

Abstract
Peptidoglycan is the major structural constituent of the bacterial cell wall, forming a meshwork outside the cytoplasmic membrane that maintains cell shape and prevents lysis. In Gram-negative bacteria, peptidoglycan is located in the periplasm, where it is protected from exogenous lytic enyzmes by the outer membrane. Here we show that the type VI secretion system (T6SS) of Pseudomonas aeruginosa breaches this barrier to deliver two effector proteins, Tse1 and Tse3, to the periplasm of recipient cells. In this compartment, the effectors hydrolyze peptidoglycan, thereby providing a fitness advantage for P. aeruginosa cells in competition with other bacteria. To protect itself from lysis by Tse1 and Tse3, P. aeruginosa utilizes specific periplasmically-localized immunity proteins. The requirement for these immunity proteins depends on intercellular self-intoxication through an active T6SS, indicating a mechanism for export whereby effectors do not access donor cell periplasm in transit.
Competition among bacteria for niches is widespread, fierce and deliberate. These organisms elaborate factors ranging in complexity from small diffusible molecules, to exported proteins, to multicomponent machines, in order to inhibit the proliferation of rival cells1,2. A common target of such factors is the peptidoglycan cell wall3-6. The conserved, essential, and accessible nature of this molecule makes it an Achilles heel of bacteria.
The T6SS is a complex and widely distributed protein export machine capable of cell contact-dependent targeting of effector proteins between Gram-negative bacterial cells7-10. However, the mechanism by which effectors are delivered via the secretory apparatus, and the function(s) of the effectors within recipient cells, have remained elusive. Current models of the T6SS derive from the observation that several of its components share structural homology to bacteriophage proteins11-13; it has been proposed that target cell recognition and effector delivery occur in a process analogous to bacteriophage entry14.
The observation that T6S can target bacteria was originally made through studies of the hemolysin co-regulated protein secretion island I (HSI-I)-encoded T6SS (H1-T6SS) of P. aeruginosa, which exports at least three proteins, Tse1-37,13. These proteins are unrelated to each other and lack significant primary sequence homology to characterized proteins. One substrate, Tse2, is toxic by an unknown mechanism in the cytoplasm of recipient cells lacking Tsi2, a Tse2-specific immunity protein. Here we show that Tse1 and Tse3 are lytic enzymes that degrade peptidoglycan via amidase and muramidase activity, respectively. Unlike related enzymes associated with other secretion systems15, these proteins are not required for the assembly of a functional secretory apparatus. Instead, Tse1 and Tse3 function as lytic antibacterial effectors that depend upon T6S to breach the barrier imposed by the Gram-negative outer membrane.
Contacting P. aeruginosa cells actively intoxicate each other with Tse1 and Tse3. However, the peptidoglycan of P. aeruginosa is not inherently resistant to the activities of these enzymes. To protect itself, the bacterium synthesizes immunity proteins – type VI secretion immunity 1 and 3 (Tsi1 and Tsi3) – that specifically interact with and inactivate cognate toxins in the periplasm. Orthologs of tsi1 and tsi3 appear restricted to P. aeruginosa, therefore the species is able to exploit the H1-T6SS to target closely related organisms that are likely to compete for overlapping niches, while minimizing the fitness cost associated with self-targeting.
Tse1 and Tse3 are lytic enzymes
To identify potential functions of Tse1 and Tse3, we searched their sequences for catalytic motifs using structure prediction algorithms16. Interestingly, motifs present in peptidoglycan degrading enzymes were apparent in both proteins (Fig. 1a and Supplementary Fig. 1). Tse1 contains invariant catalytic amino acids present in cell wall amidases (DL-endopeptidases)17, whereas Tse3 possesses a motif that includes a catalytic glutamic acid found in muramidases18,19.
Figure 1. Tse1 and Tse3 are lytic proteins belonging to amidase and muramidase enzyme families.

a. Genomic organization of tse1 and tse3 and their homology with characterized amidase and muramidase enzymes, respectively. Highly conserved (boxed) and catalytic (starred) residues of the respective enzyme families are indicated. SWISS-PROT entry names for the proteins shown are: Tse1 (Q9I2Q1_PSEAE), Spr (SPR_ECOLI), P60 (P60_LISIN), Tse3 (Q9HYC5_PSEAE), GEWL (LYG_ANSAN), Slt70 (SLT_ECOLI). See Supplementary Fig. 1 for full alignments.
b,d. Partial HPLC chromatograms of sodium borohydride-reduced soluble E. coli peptidoglycan products resulting from (b) digestion with Tse1 and subsequent cleavage with cellosyl or (d) digestion with Tse3 alone. Peak assignments were made based on MS; predicted structures are shown schematically with hexagons and circles corresponding to sugars and amino acid residues, respectively. Reduced sugar moieties are shown with grey fill. Full chromatograms and MS data are provided in the supplement (Supplementary Fig. 3 and Supplementary Table 1).
c. Simplified representation of Gram-negative peptidoglycan showing cleavage sites of Tse1 and Tse3 based on data summarized in b and d.
e. Growth in liquid media of E. coli producing the indicated peri-Tse proteins. Periplasmic localization was achieved by fusion to the PelB leader sequence35. Cultures were induced at the indicated time (arrow). Error bars ± s.d. (n=3).
f. Representative micrographs of strains shown in e acquired prior to complete lysis. The lipophilic dye TMA-DPH is used to highlight the cellular membranes. Supplementary Fig. 5 contains the full microscopic fields from which these images were derived. All images were acquired at the same magnification. Scale bar = 2 μm.
To test our predictions, we incubated purified Tse1 and Tse3 (Supplementary Fig. 2) with isolated E. coli peptidoglycan sacculi. Soluble products released by the enzymes were separated by high performance liquid chromatography (HPLC) and analyzed by mass spectrometry (MS). To generate separable fragments, Tse1-treated samples were digested with cellosyl, a muramidase, prior to HPLC. The observed absence of the major crosslinked fragment, and the formation of two Tse1-specific products, is consistent with enzymatic cleavage of an amide bond in the peptidoglycan peptide crosslink (Fig. 1b and Supplementary Fig. 3). Moreover, our MS data suggest that the enzyme possesses specificity for the γ-D-glutamyl-L-meso-diaminopimelic acid bond in the donor peptide stem (Fig. 1c and Supplementary Table 1). A variant of Tse1 containing an alanine substitution in its predicted catalytic cysteine ((C30A), Tse1*) did not degrade peptidoglycan (Fig. 1b).
Soluble peptidoglycan fragments released by Tse3 confirmed our prediction that the enzyme cleaves the glycan backbone between N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc) residues (Fig. 1d and Supplementary Fig. 3). Enzymes that cleave this bond can do so hydrolytically (lysozymes) or non-hydrolytically (lytic transglycosylases); the latter results in the formation of 1,6-anhydroMurNAc. Our analyses showed that Tse3 possesses lysozyme-like activity and furthermore suggest that its activity is limited to a fraction of the MurNAc-GlcNAc bonds. The enzyme solubilized a significant proportion of the sacculi to release non-crosslinked peptidoglycan fragments and high molecular weight, soluble peptidoglycan fragments (Fig. 1c, Supplementary Fig. 3 and Supplementary Table 1). A Tse3 protein with glutamine substituted at the site of the predicted catalytic glutamic acid ((E250Q), Tse3*) displayed significantly diminished activity.
If Tse1 and Tse3 degrade peptidoglycan, we reasoned the enzymes might have the capacity to lyse bacterial cells. Ectopic expression of Tse1 and Tse3 in the cytoplasm of Escherichia coli resulted in no significant lysis (Supplementary Fig. 4a,b). However, periplasmically-localized forms of both proteins (peri-Tse1, peri-Tse3) abruptly lysed cells following induction (Fig. 1e and Supplementary Fig. 4c). In accordance with our in vitro studies, peri-Tse1* and peri-Tse3* did not induce lysis at expression levels equivalent to those of the native enzymes (Supplementary Fig. 4d). We also examined cells producing the periplasmically localized enzymes using fluorescence microscopy. Consistent with our biochemical data, cells producing peri-Tse1 were amorphous or spherical, while those producing peri-Tse3 were swollen and filamentous (Fig. 1f and Supplementary Fig. 5). In total, these data demonstrate that Tse1 and Tse3 are enzymes that degrade peptidoglycan in vivo, and that, unlike related enzymes involved in cell wall metabolism, they possess no inherent means of accessing their substrate in the periplasmic space.
T6S function does not require Tse1&3
Since the Tse enzymes alone are unable to reach their target cellular compartment, we hypothesized that their function must be linked to export by the T6SS. In this regard, they could: 1) remodel donor peptidoglycan to allow for the assembly of the mature T6S apparatus, 2) remodel recipient cell peptidoglycan to facilitate the passage of the T6S apparatus through the recipient cell wall, or 3) act as antibacterial effectors that compromise recipient cell wall integrity. To determine if Tse1 and Tse3 are essential for T6S apparatus assembly, we examined whether the enzymes are required for export of the third effector, Tse2. The secretion of Tse2 was not diminished in a strain lacking tse1 and tse3, suggesting that assembly of the T6S apparatus is unhindered by their absence (Fig. 2a). If Tse1 and Tse3 act as enzymes that remodel recipient cell peptidoglycan to facilitate effector translocation, Tse2 action on recipient cells should be severely impaired or nullified in the Δtse1 Δtse3 background. Instead, we found that this strain retained the ability to functionally target Tse2 to recipient cells (Fig. 2b). These findings led us to further examine the hypothesis that Tse1 and Tse3 are effector proteins rather than accessory enzymes of the T6S apparatus.
Figure 2. Tse1 and Tse3 are not required for Tse2 export or transfer to recipient cells via the T6S apparatus.
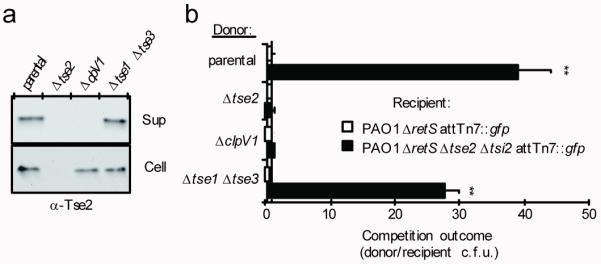
a. Western blot analysis of supernatant (Sup) and cell-associated (Cell) fractions of the indicated P. aeruginosa strains. The parental background for all experiments represented in this figure is PAO1 ΔretS, a strain in which the H1-T6SS is activated constitutively13,36.
b. Growth competition assays between the indicated donor and recipient strains under T6S-conducive conditions. Experiments were initiated with equal colony forming units (c.f.u.) of donor and recipient bacteria as denoted by the dashed line. The ΔclpV1 strain is a T6S-deficient control. Asterisks indicate significant differences in competition outcome between recipient strains against the same donor strain. **P < 0.01. Error bars ± s.d. (n=3).
Immunity proteins inhibit Tse1&3
Previous data indicate that P. aeruginosa can target itself via the T6SS7. If Tse1 and Tse3 act as antibacterial effectors, it follows that P. aeruginosa must be immune to their toxic effects. The tse1 and tse3 genes are each found in predicted bicistronic operons with a hypothetical gene, henceforth referred to as tsi1 and tsi3, respectively. Immunity proteins often inactivate their cognate toxin by direct interaction20; therefore, as a first step toward defining a functional link between cognate Tsi and Tse proteins, we asked whether they physically associate. A solution containing a mixture of purified Tse1 and Tse3 was mixed with E. coli lysates containing either Tsi1 or Tsi3. Co-immunoprecipitation studies indicated that Tsi1 and Tsi3 interact specifically with Tse1 and Tse3, respectively, and interactions between non-cognate pairs were not detected (Fig. 3a). To investigate the immunity properties of the Tsi proteins, we measured their ability to inhibit toxicity of peri-Tse1 and peri-Tse3 in E. coli. Both Tsi1 and Tsi3 significantly decreased the toxicity of cognate, but not non-cognate Tse proteins (Fig. 3b). These results show that the activity of periplasmic Tse1 and Tse3 is specifically inhibited by cognate Tsi proteins.
Figure 3. Tsi1 and Tsi3 provide immunity to cognate toxins.

a. Western blot analysis of hexahistidine-tagged Tse proteins (–His6) in total and bead-associated fractions of an α-VSV–G (vesicular stomatitis virus glycoprotein) immunoprecipitation of VSV–G epitope fused Tsi proteins (–V) from E. coli.
b. Growth of E. coli harboring a vector expressing the indicated tse gene (top panels) or vectors expressing the indicated tse and tsi genes (bottom panels). Numbers at top indicate 10-fold serial dilutions.
c. Fluorescence micrographs showing colony growth of the indicated strains. The parental background for this experiment was PAO1 ΔretS attTn7::gfp. Growth of the Δtsi strains was rescued by the addition of 1.0% w/v NaCl to the underlying medium. For quantification of data and complementation analyses see Supplementary Fig. 7.
d. Replication rates of the indicated P. aeruginosa strains in liquid medium of low osmolarity formulated as in c. The parental strain used in this experiment was PAO1 ΔretS. Error bars ± s.d. (n=3).
T6S delivers Tse1&3 to the periplasm
Most genes encoding immunity functions are essential in the presence of their cognate toxins. However, mutations that inactivate tsi1 and tsi3 are readily generated in P. aeruginosa strains that constitutively express and export Tse1 and Tse3. Based on this observation, we hypothesized that under standard laboratory conditions, the Tse proteins do not efficiently access their substrate in the periplasm. This suggests that T6S occurs by a mechanism wherein effectors are denied access to donor cell periplasm and are instead released directly to the periplasm of the recipient cell. According to this mechanism, the tsi genes would only be essential when a strain is grown under conditions that permit intercellular transfer of effectors between neighboring cells by the T6SS. As predicted, deletions in tsi1 and tsi3 severely impaired the growth of P. aeruginosa on a solid substrate, a condition conducive to T6S-based effector delivery (Fig. 3c and Supplementary Fig. 6)21,22. In contrast, this growth inhibition did not occur in liquid media, which is not conducive to effector delivery by the T6SS (Fig. 3d). The growth inhibition phenotype required a functional T6SS and intact cognate effector genes, and consistent with the proposed functions of Tse1 and Tse3 in compromising cell wall integrity, growth of immunity deficient strains was fully rescued by increasing the osmolarity of the medium (Fig. 3c).
Bioinformatic analyses suggested that the Tsi proteins reside in the periplasm – Tsi1 as a soluble periplasmic protein and Tsi3 as an outer membrane lipoprotein. These predictions were confirmed by subcellular fractionation experiments, which indicated enrichment of the proteins in the periplasmic compartment (Fig 4a). This result, taken together with the observation that the Tsi proteins interact directly with their cognate Tse proteins (Fig. 3a), provided us with a means of addressing whether the T6SS delivers Tse proteins intercellularly to the periplasm. We reasoned that if the Tse proteins are indeed delivered to the periplasm of another bacterial cell, not only should we be able to observe intoxication between distinct donor and recipient strains of P. aeruginosa, but the production of an otherwise competent immunity protein that is mislocalized to the cytoplasm should not be able to prevent such intoxication.
Figure 4. Tse1 and Tse3 delivered to the periplasm provide a fitness advantage to donor cells.

a. Western blot analyses of cytoplasmic (Cyto) and periplasmic (Peri) fractions of P. aeruginosa strains producing Tsi1–V, Tsi3–V or Tsi3–SS–V. Equivalent ratios of the Cyto and Peri samples were loaded in each panel. RNA polymerase (RNAP) and β-lactamase (β-lac) enzymes were used as cytoplasmic and periplasmic fractionation controls, respectively. The presence of Tsi3–a predicted outer membrane lipoprotein–in the periplasmic fraction is consistent with previous studies utilizing this method of fractionation37.
b. Growth competition assays between the indicated donor and recipient strains under T6S-conducive conditions. Experiments were initiated with equal c.f.u. of donor and recipient bacteria as denoted by the dashed line. The parental strain used in this experiment was PAO1 ΔretS. All donor strains were modified at the attB site with lacZ. Asterisks indicate outcomes significantly different than parental versus Δtse3 Δtsi3 (top bar). Error bars ± s.d. (n=4). **P < 0.01.
c. Lysis of EDTA-permeabilized or intact P. aeruginosa cells with equal quantities of Tse1, Tse1*, or Lysozyme (Ly). Lysis was normalized to a buffer control. Error bars ± s.d. (n=3).
d. Competitive growth of P. aeruginosa against P. putida on solid (open circles) or in liquid (filled circles) medium. Competition outcome was defined as the c.f.u. ratio (P. aeruginosa/P. putida) divided by the initial ratio. The dotted line represents the boundary between competitions that increase in P. aeruginosa relative to P. putida (above the line) and those that increase in P. putida relative to P. aeruginosa (below the line). The parental strain used in this experiment was P. aeruginosa PAO1. Asterisks above competitions denote those where the outcome (P. aeruginosa/P. putida) was significantly less than the parental (P < 0.05). Horizontal bars denote the average value for each dataset (n=5).
In growth competition assays between distinct donor and recipient strains of P. aeruginosa, we found that recipient cells that lack Tse3 immunity and are incapable of self-intoxication (Δtse3 Δtsi3), display a growth disadvantage against donor bacteria. This phenotype depends on H1-T6SS function and Tse3 in the donor strain. In the recipient strain, ectopic expression of wild-type tsi3, but not an allele encoding a signal sequence-deficient protein (Tsi3–SS), rescues the fitness defect (Fig. 4b). Importantly, the Tsi3–SS protein used in this experiment does not reach the periplasm, and retains activity in vitro as judged by interaction with Tse3 (Fig. 4a and Supplementary Fig. 7). The Tsi3–SS protein also fails to rescue the intercellular self-intoxication growth phenotype of Δtsi3 (Supplementary Fig. 6). Analogous experiments with Tsi1 were not feasible, as the protein was unstable in the cytoplasm.
The most parsimonious explanation for T6S-mediated intercellular toxicity by Tse1 and Tse3 is that the apparatus provides a conduit for the effectors through the outer membrane of recipient cells. This led us to predict that exogenous Tse1 and Tse3 would not lyse intact P. aeruginosa. Furthermore, we posited that if the outer membrane was the relevant barrier to Tse1 and Tse3 toxicity, compromising its integrity should render P. aeruginosa susceptible to exogenous administration of the enzymes.
To test these predictions, we measured lysis of permeabilized and intact P. aeruginosa following addition of exogenous Tse1. We did not test Tse3, as the filamentous phenotype induced by this enzyme would not affect non-growing, permeabilized cells. Intact P. aeruginosa cells were not affected by the addition of exogenous Tse1; conversely, permeabilized P. aeruginosa was highly susceptible to lysis by the enzyme (Fig. 4c). Lysis induced by Tse1 is linked to its enzymatic function, as Tse1* failed to significantly lyse cells. In total, our data show that the T6SS breaches the outer membrane to deliver lytic effector proteins directly to recipient cell periplasm.
To determine whether the T6SS can target the Tse proteins to cells of another Gram-negative organism, we conducted growth competition assays between P. aeruginosa and P. putida. These bacteria can be co-isolated from the environment23 and are likely to compete for niches24. While inactivation of either tse1 or tse3 only modestly affected the outcome of P. aeruginosa-P. putida competition assays, the fitness of P. aeruginosa lacking both genes or a functional T6SS was dramatically impaired (Fig. 4a). This partial redundancy is congruent with the enzymes exerting their effects through a single target–peptidoglycan–in the recipient cell. The fitness advantage provided by Tse1 and Tse3 was lost in liquid medium, consistent with cell contact-dependent delivery of the proteins to competitor cells (Fig. 4d). These data indicate that the T6SS targets its effectors to other species of bacteria and that these proteins can be key determinants in the outcome of interspecies bacterial interactions. In contrast with intraspecies intoxication, interspecies intoxication via the T6SS does not require the inactivation of a negative regulator of the system (eg. ΔretS), suggesting that T6S function is stimulated in response to rival bacteria.
Discussion
Our data lead us to propose a model for T6S-catalyzed translocation of effectors to the periplasm of recipient bacteria (Fig. 5). This model provides a mechanistic framework for understanding the form and function of this complex secretion system. Our findings strengthen the existing hypothesis that the T6SS is evolutionarily and functionally related to bacteriophage8,14,25. Neither the T4 bacteriophage tail spike nor other components of the puncturing device are thought to cross the inner membrane; instead, bacteriophage DNA is released to the periplasm and subsequently enters the cytoplasmic compartment using another pathway26. By analogy, the Tse proteins would utilize T6S components as a puncturing device to gain access to the periplasm, whereupon Tse2 may then utilize an independent route to access the cytoplasm (Fig. 5).
Figure 5. Proposed mechanism of T6S-dependent delivery of effector proteins.

The schematic depicts the junction between competing bacteria, with a donor cell delivering the Tse effector proteins through the T6S apparatus (grey tube) to recipient cell periplasm. Effector and immunity proteins are shown as circles and rounded rectangles, respectively. Bonds in the peptidoglycan that are predicted targets of the effector proteins are highlighted (red). Cytoplasm (C), inner membrane (IM), periplasm (P), and outer membrane (OM) of both bacteria are shown.
Niche competition in natural environments has clearly selected for potent antibacterial processes; however, the human body is also home to a complex and competitive microbiota27,28. Commensal bacteria form a protective barrier, and the ability of pathogens to colonize the host is not only dependent upon suppression or subversion of host immunity, but also can depend on their ability to displace these more innocuous organisms29-31. In polymicrobial infections, Gram-negative bacteria, including P. aeruginosa, often vie with other Gram-negative bacteria for access to nutrient-rich host tissue32. Factors such as the T6SS, that influence the relative fitness of these organisms, are thus likely to impact disease outcome.
Methods Summary
P. aeruginosa strains used in this study were derived from the sequenced strain PAO133. All deletions were in-frame and unmarked, and were generated by allelic exchange. E. coli growth curves were conducted using BL21 pLysS cells harboring expression plasmids for tse and tsi genes. Intercellular self-intoxication and interbacterial competition assays were performed by spotting mixed overnight cultures on a nitrocellulose membrane placed on a 3% agar growth medium. Samples were incubated at 37°C (P. aeruginosa-P. aeruginosa) or 30°C (P. aeruginosa-P. putida) for 12 or 24 hours. Tse1-catalyzed P. aeruginosa lysis was measured by placing cells in a minimal buffer ± 1.5 mM EDTA containing either Tse1, Tse1* or lysozyme. The change in optical density at 600 nm following 5 min of incubation was used to calculate lysis. For determination of Tse1 and Tse3 activity, isolated E. coli peptidoglycan sacculi were incubated with the purified enzymes (100 μg/mL). The resulting peptidoglycan and soluble fragments released by the enzymes were separated by HPLC and their identities were determined using MS as described previously34.
Supplementary Material
Acknowledgements
We thank P. Singh, E. Nester, H. Kulasekara, N. Salama, E.P. Greenberg, L. Ramakrishnan and members of the Mougous laboratory for insightful discussions and critical reading of the manuscript, the Harwood laboratory for use of their microscope, and Joe Gray of the Pinnacle Laboratory of Newcastle University for MS analysis. This work was supported by the National Institutes of Health (J.D.M.; RO1 AI080609) and the European Commission within the DIVINOCELL programme (W.V.). A.B.R was supported by a Graduate Research Fellowship from the National Science Foundation.
APPENDIX
Online-only Methods
Bacterial strains, plasmids, and growth conditions
P. aeruginosa strains used in this study were derived from the sequenced strain PAO133. P. aeruginosa strains were grown on either Luria-Bertani media (LB), or the equivalent lacking additional NaCl (LB low salt (LB-LS): 10 g bactopeptone and 5 g yeast extract per liter) at 37 °C supplemented with 30 μg ml−1 gentamycin, 25 μg ml−1 irgasan, 5% w/v sucrose, 40μg/ml X-gal, and stated concentrations of IPTG as required. E. coli strains included in this study included DH5α for plasmid maintenance, SM10 for conjugal transfer of plasmids into P. aeruginosa, BL21 pLysS for expression of Tse1 and Tse3 for toxicity and lysis, and Shuffle® T7 pLysS Express (New England Biolabs), for purification of Tse1 and Tse3. All E. coli strains were grown on either LB or LB-LS at 37 °C supplemented with 15 μg ml−1 gentamycin, 150 μg ml−1 carbenicillin, 50 μg ml−1 kanamycin, 30 μg ml−1 chloramphenicol, 200 μg ml−1 trimethoprim, 0.1% rhamnose, and stated concentrations of IPTG as required. P. putida used in this study was the sequenced strain, KT244024. P. putida was grown on LB or LB-LS at 30°C. In all experiments where expression from a plasmid was required, strains were grown on media supplemented with required antibiotics to select for plasmid maintenance.
Plasmids used for inducible expression were pPSV35CV for P. aeruginosa and pET29b+ (Novagen), pET22b+ (Novagen), pSCrhaB238 and pPSV35CV for E. coli39. Chromosomal deletions were made as described previously40.
DNA manipulations
The creation, maintenance, and transformation of plasmid constructs followed standard molecular cloning procedures. All primers used in this study were obtained from Integrated DNA Technologies. DNA amplification was carried out using either Phusion® (New England Biolabs) or Mangomix™ (Bioline). DNA sequencing was performed by Genewiz® Incorporated. Restriction enzymes were obtained from New England Biolabs. SOE PCR was performed as previously described41.
Plasmid construction
pPSV35CV, pEXG2, and pSCrhaB2 have been described previously38-40. E. coli pET29+ expression vectors for Tse1 and Tse3 were constructed by standard cloning techniques following amplification from PAO1 chromosomal DNA using the primer pairs 1289/1290 and 1291/1292, respectively. E. coli pET22b+ expression vectors for Tse1 and Tse3 were constructed in a similar manner using primer pairs 1477/1478 and 1475/1476. Point mutations were introduced using Quikchange (Stratagene) with primer pairs 1479/1480 and 1481/1482 for the production of tse1(C30A) and tse3(E250Q), respectively.
pPSV35CV expression vectors for Tsi1 and Tsi3 were generated by amplifying the genes from genomic DNA using primer pairs 1469/1470 and 1472/1473, respectively. The Tsi3-SS pPSV35CV expression vector was generated from a product amplified using the primer pair 1522/1473. The pSCrhaB2 vectors for expressing Tsi proteins in E. coli were produced by amplifying the genes using primer pairs 1470/1497 for tsi1 and 1473/1498 for tsi3. A VSV-epitope tag was then cloned downstream of these two genes for the purpose of tagged-expression.
All deletions were in-frame and were generated by exchange with deletion alleles constructed by SOE PCR. For tse1, tse3, tsi1, and tsi3 deletion constructs, upstream DNA flanking sequences were amplified by 628/629, 735/736, 721/722, and 1485/1486, respectively. Downstream flanking DNA sequences were amplified by 630/631, 737/738, 723/724, and 1487/1488, respectively. Deletions of both effector and immunity protein were accomplished by amplifying upstream regions of tse1-tsi1 and tse3-tsi3 with 721/722 and 735/736 respectively and downstream regions with 628/629 and 835/836 respectively.
Growth curves
For E. coli growth curves BL21 pLysS cells harboring expression plasmids were grown overnight in liquid LB shaking at 37°C and subinoculated to a starting optical density at 600 nm (OD600) of between 0.01 and 0.02 in LB-LS. Cultures were grown to OD600 0.1-0.2 and induced with 0.1mM IPTG. The vector pET29b+ was used for expression of native Tse1 and Tse3, and the pET22b+ vector was used for expression of periplasmic Tse1 and Tse3, and catalytic amino acid substitutions thereof. Both vectors added a C-terminal hexahistidine tag to expressed proteins, allowing for western blot analysis of expression. Samples for western blot analysis were taken 30 minutes after induction for Tse1, peri-Tse1, and peri-Tse1* and 45 minutes after induction for Tse3, peri-Tse3, and peri-Tse3*.
For P. aeruginosa growth curves, cells were grown overnight at 37 °C in liquid LB with shaking and sub-inoculated 1:1000 into LB-LS. Growth was measured by enumerating c.f.u. from plate counts of samples taken at the indicated time points.
E. coli toxicity measurements
Overnight LB cultures of E. coli harboring pET22b+ expression vectors and E. coli harboring both pET22b+ and pSCrhaB2 expression vectors were serially diluted in LB to 106 as 10-fold dilutions. These dilutions were spotted onto LB-LS agar with the following concentrations of inducer molecules: 0.075mM IPTG for pET22b+::tse1, pET22b+::tse3 and the associated vector control, 0.02 mM IPTG and 0.1% rhamnose for pET22b+::tse1 pSCrhaB2::tsi1 and all associated controls, and 0.05mM IPTG and 0.1% rhamnose for pET22b+::tse3 pSCRhaB2::tsi3 and all associated controls. Pictures were taken between 20 and 26 hours after plating.
Subcellular fractionation
P. aeruginosa ΔretS cells harboring expression vectors for Tsi1-V, Tsi3-V, or Tsi3–SS-V and an additional vector expressing TEM-1 (pPSV18) were grown overnight. This overnight culture was sub-inoculated into LB supplemented with 0.1mM IPTG and grown to late logarithmic phase. Periplasmic and cytoplasmic fractions were prepared as described37,42.
E. coli BL21 cells harboring expression vectors for Tse1*, Tse3*, peri-Tse1*, and peri-Tse3* were grown overnight and sub-inoculated into LB. For Tse1* and Tse3* fractionation cells also carried an empty pET22b vector to provide expression of TEM-1. Cells were grown to an OD600 of 0.1 and induced with either 0.1 mM IPTG (Tse1* and peri-Tse1*) or 0.5 mM IPTG (Tse3* and peri-Tse3*). Cells were then harvested and fractionated as described43.
Preparation of proteins and Western blotting
Cell-associated and supernatant samples were prepared as described previously39. Western blotting was performed as described previously for α-VSV–G and α-RNA polymerase13 with the modification that α-VSV–G antibody probing was performed in 5% BSA in Tris-buffered saline containing 0.05% v/v Tween 20. The α-Tse2 polyclonal rabbit antibody was raised against the peptide YDGDVGRYLHPDKEC (GenScript). Western blots using both this antibody and the α-β-lactamase antibody(QED Biosciences Inc.) were performed identically to those using α-VSV–G. The α-His5 Western blots were performed using the Penta-His HRP Conjugate Kit according to manufacturer’s instructions (Qiagen).
Immunoprecipitation
BL21 pLysS cells expressing VSV-G-tagged Tsi1, Tsi3, or Tsi3–SS were pelleted and resuspended in lysis buffer (20mM Tris-Cl pH 7.5, 50 mM KCl, 8.0% v/v glycerol, 0.1% v/v NP 40, 1.0% v/v triton, supplemented with Dnase I (Roche), lysozyme (Roche), and Sigmafast™ protease inhibitor (Sigma) according to manufacturer instructions). Cells were disrupted by sonication to release VSV–G-tagged Tsi proteins into solution. To this suspension, Tse1 and Tse3 were added to concentrations of 30 μg ml−1 and 25 μg ml−1, respectively. This mixture was clarified by centrifugation, and a sample of the supernatant was taken as a pre-immunoprecipitation sample. The remainder of the supernatant was incubated with 100 μL α-VSV–G agarose beads (Sigma) for 2 hr at 4°C. Beads were washed three times with IP-wash buffer (100mM NaCl, 25mM KCl, 0.1% v/v triton, 0.1% v/v NP-40, 20mM Tris-Cl pH 7.5, and 2% v/v glycerol). Proteins were removed from beads with SDS loading buffer (125 mM Tris, pH 6.8, 2% (w/v) 2-Mercaptoethanol, 20% (v/v) Glycerol, 0.001% (w/v) Bromophenol Blue and 4% (w/v) SDS) and analyzed by Western blot.
Interbacterial competition assays
The inter-P. aeruginosa competitions were performed as described previously with minor modifications7. For experiments described in both Figure 2b and Figure 4b, competition assays were performed on nitrocellulose on LB or LB-LS 3% agar, respectively. Plate counts were taken of the initial inoculum to ensure a starting c.f.u. ratio of 1:1, and again after either 24 hours (Figure 2b) or 12 hours (Figure 4b) to obtain a final c.f.u. ratio. Donor and recipient colonies were disambiguated through fluorescence imaging (Figure 2e) or through the activity of a β–galactosidase reporter as visualized on plates containing 40μg/ml X-gal (Figure 4b)5. Data were analyzed using a two-tailed Student’s T-Test.
For interspecies competition assays, overnight cultures of P. aeruginosa and P. putida were grown overnight in LB broth at 37°C and 30°C, respectively. Cultures were then washed in LB and resuspended to an OD600 of 4.0 for P. aeruginosa and 4.5 for P. putida. P. putida and P. aeruginosa were mixed in a one-to-one ratio by volume, this mixture was spotted on a nitrocellulose membrane placed on LB-LS 3% agar, and the c.f.u. ratio of the organisms was measured by plate counts. The assays were incubated for 24 hours at 30°C, after which the cells were resuspended in LB broth and the final c.f.u. ratio determined through plate counts. Data were analyzed using a one-tailed Student’s T-Test.
Purification of Tse1 and Tse3
For purification, Tse1, Tse3, Tse1*, and Tse3* were expressed in pET29b+ vectors in Shuffle® Express T7 lysY cells (New England Biolabs). The proteins were purified to homogeneity using previously reported methods44, except that in all steps no reducing agents or lysozyme were used.
Bioinformatic analyses
Predicted structural homology was queried using PHYRE16. Alignments were performed using T-Espresso45. Sequences of cell wall amidases and muramidases for alignments were obtained from seed sequences from PFAM46. Critical motifs were defined by previous work in the study of NlpC/P60 and lytic transglycosylase/GEWL enzymes17,18.
Enzymatic assays
Tse1 and Tse1*
Purified peptidoglycan sacculi (300 μg) from E. coli MC106147 were incubated with Tse1 or Tse1* (100 μg/ml) in 300 μl of 20 mM Tris/HCl, pH 8.0 for 4 h at 37°C. A sample with enzyme buffer instead of Tse1 served as a control. The pH was adjusted to 4.8 and the sample was incubated with 40 μg/ml of the muramidase cellosyl (kindly provided by Höchst AG, Frankfurt, Germany) for 16 h at 37°C to convert the residual peptidoglycan or solubilized fragments into muropeptides. The sample was boiled for 10 min and insoluble material was removed by brief centrifugation. The reduced muropeptides were reduced with sodium borohydride and analysed by HPLC as described47. Fractions 1 and 2 were collected, concentrated in a SpeedVac, acidified by 1% trifluoroacetic acid and analysed by offline electrospray mass spectrometry on a Finnigan LTQ-FT mass spectrometer (ThermoElectron, Bremen, Germany) as described34.
Tse3 and Tse3*
Purified peptidoglycan sacculi (300 μg) from E. coli MC1061 were incubated with Tse3 or Tse3* (100 μg/ml) in 300 μl of 20 mM sodium phosphate, pH 4.8 for 20 h at 37°C. A sample with enzyme buffer instead of Tse3 served as a control. The samples were boiled for 10 min and centrifuged for 15 min (16,000×g). The supernatant was reduced with sodium borohydride and analysed by HPLC as described above (supernatant samples). The pellet was resuspended in 20 mM sodium phosphate, pH 4.8 and incubated with 40 μg/ml cellosyl for 14 h at at 37°C. The samples were boiled for 10 min, cleared by brief centrifugation and analysed by HPLC as described above (pellet samples). Fractions 3, 4 and 5 were collected and analysed by mass spectrometry as described above.
Self-intoxication assays
PAO1 ΔretS attTn7::gfp cells bearing the indicated gene deletions were grown overnight in LB broth at 37°C. Cells were then diluted to 103 c.f.u./mL and 20 μL of this solution was placed on a nitrocellulose membrane placed on LB-LS 3% agar or LB 3% agar (contains 1.0% w/v NaCl). Fluorescence images were acquired following 23 hours of incubation at 37°C. For quantification and complementation, non-fluorescent strains were used and 1 mM IPTG was included for induction of all strains – except for the tsi3-complemented strain, for which no IPTG was required to achieve comparable levels of expression to the tsi3–SS-complemented strain. At 23 hours cells were resuspended in LB. Plate counts of the initial inoculum and the final suspension were used to determine growth. Data were analyzed using a one-tailed Student’s T-test.
Fluorescence microscopy
BL21 pLysS cells harboring periplasmic-expression vectors for Tse1, Tse3, and catalytic substitution mutants were grown in conditions identical to those in the E. coli growth curve experiments. Cells were harvested 30 minutes post-induction for Tse1 experiments and one-hour post-induction for Tse3 experiments. These cells were resuspended in PBS and incubated with 0.3 μM TMA-DPH (1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate) for 10 minutes. The stained cells were placed on 1% agarose pads containing PBS for microscopic analysis. Microscopy was performed as described previously39.
EDTA-permeabilization lysis assay
Assays were performed as previously described with minor modifications48. Cells were sub-inoculated into LB broth from overnight liquid cultures and grown to late logarithmic phase. Cells were washed in 20 mM Tris-Cl pH 7.5 and Tse1, Tse1*, or lysozyme were added to a final concentration of 0.01 mg/mL. An initial OD600 measurement was taken before EDTA pH 8.0 was added to a final concentration of 1.5 mM. Cells were incubated with shaking at 37°C for 5 minutes and a final OD600 reading was taken. P. aeruginosa undergoes rapid autolysis under these assay conditions, thus lysis was expressed as a percentage of lysis above a buffer-only control.
Online-only methods references
- 38.Cardona ST, Valvano MA. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid. 2005;54:219–228. doi: 10.1016/j.plasmid.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Hsu F, Schwarz S, Mougous JD. TagR promotes PpkA-catalysed type VI secretion activation in Pseudomonas aeruginosa. Mol Microbiol. 2009;72:1111–1125. doi: 10.1111/j.1365-2958.2009.06701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2005;102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horton RM, et al. Gene splicing by overlap extension. Methods in enzymology. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- 42.Wood PM. Periplasmic location of the terminal reductase in nitrite respiration. FEBS letters. 1978;92:214–218. doi: 10.1016/0014-5793(78)80757-1. doi:0014-5793(78)80757-1 [pii] [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Walsh CT. Peptidyl-prolyl cis-trans-isomerase from Escherichia coli: a periplasmic homolog of cyclophilin that is not inhibited by cyclosporin A. Proc Natl Acad Sci U S A. 1990;87:4028–4032. doi: 10.1073/pnas.87.11.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mougous JD, et al. Identification, function and structure of the mycobacterial sulfotransferase that initiates sulfolipid-1 biosynthesis. Nature structural & molecular biology. 2004;11:721–729. doi: 10.1038/nsmb802. [DOI] [PubMed] [Google Scholar]
- 45.Armougom F, et al. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic acids research. 2006;34:W604–608. doi: 10.1093/nar/gkl092. doi:34/suppl_2/W604 [pii] 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finn RD, et al. The Pfam protein families database. Nucleic acids research. 2010;38:D211–222. doi: 10.1093/nar/gkp985. doi:gkp985 [pii] 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glauner B. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal Biochem. 1988;172:451–464. doi: 10.1016/0003-2697(88)90468-x. [DOI] [PubMed] [Google Scholar]
- 48.Watt SR, Clarke AJ. Role of autolysins in the EDTA-induced lysis of Pseudomonas aeruginosa. FEMS Microbiol Lett. 1994;124:113–119. doi: 10.1111/j.1574-6968.1994.tb07270.x. [DOI] [PubMed] [Google Scholar]
References
- 1.Hayes CS, Aoki SK, Low DA. Bacterial contact-dependent delivery systems. Annual review of genetics. 2010;44:71–90. doi: 10.1146/annurev.genet.42.110807.091449. doi:10.1146/annurev.genet.42.110807.091449. [DOI] [PubMed] [Google Scholar]
- 2.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. doi: 10.1038/nrmicro2259. doi:nrmicro2259 [pii] 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundling A, Schneewind O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J Bacteriol. 2006;188:2463–2472. doi: 10.1128/JB.188.7.2463-2472.2006. doi:188/7/2463 [pii] 10.1128/JB.188.7.2463-2472.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vollmer W, Pilsl H, Hantke K, Holtje JV, Braun V. Pesticin displays muramidase activity. J Bacteriol. 1997;179:1580–1583. doi: 10.1128/jb.179.5.1580-1583.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brotz H, Bierbaum G, Markus A, Molitor E, Sahl HG. Mode of action of the lantibiotic mersacidin: inhibition of peptidoglycan biosynthesis via a novel mechanism? Antimicrob Agents Chemother. 1995;39:714–719. doi: 10.1128/AAC.39.3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley MA, Wertz JE. Bacteriocins: evolution, ecology, and application. Annual review of microbiology. 2002;56:117–137. doi: 10.1146/annurev.micro.56.012302.161024. [DOI] [PubMed] [Google Scholar]
- 7.Hood RD, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell host & microbe. 2010;7:25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarz S, Hood RD, Mougous JD. What is type VI secretion doing in all those bugs? Trends Microbiol. 2010 doi: 10.1016/j.tim.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cascales E. The type VI secretion toolkit. EMBO reports. 2008;9:735–741. doi: 10.1038/embor.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC genomics. 2009;10:104. doi: 10.1186/1471-2164-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ballister ER, Lai AH, Zuckermann RN, Cheng Y, Mougous JD. In Vitro Self-Assembly of Tailorable Nanotubes from a Simple Protein Building Block. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3733–3738. doi: 10.1073/pnas.0712247105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leiman PG, et al. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci U S A. 2009;106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mougous JD, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanamaru S. Structural similarity of tailed phages and pathogenic bacterial secretion systems. Proc Natl Acad Sci U S A. 2009;106:4067–4068. doi: 10.1073/pnas.0901205106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. Biogenesis, architecture, and function of bacterial type iv secretion systems. Annual review of microbiology. 2005;59:451–485. doi: 10.1146/annurev.micro.58.030603.123630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nature protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. doi:nprot.2009.2 [pii] 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 17.Anantharaman V, Aravind L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome biology. 2003;4:R11. doi: 10.1186/gb-2003-4-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheurwater E, Reid CW, Clarke AJ. Lytic transglycosylases: bacterial space-making autolysins. Int J Biochem Cell Biol. 2008;40:586–591. doi: 10.1016/j.biocel.2007.03.018. doi:S1357-2725(07)00097-0 [pii] 10.1016/j.biocel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008;32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. doi:FMR099 [pii] 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 20.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 21.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. doi:10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 22.Schwarz S, et al. Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 2010:6. doi: 10.1371/journal.ppat.1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortensen JE, Fisher MC, LiPuma JJ. Recovery of Pseudomonas cepacia and other Pseudomonas species from the environment. Infect Control Hosp Epidemiol. 1995;16:30–32. doi: 10.1086/646999. [DOI] [PubMed] [Google Scholar]
- 24.Nelson KE, et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environmental microbiology. 2002;4:799–808. doi: 10.1046/j.1462-2920.2002.00366.x. doi:366 [pii] [DOI] [PubMed] [Google Scholar]
- 25.Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A. 2007;104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rakhuba DV, Kolomiets EI, Dey ES, Novik GI. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol J Microbiol. 2010;59:145–155. [PubMed] [Google Scholar]
- 27.Nelson KE, et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. doi:328/5981/994 [pii] 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. doi:nature08821 [pii] 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brook I. Bacterial interference. Critical reviews in microbiology. 1999;25:155–172. doi: 10.1080/10408419991299211. [DOI] [PubMed] [Google Scholar]
- 30.Iwase T, et al. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010;465:346–349. doi: 10.1038/nature09074. [DOI] [PubMed] [Google Scholar]
- 31.Reid G, Howard J, Gan BS. Can bacterial interference prevent infection? Trends Microbiol. 2001;9:424–428. doi: 10.1016/s0966-842x(01)02132-1. [DOI] [PubMed] [Google Scholar]
- 32.Gjodsbol K, et al. Multiple bacterial species reside in chronic wounds: a longitudinal study. International wound journal. 2006;3:225–231. doi: 10.1111/j.1742-481X.2006.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stover CK, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 34.Bui NK, et al. The peptidoglycan sacculus of Myxococcus xanthus has unusual structural features and is degraded during glycerol-induced myxospore development. J Bacteriol. 2009;191:494–505. doi: 10.1128/JB.00608-08. doi:JB.00608-08 [pii] 10.1128/JB.00608-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei SP, Lin HC, Wang SS, Callaway J, Wilcox G. Characterization of the Erwinia carotovora pelB gene and its product pectate lyase. J Bacteriol. 1987;169:4379–4383. doi: 10.1128/jb.169.9.4379-4383.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodman AL, et al. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 37.Imperi F, et al. Analysis of the periplasmic proteome of Pseudomonas aeruginosa, a metabolically versatile opportunistic pathogen. Proteomics. 2009;9:1901–1915. doi: 10.1002/pmic.200800618. doi:10.1002/pmic.200800618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.