

Abstract
Mosaic aneuploidy and uniparental disomy (UPD) arise from mitotic or meiotic events. There are differences between these mechanisms in terms of (i) impact on embryonic development; (ii) co-occurrence of mosaic trisomy and UPD and (iii) potential recurrence risks. We used a genome-wide single nucleotide polymorphism (SNP) array to study patients with chromosome aneuploidy mosaicism, UPD and one individual with XX/XY chimerism to gain insight into the developmental mechanism and timing of these events. Sixteen cases of mosaic aneuploidy originated mitotically, and these included four rare trisomies and all of the monosomies, consistent with the influence of selective factors. Five trisomies arose meiotically, and three of the five had UPD in the disomic cells, confirming increased risk for UPD in the case of meiotic non-disjunction. Evidence for the meiotic origin of aneuploidy and UPD was seen in the patterns of recombination visible during analysis with 1–3 crossovers per chromosome. The mechanisms of formation of the UPD included trisomy rescue, with and without concomitant trisomy, monosomy rescue, and mitotic formation of a mosaic segmental UPD. UPD was also identified in an XX/XY chimeric individual, with one cell line having complete maternal UPD consistent with a parthenogenetic origin. Utilization of SNP arrays allows simultaneous evaluation of genomic alterations and insights into aneuploidy and UPD mechanisms. Differentiation of mitotic and meiotic origins for aneuploidy and UPD supports existence of selective factors against full trisomy of some chromosomes in the early embryo and provides data for estimation of recurrence and disease mechanisms.
INTRODUCTION
Aneuploidy is a significant cause of developmental disease, with frequency close to 50% in spontaneous abortions and 0.5% in live born individuals (1–3). Very few human chromosome aneuploidies are seen in liveborn individuals; however, mosaic aneuploidy is better tolerated. Uniparental disomy (UPD) is another mechanism for disturbance of human gene expression that can lead to human disease, and mosaic aneuploidy has been shown to be associated with UPD in some cases (4–7). In this work, we demonstrate the utility of a genome-wide single nucleotide polymorphism (SNP) array to identify the mechanisms causing mosaic chromosome aneuploidy and UPD. This analysis provides a window into the mechanisms of aneuploidy occurrence by observation of the genotypes in the disomic and trisomic cell lines.
Chromosomal mosaicism is defined as the presence of two or more different chromosome complements within an individual developed from a single zygote. Mosaicism has been reported for many types of chromosome abnormalities including trisomy, monosomy, triploidy, deletions, duplications, rings and other types of structural rearrangements. Mosaic aneuploidy is the most common type of mosaicism (1). Recent studies on early human embryos have demonstrated that over 50% of embryos generated by in vitro fertilization are mosaic for a chromosome anomaly, underlining the high frequency of non-disjunction (8–11). Mosaic aneuploidy can arise from meiotic events, with an abnormal zygote and loss of one copy of a trisomic chromosome in some cells during development, or mitotically, with a normal zygote, and a subsequent non-disjunction or anaphase lag during a somatic division. These different mechanisms have a profound effect on the developing fetus. In the cases where the non-disjunction occurred meiotically, it is likely that there is a trisomic constitution in the very early stages of development, where correct chromosome number might be very important (12,13). Alternatively, in the cases of mitotic origin of the trisomy, early development proceeded normally, with trisomy originating further along in development, and possibly affecting only a subset of tissues. Previous work has shown that there is a chromosome-specific bias in the proportion of meiotically to mitotically occurring non-disjunctions (12,13).
Another consequence of meiotically originating trisomies is the risk for UPD in the disomic cell line. In the case of a meiotic trisomy, with mitotic loss of one copy of the duplicated chromosome (also referred to as trisomy rescue), the cells that have lost one copy of the trisomic chromosome are at risk for UPD, where the chromosomes that remain are both from the same parent. UPD is well known to cause disease if the chromosome contains an imprinted gene, or if a recessive disease allele is uncovered. There are three primary mechanisms by which UPD can occur: (i) trisomy rescue, whereby there is mitotic loss of one of the three copies of the trisomic chromosome; (ii) monosomy duplication in which the lone copy of a chromosome pair is duplicated via non-disjunction or (iii) gamete complementation, whereby a gamete that is missing one chromosome pair unites with a gamete containing two copies of that pair, by chance (4). Each of these mechanisms have been reported, although trisomy rescue is thought to be the most common of the three mechanisms (7). UPD cannot be identified by standard cytogenetic techniques. Rather, when UPD is suspected based on clinical or cytogenetic features, analysis of specific chromosomes is undertaken using molecular markers or by analysis of methylation patterns for the chromosomal region of interest.
Chromosomal mosaicism can be identified cytogenetically, but identification of lower levels of mosaicism can be challenging, as many cells have to be counted. It has been estimated that analysis of 20 cells (standard for routine chromosome analysis) will detect 14% mosaicism (in the tissue being studied) with 95% confidence (14). The level of mosaicism detected goes down when the number of cells is increased, however analysis of more cells is not normally carried out unless there is a suspicion for chromosomal mosaicism. In addition, for some types of mosaicism, the abnormal cells as well as the normal cells may not divide, so analysis of metaphases might provide a biased view of the true chromosome constitution of this individual. This metaphase bias against abnormal cells has been conclusively demonstrated for some abnormalities, such as the isochromosome 12p seen in patients with Pallister Killian syndrome (15). Array analysis by comparative genomic hybridization or SNP array analysis offers several advantages for detection of mosaicism compared with chromosome analysis in which (i) a large number of cells can be surveyed at once, since DNA is extracted from a culture of many cells and (ii) both interphase and metaphase cells are analyzed, eliminating the culture bias introduced by analysis of metaphase cells only.
We have used a genome-wide SNP array for our genomic analyses. Genome-wide SNP arrays use a combination of intensity and genotyping data that provide high-resolution means to diagnose genomic abnormalities that cause clinical disease. The use of genome-wide SNP arrays permits the simultaneous evaluation of copy number to detect mosaic gains and losses, and UPD, in cases of isodisomy or isodisomic regions secondary to recombination. In the case of heterodisomy, UPD diagnosis by SNP array can be accomplished if parental DNA is analyzed.
Chimerism is similar to mosaicism in that it is defined by the presence of two genetically distinct cell lines; however, in the case of chimerism there is fusion of two different zygotes within a single embryo (16). Chimerism is often recognized because there are both 46,XX and 46,XY cell lines, which sometimes manifest clinically, but are readily discernable cytogenetically. Cytogenetic analysis are unable to detect chimerism without a difference in sex chromosome constitution between the two cell lines. Detection in these instances requires molecular analysis if chimerism is suspected. The use of a genome-wide SNP array makes the differentiation of chimerism and mosaicism possible, as the additional presence of extra genotypes in the chimeras is readily detectable. In this study, we analyzed a phenotypic male with multiple clinical abnormalities and 46,XX and 46,XY cell lines, and demonstrate that his genotypes are consistent with chimerism. We are able to propose a mechanism for the origin of his 46,XX cell line, which explains his clinical abnormalities.
We present data on a cohort of patients with mosaic chromosome abnormalities to provide information on the timing and origin of the mosaicism, mechanism by which the abnormality occurred, and frequency of UPD in these patients. We have also studied 11 patients with UPD, both segmental and whole chromosome, and were able to diagnose the mechanism by which these occurred, and provide information relevant to recurrence risks for these individuals.
RESULTS
We studied 2019 patients referred for clinical diagnostic testing using a genome-wide SNP array. Mosaic aneuploidy patients accounted for 1% of all patients referred to the CytoGenomics laboratory. In this cohort, we identified 21 patients with mosaic aneuploidy (three with concomitant UPD) in whom we could determine the developmental timing of the non-disjunction leading to aneuploidy. We also found an additional eight patients with UPD and one chimeric patient with complete UPD in one of the cell types.
Mosaic aneuploidy (monosomy)
Of 21 patients with mosaic aneuploidy, nine had mosaicism for a monosomic cell line, and one was a monsomy/trisomy mosaic (45,X/47,XXX). All but one of the mosaic monosomies involved the X chromosome and we observed one case of monosomy 7. The percentage of monosomic or trisomic cells could be calculated from the array data as described (see Materials and Methods). Based on the array data, the percentage of monosomic cells varied from 5 to 95% (Table 1). A spectrum of frequencies for mosaic monosomies or partial monosomies is illustrated in a composite picture in Figure 1. Mosaic monosomy is diagnosed when the log R ratio shows a decrease across the whole chromosome, which is less than the decrease seen for complete loss of one copy of the region. In addition, the B allele frequency appears altered, with values that are dependent on the percent and genotype of the remaining allele. Loss of an A allele, results in a shift of the frequency towards 0%, while loss of a B allele results in a shift towards 100%. Therefore, the percent mosaicism can be calculated from the relative shifting of the B allele frequency as discussed in the Materials and Methods section.
Table 1.
Patients with mosaic aneuploidy or chimerism
| Patient number | Type of aneuploidy | Mosaic % by array | Mosaic % by G-banded chromosomes | Mosaic % by metaphase FISH | Mosaic % by interphase FISH | Parental chromosome gained or lost | Mitosis/Meiosis | UPD | Tissue | Cell lines |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Monosomy X | 5 | 6.67 | Mitosis | Yes | Blood | 45,X/46,XX | |||
| 2 | Monosomy X | 25 | 16.00 | Paternal | Mitosis | Yes | Skin | 45,X/46,X,r(Y) | ||
| 3 | Monosomy X | 30 | 25.00 | 22.00 | Paternal | Mitosis | Yes | Blood | 45,X/46,XY | |
| 4 | Monosomy X | 30 | 40.00 | Maternal | Mitosis | Yes | Blood | 45,X/47,XXX | ||
| Trisomy X | 70 | 60.00 | Paternal | Mitosis | No | Blood | 45,X/47,XXX | |||
| 5 | Monosomy 7 | 40 | 0.00 | 7.00 | Paternal | Mitosis | Yes | Blood | 45,XY,-7/46,XY | |
| 6 | Monosomy X | 50 | 42.11 | Mitosis | Yes | Blood | 45,X/46,X,r(X) | |||
| 7 | Monosomy X | 75 | 6.67 | Mitosis | Yes | Blood | 45,X/46,X,r(X) | |||
| 8 | Monosomy X | 80 | 75.00 | Mitosis | Yes | Blood | 45,X/46,X,r(X) | |||
| 9 | Monosomy X | 80 | 76.67 | Mitosis | Yes | Blood | 45,X/46,X,r(X) | |||
| 10 | Monosomy X | 90 | Mitosis | Yes | Blood | 45,X/46,XX | ||||
| 11 | Trisomy 14 | 5 | 0.00 | Maternal | MI | No | Skin | 47,XX, + 14/46,XX | ||
| 50 | 2.56 | Maternal | MI | No | Blood | 47,XX, + 14/46,XX | ||||
| 12 | Trisomy 8 | 40 | 100.00 | MI | Yes | Blood | 47,XY, + 8/46,XY | |||
| 13 | Trisomy 9 | 50 | 0.00 | 2.50 | 24.00 | MI | Yes | Blood | 47,XX, + 9/46,XX | |
| 14 | Trisomy 18 | 10 | 15.15 | MII | No | Blood | 47,XX, + 18/46,XX | |||
| 15 | Trisomy 14 | 20 | 10.00 | Paternal | MII | Yes | Blood | 47,XX, + 14/46,XX | ||
| 16 | Trisomy 8 | 5 | 1.50 | Mitosis | No | Blood | 47,XY, + 8/46,XY | |||
| 17 | Trisomy 9 | 20 | 2.00 | Paternal | Mitosis | No | Blood | 47,XY, + 9/47,XY | ||
| 18 | Trisomy 9 | 20 | 35.00 | Mitosis | No | Blood | 47,XX, + 9/46,XX | |||
| 19 | Trisomy 8 | 20 | 12.62 | Mitosis | No | Blood | 48,XY, + 8, + 19/46,XY | |||
| Trisomy 19 | 20 | 14.00 | Mitosis | No | Blood | 48,XY, + 8, + 19/46,XY | ||||
| 20 | Trisomy 21 | 50 | 85.00 | Mitosis | No | Skin (hypo) | 48,XX, + 7, + 21/46,XX | |||
| Trisomy 7 | 50 | 85.00 | Mitosis | No | Skin (hypo) | 48,XX, + 7, + 21/46,XX | ||||
| Trisomy 21 | 60 | 50.00 | Mitosis | No | Skin (hyper) | 48,XX, + 7, + 21/46,XX | ||||
| Trisomy 7 | 60 | 50.00 | Mitosis | No | Skin (hyper) | 48,XX, + 7, + 21/46,XX | ||||
| 21 | Trisomy 17 | 50 | Mitosis | No | Skin (left) | 47,XY, + 17/46,XY | ||||
| 75 | Mitosis | No | Skin (right) | 47,XY, + 17/46,XY | ||||||
| 30 | Chimera | 20 | 45.00 | Paternal | Fertilization | Yes | Skin (hyper) | 46,XX/46,XY |
MI, meiosis I; MII, meiosis II.
Figure 1.
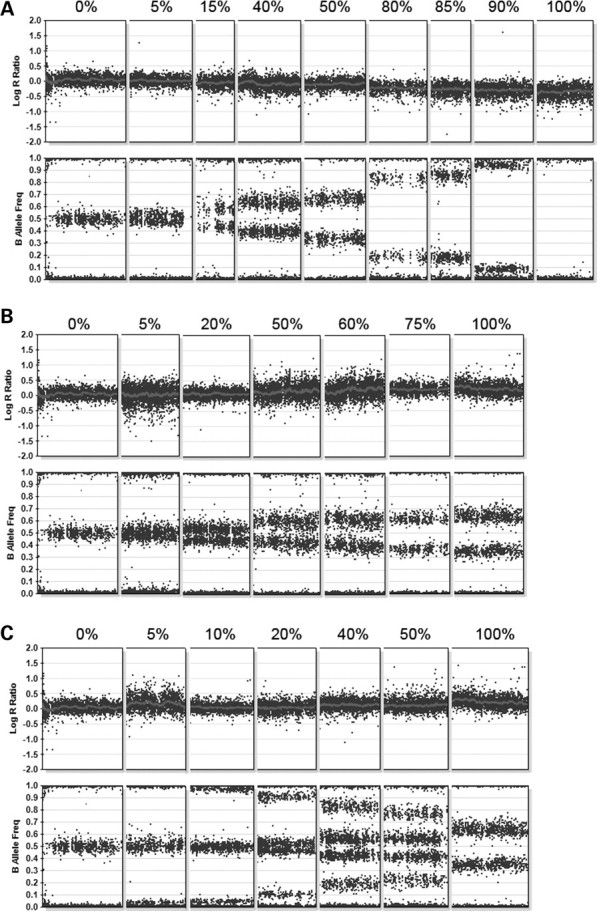
Composite array results for mosaic deletions and duplications. This figure shows segments from different chromosomes illustrating mosaicism from 0–100%. For all figure parts, the percentages above the data indicate the level of mosaicism, with 0% representing a patient with normal copy number, and 100% representing a non-mosaic patient. (A) BeadStudio output for nine patients with varying levels of mosaicism for deletions involving autosomes. (B) BeadStudio output for seven patients with varying levels of mosaicism for trisomies. The pattern of B allele frequency indicates that the same two haplotypes present in the euploid cell line are also present in the triploid cell line at altered ratios. (C) BeadStudio output from seven patients with varying levels of mosaicism for trisomies. The additional B allele frequencies in the mosaic patients represent genotypes present in the trisomic cell line that are not present in the euploid cell line, suggesting a meiotic origin of the trisomy.
In eight of the sex chromosome mosaic monosomies, the percentages of cells that were monosomic were in good agreement when array and cytogenetics were compared. The comparison could not be made in case no. 7, because the array was carried out on a lymphoblastoid cell line, and it is expected that there will be clonal selection within the cell line. In the case of the patient with mosaic monosomy 7 (case 5) detected by array analysis, we could not identify any cells with monosomy 7 by metaphase analysis of G-banded cells (Fig. 2A). However, fluorescence in situ hybridization (FISH) using a chromosome 7 centromere probe was consistent with monosomy 7 in 14 of 200 (7%) cells (Fig. 2B). In this case, there was also an altered B allele frequency indicating a higher percentage mosaicism surrounding the centromere, and we hypothesize that this monosomy may have originated as a small marker chromosome, with subsequent loss of the marker to create the monosomy (Fig. 2A and C). We were unable to identify the putative marker chromosome (or the monosomy) on analysis of G-banded chromosomes, consistent with selection against these cells in dividing cultures. In this case, it was clinically relevant to determine the parent of origin in order to assess the possibility of paternal loss associated with Russell-Silver syndrome (17). Analysis of informative SNPs on chromosome 7 in the subject and parental samples confirmed that the missing chromosome was paternal. Therefore, this patient's phenotype would be the result of the loss of paternally expressed genes, as is seen in patients with maternal UPD for chromosome 7.
Figure 2.

Mosaic monosomies. (A) BeadStudio output for patient no. 5 with a mosaic monosomy 7. Note the decreased log R ratio and altered B allele frequency. There is a somewhat lesser percent mosaicism around the centromere, which suggests that the deleted chromosome originated as a small pericentromeric marker, which was subsequently lost. (B) FISH confirmation of the monosomy 7 in interphase cells using a chromosome 7 centromere-specific probe. (C) Representation of the proposed mechanism, with the formation of a pericentromeric marker, which is subsequently lost to produce monosomy 7. (D) BeadStudio output for patient no. 4 with 45,X/47,XXX. (E) FISH confirmation of the parental origin of the X chromosomes. We used an X chromosome fosmid probe (G248P81417G5, labeled in red) within a known paternally inherited deletion within Xp22.3 indicating that the 45,X cell line contains the paternal X, while the 47,XXX cell line contains one paternal X and two non-deleted maternal X chromosomes. The X chromosome centromere is labeled in green. (F) Representation of origin of the 45,X/47,XXX showing mitotic non-disjunction.
Mosaic monosomy could either arise by mitotic non-disjunction in a diploid embryo leading to monosomy in a subset of cells, or monosomy rescue in some cells of a monosomic zygote early in development. These alternatives can be differentiated by inspection of the patterns of genotypes in the mosaic cells. All of the cases of mosaic monosomy arose by mitotic non-disjunction as we could identify mosaic loss of heterozygosity with allele frequency patterns consistent with the presence of two distinct haplotypes in all patients. In the case of monosomy rescue, we would expect duplication of the existing genotypes in the diploid cell line, with homozygosity at all loci.
Mosaic aneuploidy (trisomy)
Twelve patients had mosaicism for a trisomic cell line. These included three cases of mosaic trisomy 9; two cases each of mosaic trisomy 8 and 14; one case each of mosaic trisomy 17 and 18; two patients with double trisomies (+7, +21 and +8, +19) and one patient with a monosomy/trisomy mosaicism (45,X/47,XXX) (Table 1). As with mosaic monosomy, the percent mosaicism of the trisomies were calculated using the altered percentages of B allele frequencies observed for the abnormal chromosome. In the case of mosaic trisomy, the log R ratio indicates an increase in copy number, between two and three copies (Fig. 1B and C). The B allele frequency also shows an intermediate percentage, with additional frequencies observed between 0 and 50% (for addition of an A allele), and between 50 and 100% percent (for addition of a B allele). In the case of heterozygous alleles, the additional allele would result in a shift from 50% (for AB genotype) towards 33% (for a gain of an A allele), or towards 66% (for the gain of a B allele). In the case of homozygous alleles, the additional allele would not result in a shift of B allele frequency, unless the trisomic cell line introduces a genotype that was not present in the euploid cell line. In this case, additional shifts in the B allele frequency are observed, corresponding to a shift in B allele frequency from 0% towards 33% (in the case of AA in the euploid cell line and AAB in the trisomic cell line), and a shift from 100% toward 66% (in the case of BB in the euploid cell line and ABB in the trisomic cell line) (Fig. 1C). These additional B allele frequencies found only in the trisomic cell line would not be observed in mosaic trisomies because of mitotic non-disjunction since no new genotypes are introduced (Fig. 1B). For each of the mosaic trisomy cases, we were able to determine whether the mosaic trisomy arose by non-disjunction during meiosis, followed by mitotic loss in some cells, or mitotic non-disjunction with gain of the trisomic chromosome in some cells. The mosaic trisomies are especially informative for determination of the origin of the trisomy, as examination of the genotypes allows identification of the haplotype of the chromosome that is present in only a subset of cells. Meiotic origin of the trisomy is seen when the mosaic extra chromosome contains a genotype not present in the other two chromosomes. Meiotic crossovers can also be identified at the boundaries of these regions with three haplotypes (Fig. 3B–D).
Figure 3.
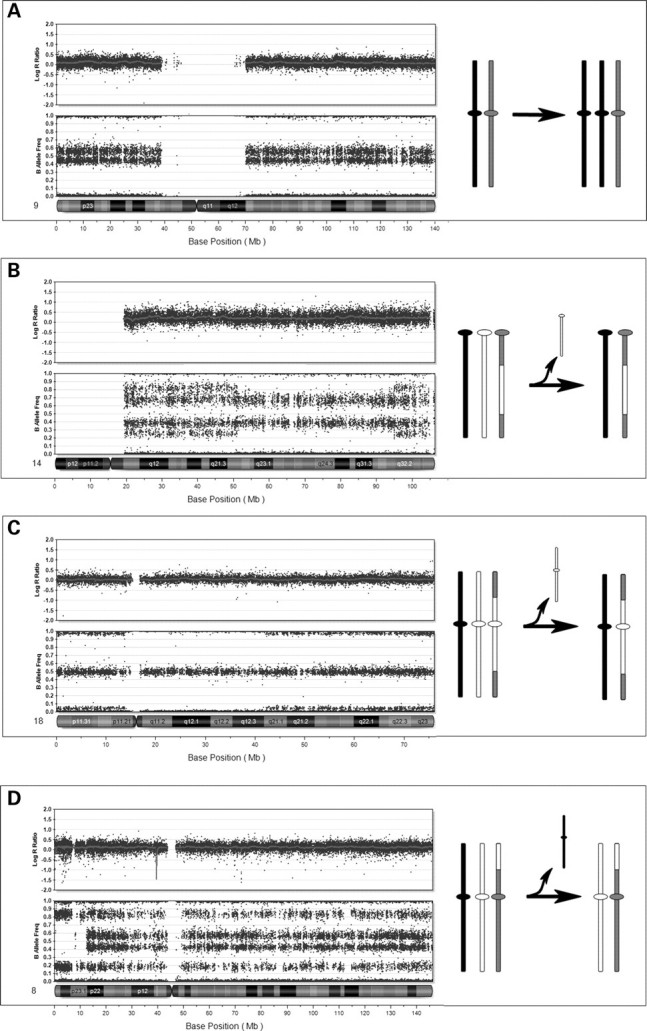
Mosaic trisomies. (A) Mosaic trisomy 9 (20%) in patient no. 17, with no evidence for recombination suggesting a mitotic origin. On the right is a representation of the mitotic event. (B) Mosaic trisomy 14 (50%) in peripheral blood from patient no. 11, with a complex pattern of genotypes consistent with non-disjunction in meiosis I. There is evidence for two recombination sites at the points where genotype complexity changes. Illustration at right shows the distribution of genotypes resulting from meiotic recombination. (C) Mosaic trisomy 18 (10%) in peripheral blood of patient no. 14 with a genotype pattern consistent with non-disjunction in meiosis II. Evidence for two recombination sites are observed. Illustration at right shows regions of crossovers and resulting genotypes across the chromosome. (D) Mosaic trisomy 8 (40%) in patient no. 12 with a genotype pattern consistent with non-disjunction in meiosis I. Altered pattern near the telomere of the p-arm demonstrates UPD (isodisomy) for this region. This is illustrated in the figure on the right.
Seven of the 12 cases of mosaic trisomy arose by mitotic non-disjunction. A mitotic origin was suggested by the absence of a third haplotype, indicated on the SNP array by additional genotypes closer to the homozygous AA or BB tracks (as illustrated in Fig. 1B) indicating that the trisomic cell line contains two identical chromosomes. However, we cannot rule out the possibility that the non-disjunction occurred in meiosis II, with no genetic recombination. This was seen in one case of +8, two cases of +9 (Fig. 3A), +17, +X in a 45,X/47,XXX individual (Fig. 2C), and both cases of the double trisomies (+7/+21 and +8/+19). These mitotic events involved chromosomes that are rarely found as trisomies (7, 17 and 19), as well as the 45,X/47,XXX. The 45,X/47,XXX patient was shown to have one paternal X chromosome in the monosomic cell line, and the same paternal X chromosome in the XXX cell line (in addition to two identical maternal Xs). This was determined using a FISH probe for a deletion that was present on the paternal chromosome only (Fig. 2D). The most rare abnormalities (double trisomies including chromosome 7 in one case and 19 in the other), mosaic trisomy 17 and the previously discussed mosaic monosomy 7, all arose mitotically, consistent with these abnormalities being lethal if they occurred during meiosis.
Five cases of mosaic trisomy arose by meiotic non-disjunction including one case of mosaic +8, one of +9, and two cases of +14, and one case of +18 (Fig. 3B–D). In three cases the non-disjunction occurred in meiosis I and in two cases in meiosis II (Table 1). Meiotic non-disjunction was recognized when an increased number of haplotypes were visualized at different locations across the chromosome. Meiosis I non-disjunction could be differentiated from meiosis II non-disjunction by the location of the regions of extra haplotypes. When the additional haplotypes are visible near the centromere, this signifies the presence of the two different homologs, consistent with a meiosis I non-disjunction (Fig. 3B–D). When the additional haplotypes are present near the telomeres, but not the centromere, this is consistent with a meiosis II origin, where it is the sister chromatids that have undergone non-disjunction (Fig. 3C). Of note, the additional haplotypes seen as a result of meiotic non-disjunction are exceedingly helpful in aiding the recognition of low-level mosaicism, even though there is a small separation between the different allele frequencies. This can be seen in the case of trisomy 18 pictured in Figure 3C.
Inspection of genotypes in our cases of mosaic trisomy caused by meiotic non-disjunction revealed UPD in three cases (UPD 8, UPD 9 and one case of UPD 14). This is shown for the individual with mosaic UPD 8 in Figure 3D. Only the patient with UPD 14 showed clinical features consistent with UPD (see below) (18). The presence of UPD in the euploid cells lines was identified by the presence of mosaic loss of heterozygosity secondary to trisomy rescue of a meiotic non-disjunction. The mosaic trisomies for chromosomes 8 and 9 suggested UPD in the euploid cell line, with a region of mosaic loss of heterozygosity at the p-arm telomere, and one crossover site. The presence of three haplotypes at the centromeres suggested meiosis I non-disjunction, with subsequent loss of the unique parental chromosome during mitosis. Parental samples were not available for these patients. One patient with mosaic trisomy 14 showed the presence of three haplotypes in approximately 20% of cells, with two haplotypes in 80% of cells in unstimulated peripheral blood. The presence of two crossover sites was observed near the centromere and telomere, and a drop out of heterozygous B allele frequencies indicated the presence of UPD in the euploid cell line. Parental genotypes were obtained, and informative SNPs revealed the presence of paternal isodisomy near the centromere and telomere, and paternal heterodisomy for the remainder of the chromosome in approximately 80% of cells. These results are consistent with paternal non-disjunction in meiosis II, followed by loss of the maternal chromosome during development, resulting in mosaic paternal UPD 14, which was consistent with the patient's phenotype.
There was no evidence for UPD in the remaining two of the patients who had undergone meiotic non-disjunction (chromosomes 14 and 18). Parental samples from the patient with mosaic trisomy 14 revealed two contributions from the maternal genome in the trisomic cell lines in 50% of cells, and biparental inheritance in the remaining cells, ruling out UPD. The pattern of B allele frequencies supports this finding, with no presence of mosaic loss of heterozygosity seen along chromosome 14 in this patient (Fig. 3B). This patient did not have clinical features consistent with trisomy 14, but showed only developmental delay, and congenital hip dysplasias.
We calculated the percent mosaicism in each case from the array data obtained by analysis of whole blood as described above, and compared these with the results of cytogenetic or molecular cytogenetic analysis of PHA-stimulated peripheral blood or cultured fibroblasts. The array results were often divergent with the data obtained by cytogenetics (Table 1). There were examples of increased frequencies in both the array and cytogenetic preparations in different cases. For example, two cases with trisomy 8 mosaicism showed opposite patterns. Case no. 12 was calculated as 40% mosaic trisomy 8 on array analysis, whereas 100% of 20 cells studied in the blood showed trisomy 8. By contrast, case no. 16 was calculated as 5% mosaic by array analysis, whereas cytogenetic analysis showed 1.5% of cells with trisomy 8. Overall, there were eight instances of increased aneuploidy frequency in the array data and four instances of increased aneuploidy detected in metaphase preparations. For the individual with the double +7, +21 trisomy, we studied two independent cultures, and calculated increased frequency of the aneuploidy by array in one, and by cytogenetics in the other (Table 1).
Uniparental disomy
We identified six patients with at least one run of homozygosity greater than 20 Mb in length (Table 2). There was no known history of parental consanguinity in these individuals, and we hypothesize that the homozygosity is explained by UPD. This group of patients did not show evidence for mosaic aneuploidy. For each of these cases, it was possible to infer the mechanism by which the UPD occurred with two cases of monosomy rescue and four cases of trisomy rescue. Monosomy rescue is hypothesized in one case of UPD14 and one case of UPD 15. In these cases, there is no evidence for recombination, as all genotypes present are homozygous (Fig. 4A). For both of these cases, there is a known phenotype associated with UPD and the phenotypes of the patients were consistent with those found in patients with paternal UPD, suggesting a meiotic non-disjunction in maternal meiosis, resulting in a nullisomic egg, with subsequent rescue after fertilization. We cannot rule out the possibility of meiosis II non-disjunction in sperm, although the complete lack of evidence for crossing over makes this unlikely. The patient with UPD15 presented with Angelman syndrome, consistent with monosomic rescue via duplication of a paternal chromosome 15. Confirmation of paternal UPD was achieved by subsequent bisulfite testing in a clinical laboratory. In the case of UPD 14, the patient presented with multiple anomalies including ‘coat hanger ribs’, which is pathognomonic for paternal UPD14 (18). Analysis of parental samples and comparison of genotypes with those seen in the child confirmed a paternal UPD, by examination of informative SNPs across chromosome 14. This result was also validated by examination of microsatellite markers across chromosome 14 at a commercial laboratory.
Table 2.
Patients with UPD
| Patient number | Mechanism | Chromosome | Size of isodisomic region | Number of SNPs in isodisomic region | Mosaic % by array | Maternal/paternal UPD | Mitosis/Meiosis | UPD validation | Tissue |
|---|---|---|---|---|---|---|---|---|---|
| 22 | Monosomy rescue | chr14 | 88 252 956 | 19 213 | 100 | Paternal | MI or MII | Yes | Blood |
| 23 | Monosomy rescue | chr15 | 81 794 197 | 16 625 | 100 | Paternal | MI or MII | Yes | Blood |
| 24 | Segmental | chr11 | 45 296 182 | 10 755 | 10 | Mitosis | Skin | ||
| 45 296 182 | 10 755 | 30 | Mitosis | Pancreas | |||||
| 25 | Segmental | chr11 | 12 430 186 | 3580 | 10 | Mitosis | Blood | ||
| 26 | Trisomy rescue | chr2 | 107 225 556 | 20 727 | 100 | MI | Blood | ||
| 27 | Trisomy rescue | chr16 | 15 991 615 | 4202 | 100 | MI | Blood | ||
| 27 894 431 | 6970 | 100 | MI | Blood | |||||
| 28 | Trisomy rescue | chr14 | 21 117 384 | 4693 | 100 | MI | Blood | ||
| 29 | Trisomy rescue | chr15 | 5 911 145 | 877 | 100 | Maternal | MII | Blood | |
| 52 922 495 | 10 813 | 100 | Maternal | MII | Blood |
Figure 4.

Uniparental disomy (UPD). In these cases, the log R ratio is consistent with normal copy number for all cases. (A) Complete isodisomy of chromosome 14 with loss of heterozygosity (LOH) for the entire chromosome in patient no. 22. This is consistent with the mechanism of monosomy rescue. (B) UPD of chromosome 15 in patient no. 29. Note the regions of LOH near the centromere and across the middle of the chromosome, which are interrupted by regions of heterozygosity, suggesting origin in meiosis II, with evidence of regions of recombination. (C) Segmental UPD of 11p11.2 to p-terminus in the DNA from cultured skin (10%) from patient no. 24. (D) Analysis of DNA from pancreatic tissue in patient no. 24, which had 30% mosaicism for the 11p LOH. This patient has a clinical diagnosis of focal hyperinsulinism.
Trisomy rescue was hypothesized to have caused four cases of UPD (UPD 2, 14, 15 and 16), although we did not detect evidence of the trisomic cell line. These cases presented with chromosomes that showed both runs of homozygosity (minimum of 21 Mb) and heterozygosity, demonstrating results of the recombination process in meiosis. For example, the patient with UPD 15 showed evidence for meiosis II non-disjunction, with a run of homozygosity that included the centromere of chromosome 15 (Fig. 4B). We hypothesized that the non-disjunction occurred in maternal meiosis II, with post-zygotic loss of the paternal chromosome 15, as this patient presented with clinical features consistent with Prader–Willi syndrome (neonatal hypotonia, childhood obesity, delayed milestones), known to be caused by maternal UPD (4). Maternal UPD 15 was confirmed by follow-up methylation testing in a clinical laboratory. The three remaining UPD patients had SNP patterns consistent with non-disjunction that occurred in meiosis I, as there were heterozygous alleles near the centromere (UPD 2, 14 and 16). These three patients had genotypes that suggested UPD owing to the size of the run of homozygosity, and the paucity of such regions on other chromosomes. Crossovers were identified in all three patients, with one to three exchanges per chromosome. Parental samples were not available for these patients, and therefore the UPD could not be validated.
In addition to UPD as a result of meiotic error, two patients presented with mosaic segmental UPD, consistent with a mitotic origin. UPD for 11p15.5 was identified in two samples (skin tissue and pancreatic tissue) from a patient with focal hyperinsulinism. This diagnosis is consistent with the finding of mosaic paternal somatic UPD involving loci within 11p15 (19). In this patient, the B allele frequency pattern observed in skin tissue, which was initially studied, revealed a low percentage mosaicism for a genomic event, but we could not distinguish mosaic deletion, duplication or UPD, as there was no visible change in the log R ratio (Fig. 4C). The allele frequencies suggested a low level mosaicism for loss of heterozygosity, which is consistent with the clinical findings in the patient. Using this shift in allele frequencies, the percent mosaicism for the abnormal cell line could be estimated at 10%. When pancreatic tissue was studied, the percentage of abnormal cells increased, and it became clear that the greater difference in B allele frequency was not reflected in the log R ratio, consistent with mosaic segmental UPD (Fig. 3D). A second patient, who presented with hemihypertrophy, was also found to have mosaic loss of heterozygosity, suggesting mosaic UPD for 11p15. Similar to the previous case, the percentage of UPD was calculated to be approximately 10%. This finding is also consistent with the clinical presentation of Beckwith–Wiedemann Syndrome (20).
Mosaicism for complete uniparental (maternal) inheritance
Patient 21 was referred to the laboratory for diagnostic studies because of a history of failure to thrive in infancy, followed by childhood obesity, limb length discrepancy, pigmentary changes, hearing loss, developmental delays, and autistic spectrum disorder. While initial cytogenetic analysis of peripheral blood showed a normal 46,XY karyotype, FISH analysis of a buccal smear and subsequent chromosome analysis of a skin biopsy from a region showing pigmentary changes revealed the 46,XY/46,XX mosaicism (Fig. 5A), with 16 of 30 cells having a 46,XY karyotype and 14 of 30 with a 46,XX karyotype (Fig. 5B). Array analysis was carried out on the 46,XX/46,XY tissue. Surprisingly, we found that all of the autosomes had a similar, altered B allele frequency with a pattern consistent with two genotypes at every locus with an altered ratio between the two haplotypes, when compared with a normal diploid cell line (Fig. 5C). This finding rules out a straightforward XX/XY mosaicism in this individual since all autosomes are affected, and is consistent with chimerism. The pattern for the X chromosome was different than that for the autosomes, with only a single apparent genotype, consistent with only one X chromosome haplotype in this XX/XY chimera (Fig. 5D). This is consistent with his XX cell line demonstrating maternal UPD (parthenogenetic chimera). This could be caused by either fertilization of a diploid egg by a Y-carrying sperm, fertilization of an endoreduplicated egg or fusion of a polar body with a fertilized zygote (16). Examination of the genotypes showed that there was no evidence for genetic exchange for any of the autosomes or the X chromosome, which argues against fertilization of a diploid egg, and suggesting that the origin of the XX cell line is most likely because of endoreduplication of the egg genome. One similar parthenogenetic chimera has been reported in the literature (21). Further work is in progress to clone out the XX and XY cell lines to better understand the mechanism of formation in this individual.
Figure 5.

Analysis of a chimeric individual (patient no. 30). (A) FISH analysis of a buccal sample using centromere probe for X and Y. (B) Cytogenetic analysis of cultured cells from hyper-pigmented skin revealed both XY and XX cells. (C) BeadStudio analysis of DNA from hyperpigmented tissue demonstrates that chromosomes 1 and 2 show altered B allele frequencies and a normal log R ratio. This altered B allele frequency was seen for all autosomes. (D) BeadStudio data from the X chromosome reveals only a single genotype at all loci. Note that the log R ratio reflects a 20% increase for the normal levels expected in a male and the B allele frequency of pseudoautosomal regions appears similar to that seen with the autosomes.
DISCUSSION
We studied more than 2000 patients using a genome-wide SNP array and identified a group of patients with low-level mosaic aneuploidy, UPD and chimerism. We found a higher than expected frequency of these events. We were able to identify the mechanism, parental origin and developmental timing of these abnormalities and show that patients with mosaic trisomies, which originate meiotically, are at increased risk for UPD.
Mosaicism detection and percentage in cohort
Twenty-one patients studied had mosaic aneuploidy, which corresponds to 10% of the 210 abnormalities diagnosed in our laboratory during the same time period. We identified mosaic monosomies and trisomies that were both expected and unexpected. Expected abnormalities include common trisomies such as those for chromosomes 18 and 21, monsosomy X as well as previously described mosaic trisomies such as chromosome 8, 9, 14 and those of the sex chromosomes (X and Y). The identification of the unexpected, rare abnormalities (such as mosaic monosomy 7, trisomy 17 and double trisomies (+7, +21 and +8, +19) is likely owing to both the analysis of whole, unstimulated blood which is an advantage of all array-based studies, when compared with cytogenetics; and the increased sensitivity of the SNP array, which uses both intensity and genotyping data to identify mosaics. We therefore hypothesize that low level mosaicism may be more common than previously anticipated. Using array CGH, a mosaic aneuploidy discovery rate of 0.2% was reported (22). Previous studies of dilution series of known abnormalities using array CGH platforms or intensity data from SNP arrays have estimated the minimal detection of mosaicism to be 10–20% (22–24). We have been able to identify mosaics at levels less than 5%. In our experience and as shown in Figure 1, B allele frequency is more sensitive to the subtle loss or gain of a haplotype than the log R ratio is to the subtle shifts in intensity levels, because of the normalization and logarithmic transformation of the intensity data. Mosaicism that involves the introduction of a new haplotype in the abnormal cells is especially sensitive to detection by our analysis, as demonstrated by the patient with mosaic trisomy 14 and 18.
Mechanisms
Mosaic aneuploidy can result from meiotic or mitotic non-disjunction. In the case of meiotic non-disjunction, the trisomy or monosomy is present in the zygote, but is corrected by a subsequent mitotic event (non-disjunction or anaphase lag). Alternatively, the zygote can be normal, with a mitotic event leading to monosomy or trisomy in some cells. Analysis of the genotype patterns in the disomic and trisomic cells of a mosaic aneuploid individual can differentiate these possibilities. Differentiating between a mitotic and meiotic origin for trisomies is essential for proper counseling and determining recurrence risks, because trisomy as a result of meiotic non-disjunction is associated with a higher risk of recurrence, especially in younger women (25).
Meiosis
Five of the 12 cases of mosaic trisomy arose by meiotic non-disjunction and the remaining seven arose mitotically. The meiotically originating cases involved chromosomes 8, 9, 14 (two cases) and 18, with origins in meiosis I (chromosomes 8, 9 and one case of chromosome 14) and meiosis II (one case of chromosome 14 and one of 18). This case of meiotically originating trisomy 8 mosaicism is one of three identified in this study, with the other two having occurred mitotically. Analysis of mosaic trisomy 8 seen in liveborn individuals reported in the literature has demonstrated that most cases arise mitotically (26,27). This has lead to the hypothesis that trisomy 8 is selected against in the early embryo. However, we demonstrate one case of mosaic trisomy 8 that has occurred meiotically, indicating that there must be other factors involved in survival for these individuals. One other rare case of meiotically arising mosaic trisomy 8 has been reported (28). Mosaic trisomy 14 has been found to arise both meiotically or mitotically with equal frequency, and occurs in both maternal and paternal meiosis (2). We could determine parental origin in our two cases of chromosome 14 mosaicism and found that one originated in maternal meiosis I, while the other originated in paternal meiosis II. It is not surprising that the case of mosaic trisomy 18 originated in meiosis II, as it is well known that even full trisomy for chromosome 18 is seen in liveborn individuals and is biased for origin in maternal meiosis II (2).
Whole chromosome UPD was identified in three of the five meiotic cases of mosaic trisomy, one each of chromosomes 8, 9 and 14. This highlights the significant risk for UPD (60%) by trisomy rescue in cases of mosaic trisomy that originate meiotically. In addition, we identified six cases of whole chromosome, UPD, without evidence of mosaic trisomy [chromosomes 2, 14 (two cases), 15 (two cases) and 16]. Recognition of clinically significant UPD can be difficult as long contiguous regions of homozygosity (ROH) have been reported in the general population, with regions averaging 4 Mb in European populations (29) and 26 Mb in Han Chinese populations (30). We considered unusually long, contiguous and chromosome-specific ROH identified in patients with no history of consanguinity to be the result of UPD. Confirmation of UPD comes from correlation with clinical phenotype and validation by analysis of parental DNA. There are known imprinted genes, with a known clinical phenotype for UPD 14 and UPD 15, and the phenotypes in our patients were consistent with these. We were also able to validate UPD using parental testing in both cases of UPD 14, and one case of UPD 15.
We could identify the mechanism by which UPD occurred in each patient, either trisomy or monosomy rescue. One case of UPD 14 and one case of UPD 15 occurred via monosomy rescue, and the other four occurred by trisomy rescue, although there was no evidence for trisomy in these DNA samples. In a recent study, sex-specific recombination hotspots were identified (31). We compared the recombination sites in our patients to those previously reported, and we found concordance for the locations. This evidence for the remnants of meiotic recombination supports our interpretation of occurrence by trisomy rescue, and in addition; it is possible to predict the parent in which the non-disjunction originated based on this data.
Mitosis
All 10 cases of mosaic monosomy arose mitotically from a diploid zygote. These findings are consistent with those previously reported on 14 cases of mosaic 45,X/46,XX and two cases of 45,X/47,XXX (12). Mosaic monosomy 7 is very rare, although cases have been reported to occur somatically in association with myelodysplasia (32). Our results support the hypothesis that the presence of at least two copies of each chromosome is essential during early embryogenesis.
Seven of the trisomies as well as the two double trisomies originated mitotically (+8, +9, +9, +17, +X, +7/+21, +8/+19). Double trisomies have been identified in spontaneous abortions and were found to originate during maternal meiosis in all of these cases (33). This more severe outcome for those originating in meiosis (as evidenced by discovery in spontaneous abortions) is consistent with selection against these abnormalities during early development. Other mosaic trisomies that originated in mitosis included chromosomes that are rarely detected as trisomic in stillborns or liveborns (7,17,19), also consistent with a selective disadvantage for these trisomies early in development (1). In addition, as discussed above, mosaic trisomy 8 occurs more frequently during mitosis when it is detected in liveborns (26,27). We identified three cases of mosaic trisomy 9, one of meiotic origin and two mitotic origins, consistent with no bias in the origin for this chromosome. This is further supported by the finding of full trisomy 9 in stillborn individuals and embryos (1,8). The case of X chromosome aneuploidy was a 45,X/47,XXX mosaic, with direct evidence for a mitotic origin and it has been hypothesized that there is selection against 45,X in the early embryo (12,34).
We also identified two cases of mosaic segmental UPD for chromosome 11p. This region of the genome is known to contain several imprinted genes, and both individuals demonstrated clinical features consistent with paternal UPD (19,20). The mechanism of formation of segmental UPD is not known, although it is presumed to occur mitotically, as seen in our patients.
Chimerism
While chimerism in itself is a rare finding, we have identified an individual who is a 46,XX/46,XY chimera, with the entire 46,XX cell line derived from his mother. We hypothesize that this cell line arose by parthenogenetic development of the 46,XX line, which fused with a 46,XY cell line (16,21). This interpretation is based on analysis of the genotypes across both the autosomes and the sex chromosome in this individual. No significant genomic abnormalities were identified, beyond the complete isodisomy UPD in the XX cell line, and we hypothesize that the patient's clinical abnormalities are explained by this finding. While recognition of chimerism is difficult by cytogenetic or CGH analysis, it is straightforward with the use of an SNP array and future studies may reveal more about this unusual finding.
The use of genome-wide SNP arrays allows simultaneous evaluation of genomic dosage and genotypes. The dual property of this tool allows identification of clinically significant alterations, with simultaneous insights into the mechanisms by which these abnormalities occur. Their use in clinical diagnostics provides important information for recurrence and interpretation of the clinical effect of abnormalities.
MATERIALS AND METHODS
Patient identification and sample preparation
All patients were referred to The Children's Hospital of Philadelphia Clinical CytoGenomics Laboratory for diagnostic studies (n = 2019). Indications for the testing varied widely, including pervasive developmental delay, seizures, congenital anomalies, short stature, failure to thrive, hearing or vision loss, and various combinations of developmental and congenital issues. Of these patients, 30 (1.5%) had either a mosaic aneuploidy or UPD and studies on these patients are described here. When available, parental samples were obtained for parent of origin analysis.
Sample preparation and array analysis
DNA was extracted from peripheral blood, or cultured fibroblasts. The quality of the DNA was monitored by analysis of OD260/OD280 and OD260/OD230 ratios. Acceptable samples had values between 1.8 and 2.0 and ratios > 2.0, respectively. Thirty microliters of a 50–100 ng/µl solution of genomic DNA was aliquoted into 96-well plates and genotyped on the Illumina BeadStation. The samples were whole genome-amplified, fragmented, hybridized, fluorescently tagged and scanned, as per standard protocols (35). Initial analyses (n = 7) were carried out using the Illumina HumanHap550 BeadChip (V3), which contains 561 466 SNP probes, distributed genome-wide. All subsequent samples were analyzed using the IlluminaQuad610 array, which contains all of the SNP probes found on the Illumina HumanHap550, an additional 37 355 SNP probes, and 21 890 intensity-only probes, which were placed, in regions where SNP coverage is poor. For Quad610 analysis, we selected a subset of probes for analysis that included all intensity-only probes on the Y chromosome and in the pseudoautosomal (XY) region, but excluding these probes elsewhere in the genome, for a total of 594 906 probes. For all arrays, the call rate of the samples served as the initial screen for data quality. The B-allele frequencies for each sample were examined for imbalance of A and B alleles (AA versus BB versus AB) as indicators of suboptimal performance. Samples with call rates less than 98% were re-run, re-scanned or the DNA re-extracted. Data sets with log R ratio standard deviations above 0.35 were deemed noisy and were also re-run, re-scanned or the DNA was re-extracted.
Copy number detection and analysis
HumanHap 550 V3 and Quad610 chips use Build36 coordinates. All copy number variation calls were visually detected by using Illumina's BeadStudio software. Mosaic changes were detected by assessing for aberrations in probe intensities (as measured by log R ratios) along with a shift in genotype frequencies of the SNP probes (as measured by B allele frequencies). The expected B allele frequencies for a variety of mosaic models were calculated using the formula:
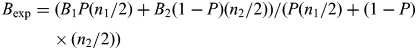 |
where B is the B allele frequency for a given SNP in cell line 1 or 2, P the percent mosaicism (in terms of cell line 1) and n the copy number for a given SNP in cell line 1 or 2. For each model (trisomy, deletion, duplication, LOH and chimerism), a table was used to calculate the expected B allele frequencies at various mosaic levels (Supplementary Material, Table S1).
Parent of origin analysis
Informative SNPs were identified using a Perl program from parental genotyping information exported from BeadStudio. Informative SNPs were then compared with genotypes for the proband to identify parent of origin for UPD cases. In cases where mosaic UPD was suspected, the genotype for each SNP of the proband's euploid cell line was modeled using the expected B allele frequency formula. The haplotype of the additional chromosome was identified, as well as the euploid cell line. Parental samples were compared with these modeled genotypes to determine parent of origin. Similar modeling was also performed in cases of mosaic monosomy.
Validation
Patient samples were validated by cytogenetics, FISH and/or clinical testing for UPD including microsatellite markers or methylation testing. FISH was carried out by standard methods using either a commercially available probe (Vysis, Inc. or Cytocell, Inc.), or using Bacterial Artificial Chromosome (BAC) or fosmid probes that were grown and labeled for this analysis. DNA clones were ordered from CHORI (bacpac.chori.org). DNA purification was carried out according to standard protocols using the PureLink HiPure Filter Maxi Kit. DNA was labeled by nick translation using a commercially available kit (Vysis, Inc.).
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by funds from The Department of Pathology, The Children's Hospital of Philadelphia, The Ring Chromosome 20 Foundation and The Foerderer Foundation to N.B.S., and a Ruth L. Kirschstein Nation Research Service Award (T32 GM008638-10) to L.K.C.
Conflict of Interest statement. No authors have any conflicts of interest to declare.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Doris DiPatri, Maria Skorski, Latricia Lewis-Bunch, Cathy Cameron, Rochelle Kline, Nancy Owens, Anna Szymanski, Adam Gleason and Surabhi Mulchandani of the clinical CytoGenomics Laboratory at The Children's Hospital of Philadelphia for patient sample analysis, and help with all aspects of this project. Genotyping was carried out in the Center for Applied Genomics at CHOP, and we thank Cecelia Kim, Joe Glessner, Ed Frackleton and Kelly Thomas. We also thank Xiaowu Gai, Mike Xie, Juan Perin and Pete White, of the Center for Biomedical Informatics for collaboration in setting up the workflow behind this project. We are grateful to Jaclyn Biegel, Tamim Shaikh and members of their group for helpful discussion. We thank the numerous clinicians and genetic counselors who referred patients for these studies.
REFERENCES
- 1.Hassold T.J., Jacobs P.A. Trisomy in man. Annu. Rev. Genet. 1984;18:69–97. doi: 10.1146/annurev.ge.18.120184.000441. [DOI] [PubMed] [Google Scholar]
- 2.Hassold T., Hall H., Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum. Mol. Genet. 2007;16(Spec No. 2):R203–R208. doi: 10.1093/hmg/ddm243. [DOI] [PubMed] [Google Scholar]
- 3.Kalousek D.K. Pathogenesis of chromosomal mosaicism and its effect on early human development. Am. J. Med. Genet. 2000;91:39–45. doi: 10.1002/(sici)1096-8628(20000306)91:1<39::aid-ajmg7>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 4.Engel E. A fascination with chromosome rescue in uniparental disomy: Mendelian recessive outlaws and imprinting copyrights infringements. Eur. J. Hum. Genet. 2006;14:1158–1169. doi: 10.1038/sj.ejhg.5201619. [DOI] [PubMed] [Google Scholar]
- 5.Kotzot D. Complex and segmental uniparental disomy updated. J. Med. Genet. 2008;45:545–556. doi: 10.1136/jmg.2008.058016. [DOI] [PubMed] [Google Scholar]
- 6.Robinson W.P. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays. 2000;22:452–459. doi: 10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 7.Ledbetter D.H., Engel E. Uniparental disomy in humans: development of an imprinting map and its implications for prenatal diagnosis. Hum. Mol. Genet. 1995;4 Spec No:1757–1764. doi: 10.1093/hmg/4.suppl_1.1757. [DOI] [PubMed] [Google Scholar]
- 8.Munne S. Chromosome abnormalities and their relationship to morphology and development of human embryos. Reprod. Biomed. Online. 2006;12:234–253. doi: 10.1016/s1472-6483(10)60866-8. [DOI] [PubMed] [Google Scholar]
- 9.Delhanty J.D. Mechanisms of aneuploidy induction in human oogenesis and early embryogenesis. Cytogenet. Genome. Res. 2005;111:237–244. doi: 10.1159/000086894. [DOI] [PubMed] [Google Scholar]
- 10.Bielanska M., Tan S.L., Ao A. Chromosomal mosaicism throughout human preimplantation development in vitro: incidence, type, and relevance to embryo outcome. Hum. Reprod. 2002;17:413–419. doi: 10.1093/humrep/17.2.413. [DOI] [PubMed] [Google Scholar]
- 11.Munne S., Bahce M., Sandalinas M., Escudero T., Marquez C., Velilla E., Colls P., Oter M., Alikani M., Cohen J. Differences in chromosome susceptibility to aneuploidy and survival to first trimester. Reprod. Biomed. Online. 2004;8:81–90. doi: 10.1016/s1472-6483(10)60501-9. [DOI] [PubMed] [Google Scholar]
- 12.Robinson W.P., Binkert F., Bernasconi F., Lorda-Sanchez I., Werder E.A., Schinzel A.A. Molecular studies of chromosomal mosaicism: relative frequency of chromosome gain or loss and possible role of cell selection. Am. J. Hum. Genet. 1995;56:444–451. [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson W.P., Bernasconi F., Lau A., McFadden D.E. Frequency of meiotic trisomy depends on involved chromosome and mode of ascertainment. Am. J. Med. Genet. 1999;84:34–42. [PubMed] [Google Scholar]
- 14.Hook E.B. Exclusion of chromosomal mosaicism: tables of 90, 95 and 99% confidence limits and comments on use. Am. J. Hum. Genet. 1977;29:94–97. [PMC free article] [PubMed] [Google Scholar]
- 15.Priest J.H., Rust J.M., Fernhoff P.M. Tissue specificity and stability of mosaicism in Pallister-Killian +i(12p) syndrome: relevance for prenatal diagnosis. Am. J. Med. Genet. 1992;42:820–824. doi: 10.1002/ajmg.1320420615. [DOI] [PubMed] [Google Scholar]
- 16.Malan V., Vekemans M., Turleau C. Chimera and other fertilization errors. Clin. Genet. 2006;70:363–373. doi: 10.1111/j.1399-0004.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- 17.Kotzot D. Maternal uniparental disomy 7 and Silver-Russell syndrome: clinical update and comparison with other subgroups. Eur. J. Med. Genet. 2008;51:444–451. doi: 10.1016/j.ejmg.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Offiah A.C., Cornette L., Hall C.M. Paternal uniparental disomy 14: introducing the "coat-hanger" sign. Pediatr. Radiol. 2003;33:509–512. doi: 10.1007/s00247-003-0931-8. [DOI] [PubMed] [Google Scholar]
- 19.Hussain K., Flanagan S.E., Smith V.V., Ashworth M., Day M., Pierro A., Ellard S. An ABCC8 gene mutation and mosaic uniparental isodisomy resulting in atypical diffuse congenital hyperinsulinism. Diabetes. 2008;57:259–263. doi: 10.2337/db07-0998. [DOI] [PubMed] [Google Scholar]
- 20.Slatter R.E., Elliott M., Welham K., Carrera M., Schofield P.N., Barton D.E., Maher E.R. Mosaic uniparental disomy in Beckwith-Wiedemann syndrome. J. Med. Genet. 1994;31:749–753. doi: 10.1136/jmg.31.10.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strain L., Warner J.P., Johnston T., Bonthron D.T. A human parthenogenetic chimera. Nat. Genet. 1995;11:164–169. doi: 10.1038/ng1095-164. [DOI] [PubMed] [Google Scholar]
- 22.Ballif B.C., Rorem E.A., Sundin K., Lincicum M., Gaskin S., Coppinger J., Kashork C.D., Shaffer L.G., Bejjani B.A. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am. J. Med. Genet. A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- 23.Cross J., Peters G., Wu Z., Brohede J., Hannan G.N. Resolution of trisomic mosaicism in prenatal diagnosis: estimated performance of a 50K SNP microarray. Prenat. Diagn. 2007;27:1197–1204. doi: 10.1002/pd.1884. [DOI] [PubMed] [Google Scholar]
- 24.Xiang B., Li A., Valentin D., Nowak N.J., Zhao H., Li P. Analytical and clinical validity of whole-genome oligonucleotide array comparative genomic hybridization for pediatric patients with mental retardation and developmental delay. Am. J. Med. Genet. A. 2008;146A:1942–1954. doi: 10.1002/ajmg.a.32411. [DOI] [PubMed] [Google Scholar]
- 25.Warburton D., Dallaire L., Thangavelu M., Ross L., Levin B., Kline J. Trisomy recurrence: a reconsideration based on North American data. Am. J. Hum. Genet. 2004;75:376–385. doi: 10.1086/423331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.James R.S., Jacobs P.A. Molecular studies of the aetiology of trisomy 8 in spontaneous abortions and the liveborn population. Hum. Genet. 1996;97:283–286. doi: 10.1007/BF02185754. [DOI] [PubMed] [Google Scholar]
- 27.Karadima G., Bugge M., Nicolaidis P., Vassilopoulos D., Avramopoulos D., Grigoriadou M., Albrecht B., Passarge E., Anneren G., Blennow E., et al. Origin of nondisjunction in trisomy 8 and trisomy 8 mosaicism. Eur. J. Hum. Genet. 1998;6:432–438. doi: 10.1038/sj.ejhg.5200212. [DOI] [PubMed] [Google Scholar]
- 28.Baidas S., Chen T.J., Kolev V., Wong L.J., Imholte J., Qin N., Meck J. Constitutional trisomy 8 mosaicism due to meiosis II non-disjunction in a phenotypically normal woman with hematologic abnormalities. Am. J. Med. Genet. A. 2004;124A:383–387. doi: 10.1002/ajmg.a.20390. [DOI] [PubMed] [Google Scholar]
- 29.McQuillan R., Leutenegger A.L., Abdel-Rahman R., Franklin C.S., Pericic M., Barac-Lauc L., Smolej-Narancic N., Janicijevic B., Polasek O., Tenesa A., et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L.H., Ho S.F., Chen C.H., Wei C.Y., Wong W.C., Li L.Y., Hung S.I., Chung W.H., Pan W.H., Lee M.T., et al. Long contiguous stretches of homozygosity in the human genome. Hum. Mutat. 2006;27:1115–1121. doi: 10.1002/humu.20399. [DOI] [PubMed] [Google Scholar]
- 31.Chowdhury R., Bois P.R., Feingold E., Sherman S.L., Cheung V.G. Genetic analysis of variation in human meiotic recombination. PLoS Genet. 2009;5:e1000648. doi: 10.1371/journal.pgen.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neas K., Peters G., Jackson J., Tembe M., Wu Z.H., Brohede J., Hannan G.N., Collins F. Chromosome 7 aberrations in a young girl with myelodysplasia and hepatoblastoma: an unusual association. Clin. Dysmorphol. 2006;15:1–8. doi: 10.1097/01.mcd.0000184966.46231.49. [DOI] [PubMed] [Google Scholar]
- 33.Diego-Alvarez D., Ramos-Corrales C., Garcia-Hoyos M., Bustamante-Aragones A., Cantalapiedra D., Diaz-Recasens J., Vallespin-Garcia E., Ayuso C., Lorda-Sanchez I. Double trisomy in spontaneous miscarriages: cytogenetic and molecular approach. Hum. Reprod. 2006;21:958–966. doi: 10.1093/humrep/dei406. [DOI] [PubMed] [Google Scholar]
- 34.Leonova J., Hanson C. A study of 45,X/46,XX mosaicism in Turner syndrome females: a novel primer pair for the (CAG)n repeat within the androgen receptor gene. Hereditas. 1999;131:87–92. doi: 10.1111/j.1601-5223.1999.00087.x. [DOI] [PubMed] [Google Scholar]
- 35.Gunderson K.L., Steemers F.J., Lee G., Mendoza L.G., Chee M.S. A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.