Research Microscope 80i, Nikon) equipped with a digital camera (DXM 1200C, Nikon).
Subcellular protein fractionation
The cytoplasmic and nuclear protein fractions of muscle tissues were extracted following the protocol as previously described (Siu & Alway, 2005c, 2006; Siu et al. 2006; Teng et al. 2011). The protein concentration was then assayed in duplicate spectrophotometrically at 595 nm (Benchmark Plus, Bio-Rad Laboratories, Hercules, CA, USA) using a commercial Bradford method (Coomassie Protein Assay, Pierce Biotechnology, Inc., Rockford, IL, USA) with bovine serum albumin used to construct a standard curve. Purity of protein fractions was assessed by immunobloting the fractions with an anti-lamin B1 (a nuclear protein), an anti-β-tubulin (a cytoplasmic protein) and an anti-CuZnSOD (a cytosolic isoform of superoxide dismutase) antibodies (Fig. 1).
Figure 1. Verification of extracted protein fraction purity.

The purity of the extracted protein fractions was examined by immunoblotting cytoplasmic protein and nuclear protein fractions with anti-CuZnSOD, anti-β-tubulin and anti-lamin B1 antibodies. The data showed that the cytoplasmic and nuclear fractions were not contaminated by other protein fractions.
Apoptotic cell death ELISA
DNA fragmentation in the cytoplasmic protein extract was examined using a commercially available ELISA kit (Cell Death Detection ELISA, Roche Diagnostics) by following the previously described procedure (Siu et al. 2009). The resulting optical density (OD) measured was normalized to the milligram of protein used in each reaction and presented as the apoptotic DNA fragmentation index (OD405 (mg protein)−1).
Fluorometric caspase-3 activity assay
The protease activity of caspase-3 was measured in the cytoplasmic fractions by a fluorometric assay as previously described (Siu & Alway, 2005b, 2006). Caspase inhibitor, z-VAD-fmk (550377, BD Pharmingen) was used to check for caspase-specific activity of the assay. The change in the fluorescence intensity after 2 h of incubation was measured on a microplate reader (Infinite F200, Tecan, Mannedorf, Switzerland) with an excitation wavelength of 405 ± 10 nm and an emission wavelength of 500 ± 25 nm. The enzymatic activity of caspase-3 in cytoplasmic fractions was presented as the change in arbitrary fluorescence intensity normalized to milligram of protein used.
Western immunoblotting
Protein expression of apoptotic factors (Bax, Bcl-2, phospho-Bcl-2, XIAP, p53, phospho-p53, AIF, EndoG), cellular stress markers (CuZnSOD, MnSOD, HSP70), and autophagic factors (LC3-I, LC3-II and FoxO3) was measured in the compressed and intra-animal control muscles. Bax, Bcl-2, phospho-Bcl-2, XIAP, p53, phospho-p53, AIF, EndoG, CuZnSOD, MnSOD, HSP70, LC3-I, LC3-II, FoxO3 and phospho-FoxO3 were determined in the cytoplasmic protein fraction whereas AIF, EndoG and FoxO3 were determined in the nuclear protein fraction. Protein extracts were boiled at 95 °C for 5 min in Laemmli buffer with 5%β-mercaptoethanol. Forty micrograms of protein was loaded on 7.5% (for FoxO3), 10% (for Bax, Bcl-2, phospho-Bcl-2, XIAP, p53, phospho-p53, AIF, EndoG, CuZnSOD, MnSOD and HSP70) or 15% (for LC3) polyacrylamide gel. After electrophoretic separation by SDS-PAGE, the proteins were transferred to PVDF membranes (Immobilon P, Millipore). Equal loading and transfer efficiency were verified by staining gels with Coomassie blue and the membranes with Ponceau S red. After the transfer, the membranes were blocked in 5% non-fat milk (for Bax, Bcl-2, XIAP, p53, AIF, EndoG, CuZnSOD, MnSOD, HSP70, LC3-I, LC3-II and FoxO3) or 5% BSA (for phospho-Bcl-2, phospho-p53 and phospho-FoxO3) in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h at room temperature and incubated overnight at 4 °C with the corresponding primary antibody (Table 1) diluted in TBST with 2% BSA. Membranes were then washed in TBST three times and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h (1:3000 dilution, 7076 for anti-mouse IgG antibody, 7074 for anti-rabbit IgG antibody, Cell Signaling Technology, Inc., Danvers, MA, USA; and sc2020 for anti-goat IgG antibody, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Finally, the luminol reagent (NEL103001EA, Perkin Elmer) for chemiluminescence detection of HRP was applied. The chemiluminescence signal was captured with a Kodak 4000R Pro camera. The resulting bands were quantified as optical density (OD) × band area and expressed as arbitrary units. β-Tubulin (1:2000 dilution, T0198, Sigma) and lamin B1 (1:500 dilution, sc 56145, Santa Cruz) were probed and used as the internal control proteins. Data were expressed by normalizing to the corresponding internal control. The reason for choosing β-tubulin and lamin B1 as the internal control proteins is that they are abundantly expressed as indispensable structural proteins (β-tubulin for microtubule and lamin B1 for nuclear lamin) in cytoplasmic and nuclear subcellular compartments, respectively. The protein content of β-tubulin and lamin B1 was examined by densitometric analysis and no significant difference in β-tubulin and lamin B1 was found between groups. This supported that β-tubulin and lamin B1 were suitable to be used as internal controls in this study.
Table 1.
Primary antibodies used in western blot
| Antibody | Dilution factor | Source |
|---|---|---|
| Bax rabbit polyclonal | 1:500 | sc493, Santa Cruz |
| Bcl-2 mouse monoclonal | 1:500 | sc7382, Santa Cruz |
| phospho-Bcl-2 rabbit polyclonal | 1:1000 | ab73985, Abcam |
| XIAP mouse monoclonal | 1:2000 | 610762, BD Transduction Laboratories |
| p53 mouse monoclonal | 1:1000 | 2524, Cell Signaling Technology |
| phospho-p53 rabbit polyclonal | 1:1000 | 9284, Cell Signaling Technology |
| AIF mouse monoclonal | 1:500 | sc13116, Santa Cruz |
| EndoG rabbit polyclonal | 1:500 | 3035, ProSci |
| CuZnSOD rabbit polyclonal | 1:200 | sc11407, Santa Cruz |
| MnSOD goat monoclonal | 1:1000 | A300–449A, Bethyl Laboratories |
| HSP70 mouse monoclonal | 1:1000 | SPA-810, Assay Designs |
| LC3 rabbit monoclonal | 1:1000 | 3868, Cell Signaling Technology |
| FoxO3 rabbit polyclonal | 1:1000 | 07–702, Millipore |
| Phospho-FoxO3 rabbit polyclonal | 1:1000 | 9466, Cell Signaling Technology |
Quantitative real time RT-PCR analysis
Total RNA was extracted from the control and compressed muscles of DMSO- and z-VAD-fmk-treated animals with TriReagent (Molecular Research, Cincinnati, OH, USA), which is based on the guanidine thiocyanate method. Frozen muscle was minced and homogenized on ice in 1 ml of ice-cold TriReagent. Total RNA was solubilized in RNase-free H2O and was quantified in duplicate by measuring the optical density (OD) at 260 nm. Purity of RNA was assured by examining the OD260/OD280 ratio. Reverse transcription was then performed with RevertAid First Strand cDNA Synthesis kit (Fermentas, Glen Bernie, USA) to generate cDNA according to the manufacturer's recommendations. Quantitative PCR (qPCR) was then performed in SYBR green/ROX qPCR Master Mix (Fermentas), 0.2 μm of forward and reverse primers (Table 2), 1 μl of cDNA on the ABI7500 real time PCR thermocycler (Applied Biosystems, Foster City, CA, USA). PCR amplification was optimized and the threshold for kinetic detection was set to occur over linear amplifications. Relative standard curve (concentration vs. threshold cycle) of target and reference genes for quantification of PCR products was generated by dilution of cDNA from the calibrator. Complementary DNA prepared from the pooled uncompressed and compressed samples was used to make the dilution series and used as the calibrator to generate the standard curve. The mRNA content of β2-microglobulin (β2M) was analysed and no significant difference was found between groups. This supported that β2M was suitable to be adopted as an internal housekeeping control gene in the present study. All samples were run in duplicate. Data are expressed as the relative mRNA expression normalized to the housekeeping gene.
Table 2.
Primers used in real time RT-PCR analysis
| Gene | Accession number | Primers (F; forward primer, R; reverse primer) |
|---|---|---|
| Beclin-1 | NM_053739 | F: 5′CGCCTCCTATTCCATCAAAA3′ |
| R: 5′AACTGTGAGGACACCCAAGC3′ | ||
| Atg5 | AM087012 | F: 5′AGGCTCAGTGGAGGCAACAG3′ |
| R: 5′CCCTATCTCCCATGGAATCTTCT3′ | ||
| Atg7 | NM_001012097 | F: 5′GCAGCCAGCAAGCGAAAG3′ |
| R: 5′TCTCATGACAACAAAGGTGTCAAA3′ | ||
| Atg12 | NM_001038495 | F: 5′GGCCTCGGAGCAGTTGTTTA3′ |
| R: 5′CAGCATCAAAACTTCTCTGA3′ | ||
| p53 | NM_030989 | F: 5′GCGTTGCTCTGATGGTGA3′ |
| R: 5′CAGCGTGATGATGGTAAGGA3′ | ||
| β2M | NM_012512 | F: 5′CGTGATCTTTCTGGTGCTTGTC3′ |
| R: 5′TTCTGAATGGCAAGCACGAC3′ |
Statistical analyses
All data are expressed as means ± standard error of the mean (SEM). Statistical analyses were performed using the SPSS 15.0 software package (SPSS Inc., Chicago, IL, USA). Two-way (2 × 2) ANOVA was performed to examine the main effects of compression, caspase inhibitor, and interaction (compression × caspase inhibitor) on the measured variables. ANOVA followed by Tukey's HSD post hoc test was used to examine the significant differences between groups. The analysis of fibre cross-sectional area distribution was performed by using Pearson's χ2 test. Statistical significance was accepted at P < 0.05.
Results
Histological analysis
In contrast to the myofibres in control uncompressed limbs of DMSO- and z-VAD-fmk-treated rats, which in general presented with an angular shape and were closely aligned to each other (Fig. 2A), compressed muscle of the DMSO group was observed to be loosely packed with massive nuclei accumulation in the interstitial space (Fig. 2A). However, these morphological degenerative characteristics were generally absent in the compressed muscle of the z-VAD-fmk-treated group. The total number of nuclei in the compressed muscle was significantly increased by 42% (P < 0.05), when compared to the intra-animal control tissues (uncompressed), in DMSO-treated animals, but the nuclei number was not significantly different (P > 0.05) between the compressed and control muscles in the z-VAD-fmk-treated group (Fig. 2B). The number of nuclei in the interstitial space of compressed muscle was significantly increased by 8.8-fold and 2.9-fold (P < 0.05) relative to the corresponding control muscle in DMSO and z-VAD-fmk groups, respectively (Fig. 2B). The number of nuclei within myofibres normalized to the number of myofibres was not found to be significantly different between compressed and control muscles in either group (data not shown). A significant effect of compression on the interstitial area was found between compressed and control tissues in the DMSO group (Fig. 2C): there was a 2.1-fold increase (P < 0.05) in the interstitial area of compressed muscle of the DMSO group but no significant difference was found between the compressed and control muscles of the z-VAD-fmk group. Muscle fibre size was approximated by measuring the major and minor axes and the fibre area. The data showed that there was no significant change in the major and minor axes of the muscle fibre between compressed and control muscles of either the DMSO or the z-VAD-fmk group (P > 0.05) (Fig. 2D). There was also no significant change of fibre area between compressed and control muscles in either group (P > 0.05) (Fig. 2E). The distribution of fibre area was not significantly different among all muscles (P > 0.05) (Fig. 2F).
Figure 2. Histological analysis.
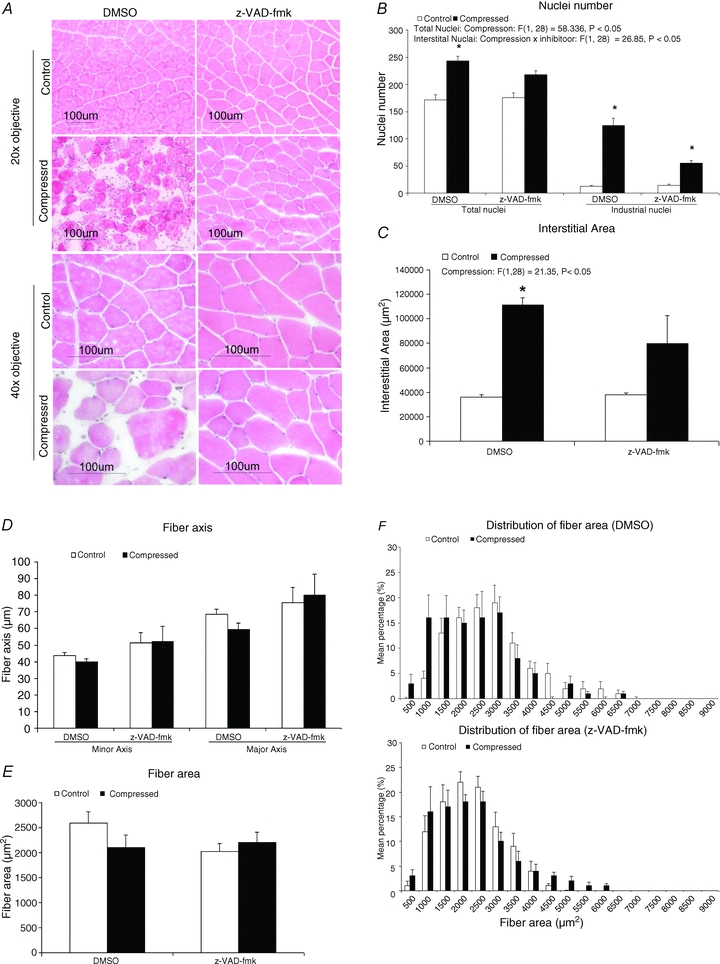
A, compressed muscles of DMSO- and z-VAD-fmk-treated animals are shown on the lower panel whereas the corresponding intra-animal control muscles are shown on the upper panel. Images were taken using objective of 20× or 40×. All control muscles of DMSO and z-VAD-fmk animals demonstrated angular shape and the myofibres were tightly packed. Compressed muscles of the DMSO group demonstrated pathohistological characteristics of degeneration including rounded myofibres and massive nuclei aggregation in interstitial space. On the other hand, these pathohistological characteristics were not found in compressed muscle of z-VAD-fmk group. B–F, numbers of total and interstitial nuclei (B), interstitial area (C), fibre axis (D), fibre area (E), and distribution of fibre area (F) are presented. Data are shown as means ± SEM. *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression × inhibitor) in these animals were analysed using a two-way 2 × 2 ANOVA.
Infiltrate identity
Sections were probed with neutrophil, macrophage and capillary markers to pinpoint the identity of infiltrates in the compressed muscle of the DMSO group. Increased immunoreactivities of neutrophil and ED1 and ED2 macrophage staining were observed in the compressed muscle of the DMSO but not the z-VAD-fmk group (Fig. 3A–C). The immunoreactivity of CD31, an endothelial marker to identify capillaries, was observed to be apparently lowered in DMSO compressed muscle when compared to all the other groups (Fig. 3D).
Figure 3. Infiltrate identity.
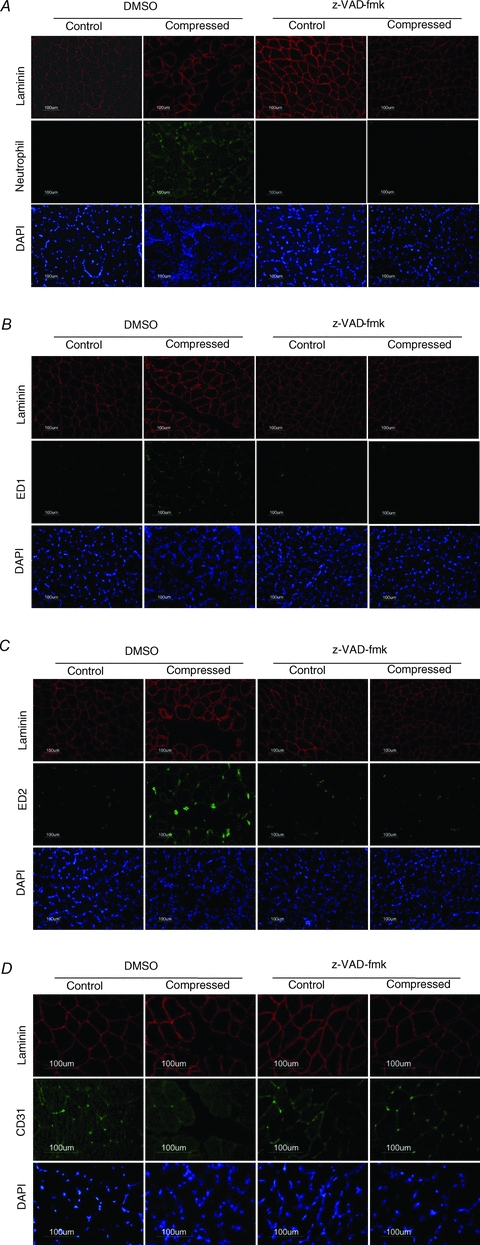
Immunocytochemical analysis was performed to evaluate the immunoreactivities of neutrophil (A), ED1 (B), ED2 (C) and CD31 (D) in the control and compressed muscles of DMSO and z-VAD-fmk rats. Compressed muscle of DMSO rats, but not z-VAD-fmk rats, showed abundant immunopositive staining of neutrophil, ED1 and ED2. The immunoreactivity of CD31 (an endothelial marker to identify capillaries) was observed to be apparently lowered in DMSO compressed muscle when compared to all the other groups (D). Images of neutrophil, ED1 and ED2 were taken using objective of 20 × whereas that of CD31 were taken using objective of 40 ×.
Molecular marker of apoptosis
There was a 1.4-fold increase in the caspase-3 protease activity in the compressed muscle, relative to the control muscle, in the DMSO group, but this increase was not found in the z-VAD-fmk group (Fig. 4A). DNA fragmentation analysis demonstrated a 16-fold elevation of the extent of apoptotic DNA fragmentation in the compressed muscle, relative to the control tissue, in the DMSO group (P < 0.05) (Fig. 4B). The TUNEL/dystrophin analysis indicated that there was a 35-fold increase (P < 0.05) in TUNEL-positive nuclei in compressed muscle, when compared to the control tissues in the DMSO group, while in contrast significant increase in TUNEL-positive nuclei was not found in the compressed muscle of the z-VAD-fmk group (Fig. 4C and D).
Figure 4. Molecular marker of apoptosis.
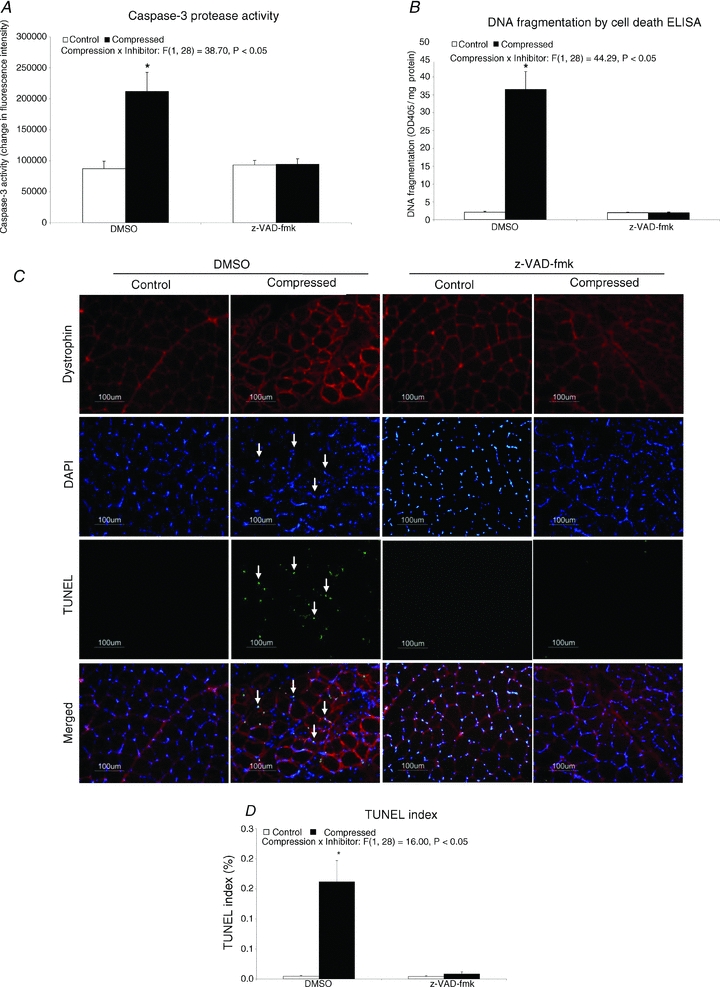
Caspase-3 protease activity is presented as a 2 h change of fluorescence intensity normalized to the total mg protein used in the assay (A). Apoptotic DNA fragmentation was measured and presented as optical density (OD) at 405 nm normalized to the total milligrams of protein used in the assay (B). Apoptotic DNA breaks in muscle tissue were determined by TUNEL staining (C) and are expressed as the TUNEL index (D). Data are presented as means ± SEM. *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression × inhibitor) in these animals were analysed using a 2 × 2 ANOVA.
Apoptotic regulatory factor: Bax, Bcl-2 and phospho-Bcl-2
Bax was found to be increased by 2.3-fold (P < 0.05) in the compressed muscle of the DMSO group, but this increase was not seen in the z-VAD-fmk group (Fig. 5A). The protein level of Bcl-2 was significantly increased by 1.2-fold (P < 0.05) in the compressed muscle relative to control muscle in the DMSO group but not in the z-VAD-fmk group (Fig. 5B). No significant difference was found in the expression ratio of Bax to Bcl-2 between the compressed and control muscles in either of the groups (P > 0.05) (Fig. 5D). Compressed muscle in the DMSO group, but not in the z-VAD-fmk group, showed a significant 60% decrease in the protein content of phosphorylated Bcl-2 (P < 0.05) (Fig. 5C).
Figure 5. Apoptotic regulatory factor: Bax, Bcl-2 and phospho-Bcl-2.
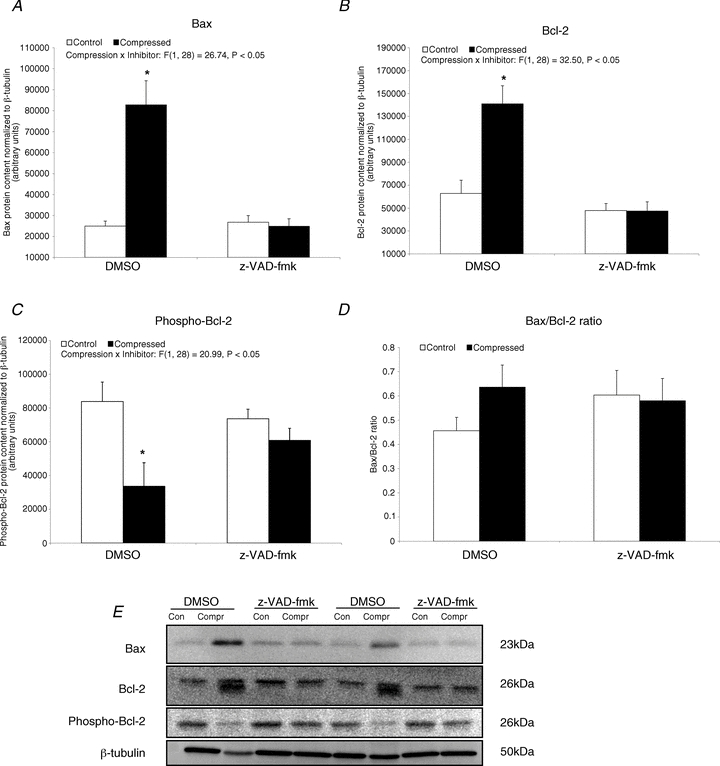
A–D, protein abundances of Bax (A), Bcl-2 (B) and phospho-Bcl-2 (C) in the cytoplasmic fraction were determined by Western blotting. The protein content ratio of Bax to Bcl-2 is presented (D). Data are presented as net intensity × resulting band area and expressed in arbitrary units. Results of Bax, Bcl-2 and phospho-Bcl-2 were normalized to corresponding β-tubulin signal. Data are presented as means ± SEM with significance set at *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression × inhibitor) in these animals were analysed using a 2 × 2 ANOVA. E, two sets of representative blots are shown. Con, control; Compr, compressed.
Apoptotic regulatory factor: XIAP, p53 and phospho-p53
Our immunoblot analysis indicated that there was a significant 62% decrease (P < 0.05) in XIAP protein content in the compressed muscle when compared to the control muscle in the DMSO group (Fig. 6A). In the DMSO-treated animals, protein expression levels of p53 and phospho-p53 were significantly increased by 21.5-fold and 15-fold, respectively, in the compressed muscle, relative to the intra-animal control tissues (P < 0.05) (Fig. 6B and C). The mRNA content of p53 was significantly increased by 1.6-fold (P < 0.05) in the compressed muscle when compared to control muscle in the DMSO group (Fig. 6D). In contrast, no significant differences in protein contents of XIAP, p53 and phospho-p53 and mRNA content of p53 were found between compressed and control muscles in the z-VAD-fmk group (P > 0.05).
Figure 6. Apoptotic regulatory factor: XIAP, p53 and phospho-p53.
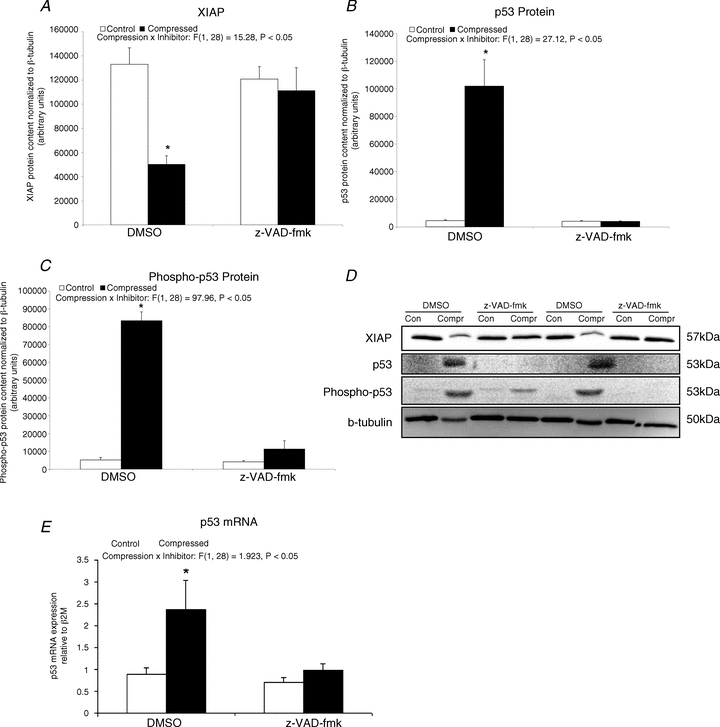
A–C, protein abundances of XIAP (A), p53 (B) and phospho-p53 (C) in the cytoplasmic fraction were determined by Western blotting. Data are presented as net intensity × resulting band area and expressed in arbitrary units. Results of XIAP, p53 and phospho-p53 were normalized to corresponding β-tubulin signal. D, two sets of representative blots are shown. Con, control; Compr, compressed. E, the mRNA content of p53 was examined by quantatitive RT-PCR. Data are presented as means ± SEM with significant levels set at *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression x inhibitor) in these animals were analysed using a 2 × 2 ANOVA.
Apoptotic caspase-independent factor: AIF and EndoG
Cytoplasmic AIF was found to be significantly down-regulated by 52% (P < 0.05) in the compressed muscle of DMSO-treated animals, but not in the z-VAD-fmk group (Fig. 7A). No significant difference in nuclear AIF was found between compressed and control muscles in either the DMSO or the z-VAD-fmk group (P > 0.05) (Fig. 7B). There was a significant 49.6-fold (P < 0.05) upregulation of cytoplasmic EndoG in the compressed muscle relative to the control muscle in the DMSO group (Fig. 7C). No detectable band of EndoG was observed in the extracted nuclear protein fractions of muscles from either of the groups.
Figure 7. Apoptotic caspase-independent factor: AIF and EndoG.
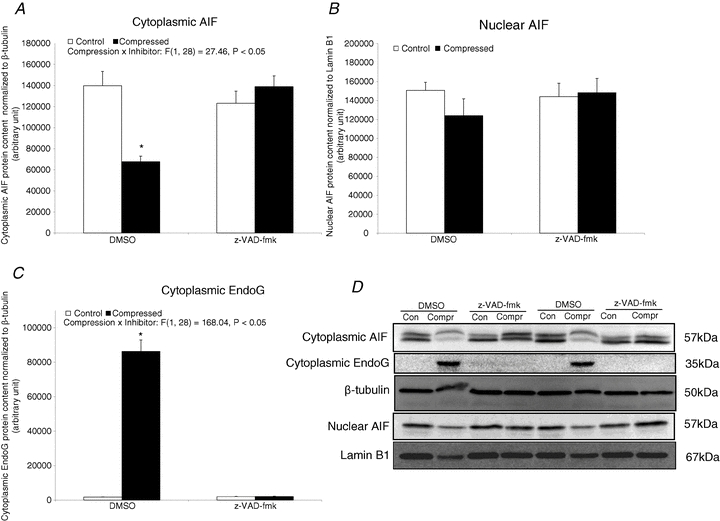
Protein abundances of AIF and EndoG were determined by Western blotting. Data are net intensity × resulting band area and expressed in arbitrary units. A–C, results of cytoplasmic AIF (A) and EndoG (C) were normalized to corresponding β-tubulin signal whereas nuclear AIF (B) were normalized to corresponding lamin B1 signal. D, two sets of representative blots are shown. Con, control; Compr, compressed. Data are presented as means ± SEM with significant levels set at *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression x inhibitor) in these animals were analysed using a 2 × 2 ANOVA.
Cellular stress protein: CuZnSOD, MnSOD and HSP70
No significant change of CuZnSOD protein content was observed between compressed and control muscles in both DMSO and z-VAD-fmk groups (P > 0.05) (Fig. 8A). There was a 97% decrease of MnSOD protein level in the compressed muscle of the DMSO group, but no significant change was found between the compressed and control muscles in the z-VAD-fmk group (Fig. 8B). Our immunoblot analysis also indicated that there was a 98% and 97% decrease of HSP70 in the compressed muscle of DMSO and z-VAD-fmk groups, respectively, relative to the corresponding controls (Fig. 8C).
Figure 8. Cellular stress protein: CuZnSOD, MnSOD and HSP70.
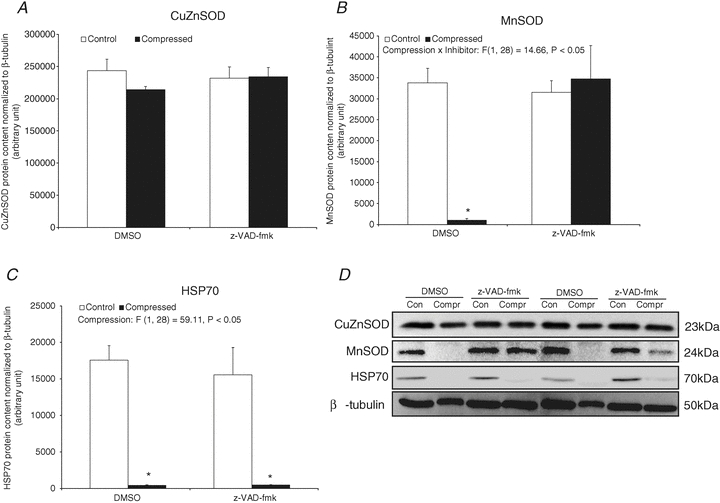
A–C, protein abundances of CuZnSOD (A), MnSOD (B), and HSP70 (C) in cytoplasmic fraction were determined by Western blotting. Data are presented as net intensity × resulting band area and expressed in arbitrary units. Results of CuZnSOD, MnSOD and HSP70 were normalized to corresponding β-tubulin signal. D, two sets of representative blots are shown. Con, control; Compr, compressed. Data are presented as means ± SEM with significant levels set at *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression × inhibitor) in these animals were analysed using a 2 × 2 ANOVA.
Autophagic regulatory factor: Beclin-1, Atg5, Atg7 and Atg12 mRNA expression
DMSO-treated animals showed that the mRNA contents of Beclin-1, Atg5 and Atg12 were significantly decreased by 75%, 55% and 72%, respectively (P < 0.05), in the compressed muscle relative to the control muscle (Fig. 9A, B and D). None of these significant changes were found in compressed muscle of animals treated with z-VAD-fmk. The mRNA content of Atg7 was not found to be significantly different between the compressed and control muscles of either the DMSO or the z-VAD-fmk group (Fig. 9C).
Figure 9. Autophagic regulatory factor: Beclin-1, Atg5, Atg7 and Atg12 mRNA expression.
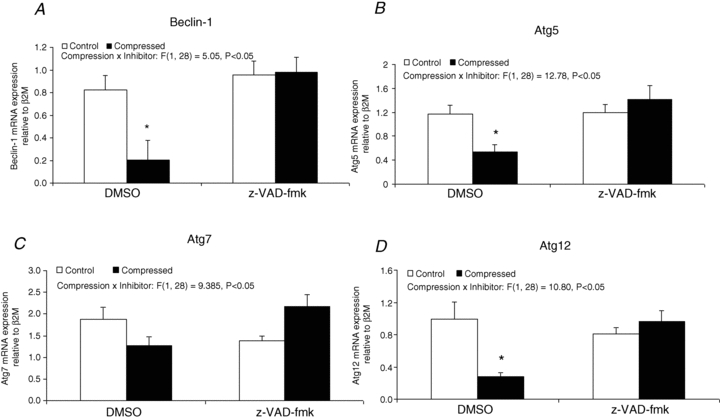
The gene expression of Beclin-1 (A), Atg5 (B), Atg7 (C), and Atg12 (D) were examined by real-time RT-PCR analysis. Results are expressed as relative mRNA level as normalized to β2M, the housekeeping gene. Data are presented as means ± SEM with significant levels set at *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression x inhibitor) in these animals were analysed using a 2 × 2 ANOVA.
Autophagic regulatory factor: LC3-I, LC3-II, FoxO3 and phospho-FoxO3 protein expression
Protein abundance of LC3-I, LC3-II, FoxO3 and phospho-FoxO3 was found to be significantly decreased in the compressed muscle relative to the intra-animal control muscle of the DMSO group (decreased by 55% for LC3-I, 82% for LC3-II, 50% for cytoplasmic FoxO3, 75% for nuclear FoxO3 and 75% for phospho-FoxO3) (Fig. 10). The ratio of LC3-II to LC3-I was not significantly different between compressed and control muscles in either the DMSO or the z-VAD-fmk group (P > 0.05) (Fig. 10C). FoxO3 protein in either nuclear or cytoplasmic fractions and the phospho-FoxO3 content were not found to be significantly different between compressed and control muscles in the z-VAD-fmk group (P > 0.05). No detectable band of phospho-FoxO3 was observed in any of the groups of muscles in our extracted nuclear protein fractions.
Figure 10. Autophagic regulatory factor: LC3-I, LC3-II, FoxO3 and phospho-FoxO3 protein expression.
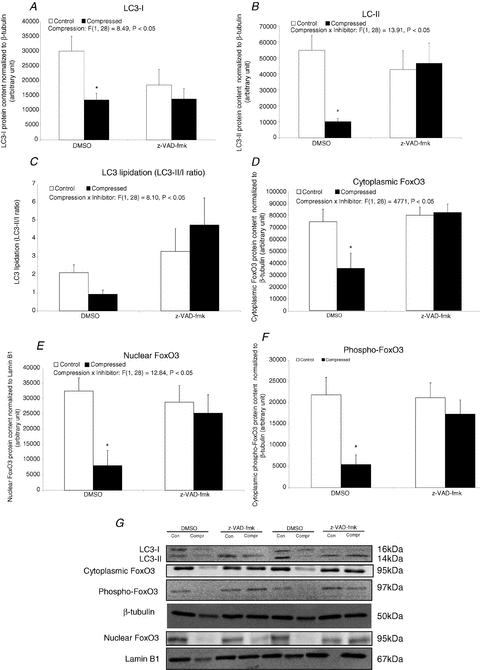
A–F, protein abundances of LC3-I (A), LC3-II (B), FoxO3 (D and E), and phospho-FoxO3 (F) were determined by Western blotting. LC3-I and LC3-II were measured in cytoplasmic fraction whereas FoxO3 and phospho-FoxO3 were determined in both cytoplasmic and nuclear fraction. LC3 lipidation is shown as the ratio of LC3-II/LC3-I (C). Data are net intensity × resulting band area and expressed in arbitrary units. Results of LC3-I and LC3-II, cytoplasmic FoxO3 and phospho-FoxO3 were normalized to corresponding β-tubulin signal whereas nuclear FoxO3 was normalized to corresponding Lamin B1 signal. G, two sets of representative blots are shown. Con, control; Compr, compressed. Data are presented as means ± SEM with significant levels set at *P < 0.05, compressed muscle compared to the corresponding control muscle in DMSO and z-VAD-fmk groups. The main effects of compression, inhibitor and interaction (compression × inhibitor) in these animals were analysed using a 2 × 2 ANOVA.
Discussion
There is a need to explore new and effective therapeutic regimens in alleviating pressure-induced deep tissue injury. In this study, we demonstrated the beneficial effect of z-VAD-fmk, a pharmacological broad spectrum caspase inhibitor, on the development of muscle pathology as induced by prolonged, moderate compression. Our data showed that z-VAD-fmk precluded the compression-induced increases in caspase-3 protease activity, TUNEL index, DNA fragmentation, activated inflammatory response and upregulation of pro-apoptotic regulatory factors including Bax, p53 and EndoG. Decreases in anti-apoptotic factors (XIAP and HSP70) that were observed in the compressed muscle of DMSO group were apparently absent in the compressed muscle of z-VAD-fmk-treated animals. Additionally, the mRNA abundance of autophagic regulatory factors (Beclin-1, Atg5 and Atg12) and the protein content of LC3, FoxO3 and phospho-FoxO3 which were found to be downregulated in compressed muscle of DMSO-treated animals were maintained at their basal level in the muscle of z-VAD-fmk-treated animals subsequent to prolonged compression. Increased immunoreactivities of markers for neutrophils and macrophages (ED1 and ED2) that were observed in the compressed muscle of DMSO animals were not found in z-VAD-fmk-treated animals. Overall, these results suggest that muscle pathohistology as induced by prolonged, moderate compression was inhibited by z-VAD-fmk treatment. Our results also indicated that the effect of caspase inhibition involved the modulation of the corresponding apoptotic and autophagic signals.
Broad spectrum caspase inhibitor suppressed caspase activation and muscle apoptosis
Caspase inhibition using z-VAD-fmk has been reported to exert its therapeutic effect in endotoxin-induced (Fauvel et al. 2001; Neviere et al. 2001; Lancel et al. 2005) or lipopolysaccharide-induced (Petillot et al. 2007) myocardial dysfunction and myocardial ischaemia–reperfusion injury (Yaoita et al. 1998) by attenuating cardiomyocyte apoptosis. In mouse skeletal muscle, caspase inhibition using a genetic approach has been shown to protect against denervation-induced muscle atrophy by impairing the downstream apoptotic signalling (Plant et al. 2009). Although the nature of the myopathic condition in pressure-induced deep tissue injury is different from the aforementioned cardiac and skeletal muscle disorders, our present findings indicated that the myopathohistology as induced by prolonged compression can be prevented by z-VAD-fmk treatment by suppressing the apoptotic signalling pathway. This suggests that the caspase-inhibiting drugs might have a potential practical application in the prevention or treatment of pressure-induced deep tissue injury and the efficacy of these drugs in clinical muscle pathology is worth further examination. Nonetheless, the pharmacological inhibition of apoptosis using a pan-caspase inhibitor, PR-03491390 (formerly named IDN-6556), has been documented in a human clinical trial in patients undergoing liver transplantation and with chronic hepatitis C (Baskin-Bey et al. 2007; Shiffman et al. 2010). Baskin-Bey and co-workers have shown that the pan-caspase inhibitor (IDN-6556), when administered in cold storage and flush solutions, provided local therapeutic protection against apoptosis and liver injury as induced by the surgical operational procedure of cold ischaemia–warm reperfusion during liver transplantation (Baskin-Bey et al. 2007). Additionally, Shiffman et al. (2010) have reported that pan-caspase inhibitor (PF-03491390) significantly decreased hepatocellular injury as indicated by the reduction of serum aspartate aminotransferase and alanine aminotransferase levels in patients with chronic hepatitis C. Shiffman et al. (2010) also reported that PF-03491390 was well tolerated over the 12-week experimental period and did not exert any significant adverse events in the patients. In general, there seems to be a great clinical potential for a pharmacological caspase inhibitor for treating muscle disorders and other apoptosis-related diseases. However, it is worth noting that the side-effects of caspase inhibitor, if any, on muscle and other tissues should be carefully considered in future trials.
In the present study, abundances of XIAP and HSP70 were found to be down-regulated concomitant with the elevation of the caspase-3 enzymatic activity in muscle following compression. These changes indicated that the XIAP-mediated caspase-dependent modulating mechanism might have been involved in the activated apoptotic signalling in the compressed muscle. It is noted that the protein expression level of some of the proteins examined in this study (e.g. XIAP and HSP70) was determined by immunobloting performed in the extracted enriched protein fraction (i.e. cytoplasmic or nuclear fractions or both). Nevertheless, our interpretation was based on the measurement in the specified subcellular protein fraction and it was not certain that the cytoplasmic fraction did not contain any of the target proteins examined in the nuclear fraction and vice versa. The role of caspases in the apoptotic signalling is well established but it has been suggested that apoptosis can be executed also by AIF and EndoG, without the involvement of caspases (Cande et al. 2002; Jaattela, 2002). Mitochondrial caspase-independent apoptotic signalling via EndoG recruitment and its nuclear translocation in rat muscle has been demonstrated in ageing and hindlimb suspension experimental conditions (Dupont-Versteegden et al. 2006; Marzetti et al. 2008). The striking elevation of EndoG protein in the cytoplasm of our examined compressed muscle suggests that the caspase-independent machinery might have contributed to the activated apoptotic signal transduction during compression. However, evidence of nuclear translocation of EndoG was not observed in this study. Therefore, it is still unclear if the increase in cytoplasmic EndoG alone is sufficient in apoptotic signalling in the absence of nuclear translocation in muscle compression and the exact role of EndoG in promoting the compression-induced apoptosis in skeletal muscle is not completely known. AIF has also been shown to mediate the caspase-independent apoptotic signalling in ageing, denervation and hindlimb suspension-mediated muscle wasting (Siu & Alway, 2005b, 2006; Siu et al. 2005a; Marzetti et al. 2008). However, in our study, a decreased level of cytoplasmic AIF protein was demonstrated concomitant with the activation of apoptosis in the compressed muscle. This finding is perhaps not surprising since the role of AIF either as a mediator of apoptosis or as a survival factor has recently been raised. It was reported by van Empel and coworkers that myocardium of the AIF-deficient mouse mutant, Harlequin, was more sensitive to ischemia–reperfusion damage and oxidative stress-related cell death, implying an important role of AIF in cardiac cell survival (van Empel et al. 2005). The putative dual role of AIF in determining the fate of a cell remains to be elucidated.
Anti-apoptotic Bcl-2 protein was found to be increased in the compressed muscle of DMSO-treated animals, consistent with our previously reported Bcl-2 response to sustained compression (Siu et al. 2009) and in other muscle wasting models such as denervation (Siu & Alway, 2005b) and ageing (Braga et al. 2008). We interpreted the elevation of Bcl-2 in the compressed muscle as an adaptive mechanism which attempted to counteract or decelerate the accelerating pro-apoptotic stimulus as induced by sustained compression. In cardiac myocytes, Bcl-2 downregulation and an increase in its phosphorylation was correlated with cardiac myocyte apoptosis after myocardial infarction (Qin et al. 2005), ischemia–reperfusion injury (Lazou et al. 2006) and H2O2-induced oxidative stress (Markou et al. 2009). These authors stipulated that cardiac apoptosis was provoked by serine phosphorylation of Bcl-2, leading to its inactivation and its disability to form dimers with Bax and, therefore, the subsequent activation of the mitochondria-dependent death pathway. Thus, the loss of the anti-apoptotic function of Bcl-2 through its serine phosphorylation suggests that phosphorylation of Bcl-2 may have a pro-apoptotic role. Phosphorylation of Bcl-2 at S70 has also been shown to be associated with the activation of caspase-9-mediated apoptotic signalling in skeletal muscle of aged mice (Braga et al. 2008). However, our findings appeared to be contradictory with the observations in cardiac muscle. In our study, Bcl-2 was not found to be phosphorylated at serine 87 (S87) in compressed muscle of DMSO-treated animals, although markers of apoptosis were generally upregulated. It is possible that the survival function of Bcl-2 could be regulated by either mono- or simultaneous multi-site phosphorylation at threonine 69 (T69), as well as S70 and S87 (Deng et al. 2004). Alternatively, we interpreted that the survival function of Bcl-2 was actuated by the observed decrease of phosphorylated Bcl-2 that hindered the inactivation of Bcl-2. The definitive role of phosphorylation in regulating the anti-apoptotic function of Bcl-2 in compressed-induced muscle damage requires further study. Nonetheless, the anti-apoptotic adaptation as demonstrated by increased Bcl-2 and the decrease in its phosphorylation did not appear to affect the elevated apoptotic cell death in the muscle following sustained compression according to our TUNEL and DNA fragmentation data.
Tumour suppressor p53 has been reported to contribute to the modulation of apoptotic signalling in the muscle of aged rat (Tamilselvan et al. 2007), unloading-induced muscle atrophy in quail muscle (Siu & Alway, 2005a; Siu et al. 2005b), denervation in rat muscle (Siu & Alway, 2005b) and rat muscles in response to ischaemia–reperfusion injury (Hatoko et al. 2002) and endurance exercise (Saleem et al. 2009). In this study, p53 protein was significantly elevated in parallel with the increase in Bax in the compressed muscle of the DMSO group, probably through the regulation of p53 phosphorylation at S15, which was also shown to be markedly upregulated in the compressed muscle. Consistent with these observations, doxorubicin-induced apoptosis in H9c2 rat cardiac myoblasts was observed to be modulated by the upregulation of p53, phospho-p53 (S15) and Bax (Chua et al. 2006; Choi et al. 2008). Evidence suggested that p53 was phosphorylated at serine 15 after DNA damage and that this phosphorylation impaired the ability of p53 to interact with its negative protein regulator, MDM2, which prevented the degradation of p53 and in turn stimulated the transcription of pro-apoptotic genes including Bax (Shieh et al. 1997). Regulatory factors that were shown to have roles in oxidative stress-related cell death including CuZnSOD and MnSOD were examined. Protein content of CuZnSOD protein was not altered whereas MnSOD protein was downregulated by compression. This is similar to the findings of previous studies showing no changes in CuZnSOD protein level in muscle of hypertensive rats (Quadrilatero & Rush, 2008) and in denervated muscle (Siu & Alway, 2005b). Similarly, diminished MnSOD was demonstrated in muscle following hindlimb suspension (Siu et al. 2008) and denervation-induced muscle wasting (Siu & Alway, 2006). We interpreted this finding as due to, at least in part, a disruption or augmentation of oxidative balance in the compressed muscle. The possible role of oxidative stress in regulating the signalling mechanisms that mediate compression-induced muscle damage is a worthwhile area of future research.
Caspase inhibition maintained compression-induced muscle autophagy at basal level
Our previous work has documented the response of autophagic factors to moderate compression by showing down-regulation of Beclin-1, FoxO3, LC3-I and LC3-II proteins in muscle following compression (Teng et al. 2011). This decreasing autophagic response to compression was further confirmed by the present demonstration of the diminished mRNA levels of Beclin-1, Atg5 and Atg12. Although these changes observed at the mRNA level may not be necessarily correlated with the protein expression levels of Beclin-1, Atg5 and Atg12, the down-regulation of autophagic signalling was in agreement with the shown reduction of protein content of FoxO3, LC3-I and LC3-II in the compressed muscle. Autophagy can be either lethal or cytoprotective depending on the cellular context. In support, studies conducted by Hara et al. (2006) showed that deletion of a crucial autophagy gene Atg5 specifically in mice neural cells resulted in a progressive deficit in motor function that was accompanied by abnormal intracellular protein accumulation. Knockout of cardiac-specific Atg5 in adulthood has also been shown to result in cardiac hypertrophy and contractile dysfunction accompanied by abnormal mitochondrial aggregation (Nakai et al. 2007). Muscle-specific Atg5 and Atg7 knockout mice have been demonstrated to result in considerable muscle atrophy, decrease in force production and induction of morphological features of myopathy including the presence of protein aggregates and abnormal mitochondria (Raben et al. 2008; Masiero et al. 2009; Masiero & Sandri, 2010). All these reports highlighted the essential role of basal level of autophagy in the maintenance of cellular survival of myocytes. Protein degradation by autophagy generates free acids and amino acids that can be reused by the cell to maintain ATP energy production and protein synthesis and therefore promotes cell survival in maintaining basal homeostatic level (Levine & Yuan, 2005). From the current context, a basal level of autophagy was interpreted to be important for maintaining cellular metabolism depending on the experimental settings. Based on this, the observation of the maintenance of basal autophagy as a result of pharmacological caspase inhibition would likely be beneficial in our experimental setting.
The basal autophagy that was restored by inhibition of caspases further substantiated the recent findings indicating the crosstalk between apoptosis and autophagy (Vandenabeele et al. 2006). In a recent report by Cho and colleagues, Beclin-1/Atg6 was undetectable in tumour necrosis factor related apoptosis-inducing ligand (TRAIL)-treated HeLa cells and it was restored by z-VAD-fmk treatment (Cho et al. 2009). This indicates that Beclin-1 might be cleaved in a caspase-dependent manner, thereby destroying its pro-autophagic activity, in a process inhibited by pan-caspase inhibitor, suggesting that Beclin-1 is a novel substrate for caspase (Cho et al. 2009). Also, knockdown of Atg6 was shown to further sensitize cells to TRAIL-induced cell death (Cho et al. 2009). This prompts the idea that Atg6 has a protective effect by suppressing cell death, at least in TRAIL-treated cells. Additionally, recent evidence also showed that Atg5 could function as a switch and that its cleavage by calpain increases pro-apoptotic signalling, turning the process of cell death from autophagy to apoptosis (Yousefi et al. 2006). These results are in line with our data demonstrating that Beclin-1 and Atg5 were downregulated in apoptotic muscles and that this decrease was prevented by pan-caspase inhibitor treatment. Our data are in support of a connection between the autophagic signalling pathway and cellular apoptosis in muscle cells. We speculate that Beclin-1 and Atg5 could be the elements interconnecting apoptosis and autophagy in muscle undergoing prolonged compression, though their mechanistic role in this model has yet to be fully elucidated.
In conclusion, our results demonstrated that caspase inhibition attenuated compression-induced muscle apoptosis and maintained basal autophagy during compression muscle injury. Our data provide evidence that pharmacological caspase inhibition might be of potential therapeutic value in alleviating muscle damage as induced by prolonged compression by modulating the corresponding apoptotic and autophagic signals. These findings provide the basis for additional research on drug or other intervention development targeting caspase/apoptosis in compression-induced muscle disorders such as pressure-induced deep tissue injury.
Acknowledgments
This study was supported by the General Research Fund (PolyU 5632/10M) from the Hong Kong Research Grants Council, Hong Kong SAR and The Hong Kong Polytechnic University Research Fund A-PH69 and G-U645. The authors gratefully acknowledged the critical reading of S. W. Choi.
Glossary
Abbreviations
- AFC
7-amino-4-trifluoromethyl coumarin
- AIF
apoptosis inducing factor
- Atg
autophagy
- EndoG
endonuclease G
- FoxO3
forkhead box O3
- HSP
heat shock protein
- LC3
microtubule-associated protein 1 light chain 3
- SOD
superoxide dismutase
- XIAP
X-linked inhibitor of apoptosis protein
- z-VAD-fmk
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone
Author contributions
Conception and design of experiments: B.T.T., E.W.T. and P.M.S. Collection, data analysis and interpretation of data: B.T.T. and P.M.S. Drafting the article or revising it critically for important intellectual content: B.T.T., I.F.B. and P.M.S. All authors approved the final version for publication.
Supplementary material
Figure 1
Figure 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Ankrom MA, Bennett RG, Sprigle S, Langemo D, Black JM, Berlowitz DR, Lyder CH. Pressure-related deep tissue injury under intact skin and the current pressure ulcer staging systems. Adv Skin Wound Care. 2005;18:35–42. doi: 10.1097/00129334-200501000-00016. [DOI] [PubMed] [Google Scholar]
- Baskin-Bey ES, Washburn K, Feng S, Oltersdorf T, Shapiro D, Huyghe M, Burgart L, Garrity-Park M, van Vilsteren FG, Oliver LK, Rosen CB, Gores GJ. Clinical trial of the pan-caspase inhibitor, IDN-6556, in human liver preservation injury. Am J Transplant. 2007;7:218–225. doi: 10.1111/j.1600-6143.2006.01595.x. [DOI] [PubMed] [Google Scholar]
- Black J, Baharestani MM, Cuddigan J, Dorner B, Edsberg L, Langemo D, Posthauer ME, Ratliff C, Taler G. National Pressure Ulcer Advisory Panel's updated pressure ulcer staging system. Adv Skin Wound Care. 2007;20:269–274. doi: 10.1097/01.ASW.0000269314.23015.e9. [DOI] [PubMed] [Google Scholar]
- Bours GJ, Halfens RJ, Abu-Saad HH, Grol RT. Prevalence, prevention, and treatment of pressure ulcers: descriptive study in 89 institutions in the Netherlands. Res Nurs Health. 2002;25:99–110. doi: 10.1002/nur.10025. [DOI] [PubMed] [Google Scholar]
- Braga M, Sinha Hikim AP, Datta S, Ferrini MG, Brown D, Kovacheva EL, Gonzalez-Cadavid NF, Sinha-Hikim I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–832. doi: 10.1007/s10495-008-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuls RG, Bouten CV, Oomens CW, Bader DL, Baaijens FP. Compression induced cell damage in engineered muscle tissue: an in vitro model to study pressure ulcer aetiology. Ann Biomed Eng. 2003;31:1357–1364. doi: 10.1114/1.1624602. [DOI] [PubMed] [Google Scholar]
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- Cho DH, Jo YK, Hwang JJ, Lee YM, Roh SA, Kim JC. Caspase-mediated cleavage of ATG6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett. 2009;274:95–100. doi: 10.1016/j.canlet.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Seon MR, Lim SS, Kim JS, Chun HS, Park JHY. Hexane/ethanol extract of glycyrrhiza uralensis licorice suppresses doxorubicin-induced apoptosis in H9c2 rat cardiac myoblasts. Exp Biol Med. 2008;233:1554–1560. doi: 10.3181/0807-RM-221. [DOI] [PubMed] [Google Scholar]
- Chua CC, Liu XW, Gao JP, Hamdy RC, Chua BHL. Multiple actions of pifithrin-α on doxorubicin-induced apoptosis in rat myoblastic H9c2 cells. Am J Physiol Heart Circ Physiol. 2006;290:H2606–H2613. doi: 10.1152/ajpheart.01138.2005. [DOI] [PubMed] [Google Scholar]
- Deng XM, Gao FQ, Flagg T, May WS. Mono- and multisite phosphorylation enhances Bcl2's antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A. 2004;101:153–158. doi: 10.1073/pnas.2533920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont-Versteegden EE, Strotman BA, Gurley CM, Gaddy D, Knox M, Fluckey JD, Peterson CA. Nuclear translocation of EndoG at the initiation of disuse muscle atrophy and apoptosis is specific to myonuclei. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1730–R1740. doi: 10.1152/ajpregu.00176.2006. [DOI] [PubMed] [Google Scholar]
- Fauvel H, Marchetti P, Chopin C, Formstecher P, Neviere R. Differential effects of caspase inhibitors on endotoxin-induced myocardial dysfunction and heart apoptosis. Am J Physiol Heart Circ Physiol. 2001;280:H1608–1614. doi: 10.1152/ajpheart.2001.280.4.H1608. [DOI] [PubMed] [Google Scholar]
- Gawlitta D, Li W, Oomens CW, Baaijens FP, Bader DL, Bouten CV. The relative contributions of compression and hypoxia to development of muscle tissue damage: an in vitro study. Ann Biomed Eng. 2007;35:273–284. doi: 10.1007/s10439-006-9222-5. [DOI] [PubMed] [Google Scholar]
- Gorecki C, Brown JM, Nelson EA, Briggs M, Schoonhoven L, Dealey C, Defloor T, Nixon J. Impact of pressure ulcers on quality of life in older patients: a systematic review. J Am Geriatr Soc. 2009;57:1175–1183. doi: 10.1111/j.1532-5415.2009.02307.x. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hatoko M, Tanaka A, Kuwahara M, Yurugi S, Iioka H, Niitsuma K. Difference of molecular response to ischemia-reperfusion of rat skeletal muscle as a function of ischemic time: Study of the expression of p53, p21(WAF-1), Bax protein, and apoptosis. Ann Plast Surg. 2002;48:68–74. doi: 10.1097/00000637-200201000-00010. [DOI] [PubMed] [Google Scholar]
- Jaattela M. Programmed cell death: many ways for cells to die decently. Ann Med. 2002;34:480–488. doi: 10.1080/078538902321012423. [DOI] [PubMed] [Google Scholar]
- Kwan MP, Tam EW, Lo SC, Leung MC, Lau RY. The time effect of pressure on tissue viability: investigation using an experimental rat model. Exp Biol Med (Maywood) 2007;232:481–487. [PubMed] [Google Scholar]
- Lancel S, Joulin O, Favory R, Goossens JF, Kluza J, Chopin C, Formstecher P, Marchetti P, Neviere R. Ventricular myocyte caspases are directly responsible for endotoxin-induced cardiac dysfunction. Circulation. 2005;111:2596–2604. doi: 10.1161/CIRCULATIONAHA.104.490979. [DOI] [PubMed] [Google Scholar]
- Lazou A, Iliodromitis EK, Cieslak D, Voskarides K, Mousikos S, Bofilis E, Kremastinos DT. Ischemic but not mechanical preconditioning attenuates ischemia/reperfusion induced myocardial apoptosis in anaesthetized rabbits: The role of Bcl-2 family proteins and ERK1/2. Apoptosis. 2006;11:2195–2204. doi: 10.1007/s10495-006-0292-5. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan JY. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder-Ganz E, Engelberg S, Scheinowitz M, Gefen A. Pressure-time cell death threshold for albino rat skeletal muscles as related to pressure sore biomechanics. J Biomech. 2006;39:2725–2732. doi: 10.1016/j.jbiomech.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Linder-Ganz E, Gefen A. Mechanical compression-induced pressure sores in rat hindlimb: muscle stiffness, histology, and computational models. J Appl Physiol. 2004;96:2034–2049. doi: 10.1152/japplphysiol.00888.2003. [DOI] [PubMed] [Google Scholar]
- Markou T, Dowling AA, Kelly T, Lazou A. Regulation of Bcl-2 phosphorylation in response to oxidative stress in cardiac myocytes. Free Radic Res. 2009;43:809–816. doi: 10.1080/10715760903071649. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech Ageing Dev. 2008;129:542–549. doi: 10.1016/j.mad.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010;6:307–309. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Neviere R, Fauvel H, Chopin C, Formstecher P, Marchetti P. Caspase inhibition prevents cardiac dysfunction and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care Med. 2001;163:218–225. doi: 10.1164/ajrccm.163.1.2003109. [DOI] [PubMed] [Google Scholar]
- NPUAP. Pressure ulcers in America: prevalence, incidence, and implications for the future. An executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care. 2001;14:208–215. doi: 10.1097/00129334-200107000-00015. [DOI] [PubMed] [Google Scholar]
- Petillot P, Lahorte C, Bonanno E, Signore A, Lancel S, Marchetti P, Vallet B, Slegers G, Neviere R. Annexin V detection of lipopolysaccharide-induced cardiac apoptosis. Shock. 2007;27:69–74. doi: 10.1097/01.shk.0000235085.56100.38. [DOI] [PubMed] [Google Scholar]
- Plant PJ, Bain JR, Correa JE, Woo M, Batt J. Absence of caspase-3 protects against denervation-induced skeletal muscle atrophy. J Appl Physiol. 2009;107:224–234. doi: 10.1152/japplphysiol.90932.2008. [DOI] [PubMed] [Google Scholar]
- Qin FZ, Liang MC, Liang CS. Progressive left ventricular remodeling, myocyte apoptosis, and protein signaling cascades after myocardial infarction in rabbits. Biochim Biophys Acta. 2005;1740:499–513. doi: 10.1016/j.bbadis.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Quadrilatero J, Rush JWE. Evidence for a pro-apoptotic phenotype in skeletal muscle of hypertensive rats. Biochem Biophys Res Commun. 2008;368:168–174. doi: 10.1016/j.bbrc.2008.01.067. [DOI] [PubMed] [Google Scholar]
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotz P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17:3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics. 2009;37:58–66. doi: 10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G. Clinical trial: the efficacy and safety of oral PF-03491390, a pancaspase inhibitor – a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther. 2010;31:969–978. doi: 10.1111/j.1365-2036.2010.04264.x. [DOI] [PubMed] [Google Scholar]
- Siu PM, Alway SE. Id2 and p53 participate in apoptosis during unloading-induced muscle atrophy. Am J Physiol Cell Physiol. 2005a;288:C1058–C1073. doi: 10.1152/ajpcell.00495.2004. [DOI] [PubMed] [Google Scholar]
- Siu PM, Alway SE. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J Physiol. 2005b;565:309–323. doi: 10.1113/jphysiol.2004.081083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM, Alway SE. Subcellular responses of p53 and Id2 in fast and slow skeletal muscle in response to stretch-induced overload. J Appl Physiol. 2005c;99:1897–1904. doi: 10.1152/japplphysiol.00374.2005. [DOI] [PubMed] [Google Scholar]
- Siu PM, Alway SE. Deficiency of the Bax gene attenuates denervation-induced apoptosis. Apoptosis. 2006;11:967–981. doi: 10.1007/s10495-006-6315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM, Pistilli EE, Alway SE. Apoptotic responses to hindlimb suspension in gastrocnemius muscles from young adult and aged rats. Am J Physiol Regul Integr Comp Physiol. 2005a;289:R1015–R1026. doi: 10.1152/ajpregu.00198.2005. [DOI] [PubMed] [Google Scholar]
- Siu PM, Pistilli EE, Alway SE. Age-dependent increase in oxidative stress in gastrocnemius muscle with unloading. J Appl Physiol. 2008;105:1695–1705. doi: 10.1152/japplphysiol.90800.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM, Pistilli EE, Butler DC, Alway SE. Aging influences cellular and molecular responses of apoptosis to skeletal muscle unloading. Am J Physiol Cell Physiol. 2005b;288:C338–C349. doi: 10.1152/ajpcell.00239.2004. [DOI] [PubMed] [Google Scholar]
- Siu PM, Pistilli EE, Murlasits Z, Alway SE. Hindlimb unloading increases muscle content of cytosolic but not nuclear Id2 and p53 proteins in young adult and aged rats. J Appl Physiol. 2006;100:907–916. doi: 10.1152/japplphysiol.01012.2005. [DOI] [PubMed] [Google Scholar]
- Siu PM, Tam EW, Teng BT, Pei XM, Ng JW, Benzie IF, Mak AF. Muscle apoptosis is induced in pressure-induced deep tissue injury. J Appl Physiol. 2009;107:1266–1275. doi: 10.1152/japplphysiol.90897.2008. [DOI] [PubMed] [Google Scholar]
- Stekelenburg A, Oomens CW, Strijkers GJ, Nicolay K, Bader DL. Compression-induced deep tissue injury examined with magnetic resonance imaging and histology. J Appl Physiol. 2006;100:1946–1954. doi: 10.1152/japplphysiol.00889.2005. [DOI] [PubMed] [Google Scholar]
- Tamilselvan J, Jayaraman G, Sivarajan K, Panneerselvam C. Age-dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: role of carnitine and lipoic acid. Free Radic Biol Med. 2007;43:1656–1669. doi: 10.1016/j.freeradbiomed.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Teng BT, Pei XM, Tam EW, Benzie IF, Siu PM. Opposing responses of apoptosis and autophagy to moderate compression in skeletal muscle. Acta Physiol (Oxf) 2011;201:239–254. doi: 10.1111/j.1748-1716.2010.02173.x. [DOI] [PubMed] [Google Scholar]
- van Empel VP, Bertrand AT, Van Der Nagel R, Kostin S, Doevendans PA, Crijns HJ, de Wit E, Sluiter W, Ackerman SL, De Windt LJ. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96:e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;2006:pe44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- Wang YN, Bouten CV, Lee DA, Bader DL. Compression-induced damage in a muscle cell model in vitro. Proc Inst Mech Eng H. 2005;219:1–12. doi: 10.1243/095441105X9246. [DOI] [PubMed] [Google Scholar]
- Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–281. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.