

Abstract
To determine the role of Gq signaling and calcineurin (CN) activation in promoting apoptosis of glomerular podocytes, constitutively active Gq [Gq(+)] or CN [CN(+)] proteins were introduced into cultured podocytes using protein transduction by tagging the proteins with the transactivator of transcription peptide. To investigate the role of CN in promoting podocyte apoptosis in vivo, a genetic model of type 1 diabetes mellitus (Akita mice) was treated with the CN inhibitor FK506. In cultured podocytes, Gq(+) stimulated nuclear translocation of nuclear factor of activated T cells (NFAT) family members, activated an NFAT reporter construct, and enhanced podocyte apoptosis in a CN-dependent fashion. CN(+) similarly promoted podocyte apoptosis, and apoptosis induced by either angiotensin II or endothelin-1 was blocked by FK506. Induction of apoptosis required NFAT-induced gene transcription because apoptosis induced by either Gq(+) or CN(+) was blocked by an inhibitor that prevented CN-dependent NFAT activation without affecting CN phosphatase activity. Podocyte apoptosis was mediated, in part, by the NFAT-responsive gene cyclooxygenase 2 (COX2) and prostaglandin E2 generation because apoptosis induced by Gq(+) was attenuated by either COX2 inhibition or blockade of the Gq-coupled E-series prostaglandins receptor. The findings appeared relevant to podocyte apoptosis in diabetic nephropathy because apoptosis was significantly reduced in Akita mice by treatment with FK506. These data suggest that Gq stimulates CN and promotes podocyte apoptosis both in vitro and in vivo. Apoptosis requires NFAT-dependent gene transcription and is mediated, in part, by CN-dependent COX2 induction, prostaglandin E2 generation, and autocrine activation of the Gq-coupled E-series prostaglandins receptor.
Diabetic nephropathy (DN) is the most common cause of end-stage renal disease in developed countries including the United States (1). Although current strategies slow disease progression (2), approximately one third of patients with diabetes develop end-stage renal disease requiring renal replacement therapy (1). As a result, much effort has been devoted to understanding the mechanisms that promote glomerular damage in diabetic kidney disease with the hope of identifying new therapeutic strategies.
Accumulating evidence suggests that glomerular podocytes play a pivotal role in the pathogenesis of diabetic kidney disease (3). In this regard, podocyte number is reduced in humans with either type 1 or type 2 diabetes mellitus (3–6). Because podocytes are terminally differentiated cells with little potential for proliferation (7), podocytes that are lost cannot be effectively replaced, leading to instability of the tuft and glomerulosclerosis (7). Although recent studies suggest that a population of renal progenitor cells have some capacity to regenerate podocytes (8), this regenerative potential is insufficient to prevent a reduction in glomerular podocytes in diabetic kidney disease (3). Indirect evidence suggests that this loss of glomerular podocytes contributes to progressive deterioration in renal function in patients with DN (3–6). For example, a reduction in podocyte number is a strong predictor of progressive kidney disease in Pima Indians with type 2 diabetes mellitus and microalbuminuria (4). Similarly, albumin excretion rates negatively correlate with podocyte number in proteinuric patients with type 1 diabetes mellitus (5). Moreover, the density of glomerular podocytes is reduced to a greater extent in diabetic patients with overt proteinuria compared with patients with microalbuminuria (6).
Although the causes of podocyte loss in DN are likely multifactorial (3), several studies suggest that podocyte apoptosis contributes to the reduction in glomerular podocytes in diabetic kidney disease (9, 10). For example, Bottinger and co-workers (9) found that hyperglycemia stimulated generation of reactive oxygen species (ROS) in cultured podocytes by reduced nicotinamide adenine dinucleotide phosphate and mitochondrial pathways, inducing apoptosis. Their studies also suggested that ROS production contributed to disease pathogenesis because chronic treatment with an reduced nicotinamide adenine dinucleotide phosphate oxidase inhibitor prevented podocyte apoptosis and ameliorated glomerular injury in rodent models of type 1 and type 2 diabetes (9). Although multiple mechanisms likely contribute to ROS generation in DN, studies by Abboud and colleagues (11, 12) suggest that TGFβ stimulates ROS generation, which, in turn, increases intracellular calcium levels and promotes extracellular matrix accumulation in diabetic rodents by activation of the calcium-sensitive phosphatase calcineurin (CN). These findings may be relevant to podocyte loss in DN because TGFβ potently induces apoptosis of glomerular podocytes (13).
Angiotensin II (ANGII) also promotes podocyte apoptosis (14, 15) and plays a key role in the pathogenesis of diabetic kidney disease (2). The effects of ANGII are mediated by binding to cell surface G protein coupled receptors (GPCR) belonging to the large superfamily of heptahelical receptor systems (16). Podocytes express numerous GPCR implicated in the pathogenesis of DN including receptors for ANGII (AT1), thromboxane (TP), E-series prostaglandins (EP1), and endothelin (ETA) (2, 17–19). Although the signaling pathways activated by AT1, ETA, EP1, and TP are diverse, all these GPCR systems couple to heterotrimeric G proteins of the Gq/11 family (16). Gq activation stimulates numerous downstream signaling cascades including the calcium-sensitive serine/threonine phosphatase CN (20, 21). Stimulation of CN phosphatase activity induces downstream genes by dephosphorylation and activation of NFAT (nuclear factor of activated T cells) family members (20). In unstimulated cells, NFAT transcription factors are located in the cytoplasm and are highly phosphorylated (20). Dephosphorylation of NFAT isoforms by CN causes their translocation to the nucleus and stimulation of gene transcription. Although originally thought to be expressed only in cells of the lymphoid lineage, abundant evidence now indicates that NFAT family members are expressed in nonimmune cells with some family members expressed ubiquitously (20). Indeed, we found that Gq signaling promotes cyclooxygenase 2 (COX2) expression in podocytes by stimulating NFAT-responsive elements in the COX2 promoter and, in turn, enhances prostaglandin E2 (PGE2) generation (21).
In the present studies, we investigated the role of Gq-dependent CN activation in promoting podocyte apoptosis both in cultured podocytes and in vivo. We found that CN caused podocyte apoptosis and that this apoptotic effect required activation of NFAT transcription factor isoforms and was mediated, in part, by CN-dependent COX2 induction and, in turn, PGE2 generation and autocrine activation of the Gq-coupled EP1 receptor. CN-induced apoptosis appeared relevant to the in vivo situation because: 1) Podocyte CN activity was increased in a mouse model of type 1 diabetes mellitus (Akita mice), and 2) Treatment with a pharmacological CN inhibitor reduced podocyte apoptosis in Akita mice. These data suggest that CN is an important mediator of podocyte apoptosis both in cultured podocytes and in diabetic kidney disease. We speculate that the beneficial effects of pharmacological CN inhibitors in glomerular disease processes may be mediated, at least in part, by inhibition of podocyte apoptosis.
Results
We first determined the NFAT isoforms expressed by cultured podocytes using RT-PCR and intron spanning primers (22). Figure 1A, shows that NFAT2 (NFATc1) and NFAT4 (NFATc3) were amplified from podocyte mRNA. Sequence analysis confirmed amplification of the appropriate target sequences. Because, in our hands, cultured podocytes are difficult to transfect, we used protein transduction (23) to introduce a constitutively active Gq construct (GqQ>L) (24) into cultured podocytes by tagging GqQ>L with the transactivator of transcription (TAT) HIV protein sequence [Gq(+)]. A cell-impermeable GqQ>L lacking the TAT sequence [Gq(−)] was used as a control. In Fig. 1B, nuclear localization of NFAT isoforms was assessed by immunohistochemistry (IHC) after TAT protein treatment. Gq(+) increased nuclear localization of NFAT2 compared with Gq(−), and this increase in nuclear NFAT2 was prevented by the pharmacological inhibitor of CN FK506 (20). In Fig. 1, C and D, we assessed subcellular localization of NFAT2 and NFAT4, respectively, by immunoblotting of nuclear and cytosolic fractions. Densitometric quantitation of the immunoblots is shown in Table 1. Treatment with Gq(+) increased the quantity of both NFAT family members detected in the nucleus. As reported by other investigators, the NFAT antibodies detect multiple immunoreactive bands by immunoblotting (25), and NFAT dephosphorylation causes a decrease in the apparent molecular weight (26). We were unable to detect a change in cytosolic NFAT levels by either immunoblotting or IHC, suggesting that only a fraction of the NFAT2 and NFAT4 translocated to the nucleus. Treatment with Gq(−) had no effect on nuclear localization of NFAT2 or NFAT4 compared with untreated cells (data not shown).
Fig. 1.

Effects of Gq signaling on NFAT activation. A, NFAT 2 and NFAT4 were amplified from total cellular podocyte mRNA by RT-PCR using intron-spanning primers. B, Subcellular localization of NFAT2 was studied by staining cells with an NFAT2 antibody (fluorescein) and staining podocyte nuclei with 4′,6-diamidino-2-phenylindole (blue). Merging of the images suggested that Gq(+) enhanced nuclear localization of NFAT2 compared with cells treated with Gq(−). The increase in nuclear NFAT2 induced by Gq(+) was blocked by FK506 (1 μm). Similar results were obtained in three separate experiments. C and D, Nuclear and cytosolic fractions were prepared, and both NFAT2 and NFAT4 were detected by immunoblotting as indicated. Treatment overnight with Gq(+) enhanced nuclear localization of both NFAT2 and NFAT4. There was little change in cytosolic levels of NFAT2 or NFAT4, suggesting that only a fraction of each NFAT family member translocated to the nucleus. Actin and histone 3 were used as loading controls for cytosolic and nuclear fractions, respectively. Blotting for the HA tag in the TAT constructs confirmed that Gq(+) was efficiently transduced into the cytosolic fractions. Similar results were obtained in three separate experiments. E, Podocytes were treated overnight with Gq(+) or Gq(−) in the presence of either the calcineurin inhibitors FK506 (1 μm) or MCIP(+). Control cells were treated with either DMSO vehicle or MCIP(−). Activation of an NFAT reporter construct was then assessed by measuring luciferase activity as described in Materials and Methods. Gq(+) significantly enhanced luciferase activity compared with cells treated with Gq(−). The increase in luciferase activity induced by Gq(+) was blocked by both FK506 and MCIP(+) but not by DMSO vehicle or MCIP(−). The inset shows immunoblotting of the HA-tagged Gq proteins in either culture medium or podocyte lysates. The luciferase data are the results of four separate experiments. *, P < 0.01 vs. Gq(−); †, P < 0.005 vs. Gq(+) treated with DMSO; **, P < 0.01 vs. Gq(+) treated with MCIP(−).
Table 1.
Densitometric analysis of NFAT2 and NFAT4
| Densitometry units |
||
|---|---|---|
| Nuclear (NFAT/histone 3) | Cytosolic NFAT/actin) | |
| NFAT2 | ||
| Gq(−) | 0.0710 ± 0.0049 | 0.2926 ± 0.0266 |
| Gq(+) | 0.1528 ± 0.0053a | 0.2725 ± 0.0574 |
| NFAT4 | ||
| Gq(−) | 0.1043 ± 0.0027 | 0.4403 ± 0.0125 |
| Gq(+) | 0.1445 ± 0.0047† | 0.4228 ± 0.0157 |
Results of six experiments, a, P < 0.01; or b, P < 0.05 vs. Gq(−).
To determine whether Gq signaling induced NFAT-dependent gene transcription, we transfected podocytes with an NFAT reporter construct and then treated cells overnight with Gq(+) or Gq(−) in the presence or absence of either FK506 (20) or the protein inhibitor of CN myocyte enriched CN interacting protein 1 (MCIP1) (27) [also termed regulator of CN1 (28)]. MCIP1 was tagged with the TAT HIV sequence [MCIP(+)] to permit uptake of MCIP1 by cultured podocytes. A cell-impermeable MCIP1 protein lacking the TAT sequence [MCIP(−)] was used as a control. As shown in Fig. 1E, Gq(+) activated the reporter construct, and activation was blocked by FK506 and MCIP(+). Immunoblotting of the hemagglutinin (HA)-tagged Gq proteins in either culture medium or podocyte lysates is shown in the inset. The HA-tagged Gq(+) was effectively transduced into cultured podocytes. There is a slight difference in molecular size of Gq(+) and Gq(−) due to the presence or absence of the TAT sequence.
Glomerular podocytes express the Gq-coupled receptors for ANGII (AT1) and endothelin (ETA) (16). Moreover, ANGII and endothelin are important mediators of kidney injury in DN (2, 18), and both GPCR systems promote apoptosis of glomerular podocytes (14, 15, 29). To determine whether apoptosis induced by either ANGII or endothelin-1 (ET-1) was mediated by CN, we treated podocytes with either ANGII or ET-1 in the presence or absence of FK506 (20). As shown in Fig. 2A, both ANGII and ET-1 induced apoptosis of glomerular podocytes, and FK506 blocked this apoptotic effect. These data are consistent with the notion that the Gq-coupled receptors for ANGII and ET-1 promote podocyte apoptosis by activating CN. To determine whether Gq α-subunits activated by these receptor systems promoted podocyte apoptosis, we determined whether Gq inhibition attenuated apoptosis induced by ANGII. For these studies, cells were treated with ANGII in the presence or absence of a peptide inhibitor of Gq (30) tagged with the TAT sequence [Gqi(+)]. A Gqi protein lacking the TAT sequence was used as a control [Gqi(−)]. As shown in Fig. 2B, Gqi(+) inhibited apoptosis induced by ANGII. Immunoblotting of the HA-tagged Gqi proteins in either culture medium or podocyte lysates is shown in Fig. 2C. The HA-tagged Gqi(+) was effectively transduced into cultured podocytes. We next investigated the role of CN in the Gq-dependent apoptotic response. For these studies, we treated cells with Gq(+) and Gq(−) in the presence or absence of FK506, DMSO vehicle, MCIP(+), or MCIP(−). As shown in Fig. 2, D and E, Gq(+) enhanced apoptosis compared with Gq(−), and this apoptotic effect was blocked by both MCIP(+) and FK506.
Fig. 2.

Gq(+) causes podocyte apoptosis by CN-dependent mechanisms. A, Podocytes were treated overnight with either ANGII or ET-1 (1 μm each) in the presence or absence of DMSO vehicle or the CN inhibitor FK506 (1 μm) as indicated. Both ANGII and ET-1 promoted podocyte apoptosis, and this apoptotic effect was prevented by FK506. B, Podocytes were treated overnight with ANGII and either Gqi(+) or Gqi(−) after which podocyte apoptosis was quantitated as described in Materials and Methods. Gqi(+) inhibited apoptosis induced by ANGII. C, Immunoblotting of the HA-tagged Gqi proteins in either culture medium or podocyte lysates. The HA-tagged Gqi(+) was effectively transduced into cultured podocytes. D, Podocytes were treated overnight with Gq(+) or Gq(−) before podocyte apoptosis was assessed. Treatment with Gq(+) significantly enhanced podocyte apoptosis. E, Podocytes were incubated overnight with Gq(+) or Gq(−) in the presence or absence of DMSO vehicle, FK506 (1 μm), MCIP(−), or MCIP(+) as indicated. The CN inhibitors FK506 and MCIP(+) inhibited apoptosis induced by Gq(+). All data are expressed as the percent apoptosis above DMEM-treated control cells, and data are the results of four to nine separate experiments. *, P < 0.01 vs. cells treated with DMSO; **, P < 0.025 vs. cells treated with DMSO; †, P < 0.01 vs. cells treated with ANGII and DMSO; ƒ, P < 0.05 vs. cells treated with either ET-1 or DMSO; ‡, P < 0.01 vs. Gq(−); §, P < 0.01 vs. Gq(+) treated with DMSO; ¶, P < 0.01 vs. Gq(+) treated with MCIP(−),††, P < 0.001 vs. DMSO and either Gqi(−) or Gq(+); ƒƒ, P < 0.01 vs. ANGII and Gq(−).
The ability of CN to promote podocyte apoptosis may be dependent on either NFAT-induced gene expression or dephosphorylation of other CN substrates such as the proapoptotic protein BAD (BCL2-associated agonist of cell death) (31). To investigate the role of NFAT-dependent gene induction in CN-mediated apoptosis, we treated cells with a cell-permeable peptide inhibitor of CN (VIVIT) (32). VIVIT blocks CN-dependent dephosphorylation of NFAT without affecting dephosphorylation of other CN substrates (32). As shown in Fig. 3A, VIVIT (1 μm) blocked podocyte apoptosis induced by Gq(+). Because Gq activates multiple signaling pathways in addition to CN, we also determined whether CN directly induced podocyte apoptosis by treating podocytes with a constitutively active CN construct (33) tagged with the TAT sequence [CN(+)]. A constitutively active control CN peptide lacking the TAT sequences was used as a control [CN(−)]. As shown in Fig. 3B, CN(+) induced podocyte apoptosis, and this effect was blocked by FK506 and VIVIT. Immunoblotting of the HA-tagged TAT proteins in culture medium or podocyte lysates is shown in the inset. The HA-tagged CN(+) was effectively transduced into cultured podocytes.
Fig. 3.

Effect of VIVIT on podocyte apoptosis induced by Gq(+) and CN(+). A, Podocytes were treated overnight with Gq(+) or Gq(−) in the presence of either the cell-permeable peptide inhibitor of calcineurin VIVIT (1 μm) or DMSO vehicle. Gq(+) significantly enhanced podocyte apoptosis compared with cells treated with Gq(−). The increase in apoptosis induced by Gq(+) was blocked by VIVIT. B, Podocytes were incubated overnight with CN(+) or CN(−) in the presence or absence of DMSO vehicle, FK506 (1 μm), or VIVIT (1 μm) as indicated. The CN inhibitors FK506 and VIVIT inhibited apoptosis induced by CN(+). The inset shows immunoblotting of the HA-tagged CN proteins in either culture medium or podocyte lysates. All data are expressed as the percent apoptosis above DMEM-treated control cells, and data are the results of four to six separate experiments. *, P < 0.01 vs. Gq(−) treated with DMSO; †, P < 0.025 vs. Gq(+) treated with DMSO; **, P < 0.005 vs. CN(−) treated with DMSO; ƒ, P < 0.01 vs. CN(+) treated with DMSO.
The COX2 promoter contains two cis-acting NFAT-responsive sequences (34), and we previously found that Gq signaling up-regulated COX2 and stimulated PGE2 generation in cultured podocytes by CN-dependent mechanisms (21). We, therefore, determined the role of COX2 in the apoptotic response using the selective COX2 inhibitor CAY10404 (35). As shown in Fig. 4A, CAY10404 (100 nm) attenuated the apoptotic response. The enzymatic activity of COX2 generates both PGE2 (21) as well as ROS (36). To determine the role of PGE2 and ROS in the apoptotic response, we incubated podocytes with Gq(+) or Gq(−) in the presence or absence of either the ROS scavenger tempol (37) or a selective antagonist of the Gq-coupled EP1 receptor SC51089 (38). As shown in Fig. 4B, podocyte apoptosis was blocked by tempol (5 μm) and was attenuated by the EP1 receptor antagonist SC51089 (10 μm). In these studies, CAY10404, tempol, and SC51089 tended to reduce the basal levels of apoptosis, perhaps due to the high levels of COX2 expression and PGE2 generation in untreated cultured podocytes (21).
Fig. 4.
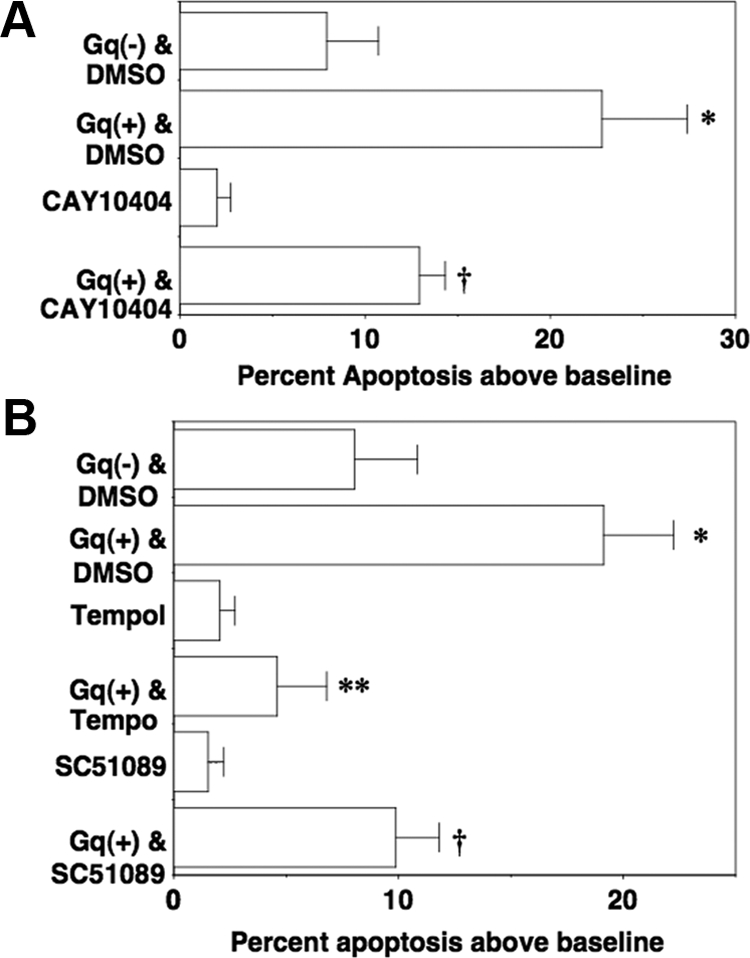
Gq(+) promotes podocyte apoptosis induced by COX2, PGE2, and ROS. A, Podocytes were incubated overnight with Gq(+) or Gq(−) in the presence or absence of the selective COX2 inhibitor CAY10404. Treatment with CAY10404 significantly attenuated podocyte apoptosis. B, Podocytes were incubated overnight with Gq(+) or Gq(−) in the presence or absence of either the ROS scavenger tempol or an antagonist of the Gq-coupled EP1 receptor SC51089. Treatment with either tempol or SC51089 significantly attenuated podocyte apoptosis induced by Gq(+). All data are expressed as the percent apoptosis above DMEM-treated control cells, and data are the results of four to six separate experiments. *, P < 0.01 vs. Gq(−) treated with DMSO vehicle; †, P < 0.05 vs. Gq(+) treated with DMSO vehicle; **, P < 0.01 vs. Gq(+) treated with DMSO vehicle.
Previous studies have demonstrated that CN phosphatase activity is increased in kidney cortices from diabetic rodents (11). To investigate CN activation in podocytes of Akita mice, we used IHC to determine nuclear localization of NFAT2 (rhodamine). For these studies, slides were also stained with an antibody to Wilms tumor antigen 1 (WT1), which specifically stains podocyte nuclei (fluorescein tag). Figure 5, A and B, shows results for diabetic Akita mice at 5 wk of age and age-matched nondiabetic controls, respectively. The left panels show immunofluorescent images for individual fluorophores and the right panels show merged images. The intensity of NFAT 2 staining tended to be enhanced in glomeruli of Akita mice (panel A) compared with controls (panel B), perhaps because NFAT2 autoregulates its own expression (39). In panel A, however, WT1 and NFAT2 share a similar subcellular distribution indicating that NFAT2 is localized to podocyte nuclei in 4- to 5-wk-old Akita mice. In Fig. 5B, NFAT2 and WT1 have different subcellular distributions in controls. In Fig. 5C, the percentage of the glomerular area that is stained for both WT1 and NFAT2 (yellow) was determined as described in Materials and Methods. There was a significant increase in the amount of NFAT2 that colocalized with WT1. Taken together, these data indicate that nuclear NFAT2 is enriched in podocyte nuclei of diabetic mouse kidneys compared with kidneys from controls. These findings are consistent with the notion that podocyte CN activity is increased in diabetic Akita mice at 5 wk of age.
Fig. 5.

Podocyte CN activity is increased in Akita mice. Frozen tissue sections were incubated with antibodies to NFAT2 to assess its subcellular localization (rhodamine) as well as WT1 to stain podocyte nuclei (fluorescein). A and B, Images from 5-wk-old Akita mice and age-matched controls, respectively. The left panels are results for the individual fluorophores, and the right panels are merged images for NFAT2 and WT1 with a magnified image of portion of the full glomerular image shown in bottom right panel. The intensity of NFAT 2 staining tending to be enhanced in glomeruli of Akita mice (panel A) compared with controls (panel B), perhaps because NFAT2 autoregulates its own expression (39). Panel A demonstrates that NFAT2 colocalizes with WT1-stained nuclei in Akita mice. In contrast, panel B demonstrates that little NFAT2 colocalizes with WT1-stained nuclei in control animals. In panel C, the amount of NFAT2 that colocalized with WT1 was assessed as described in Materials and Methods. There was a significant increase in the percentage of the glomerular area stained for NFAT2 that colocalized with WT1. These findings are consistent with the notion that podocyte CN activity is increased in diabetic Akita mice at 5 wk of age. *, P = 0.03 vs. wild type.
To determine whether enhanced podocyte CN activity in Akita mice induced podocyte apoptosis, we measured apoptosis in frozen tissue sections by terminal deoxynucleotide transferase-mediated dUTP nick end labeling (TUNEL) (rhodamine tag) as shown in Fig. 6A (left panel). Tissue sections were also stained with WT1 to stain podocyte nuclei (fluorescein tag) (Fig. 6A, middle panel). Apoptotic podocytes were quantitated by counting TUNEL-stained nuclei that colocalized with WT1 (Fig. 6A, right panel). Previous studies have shown that podocyte apoptosis is significantly increased in Akita mice compared with controls at 4–5 wk of age (9). We, therefore, determined whether FK506 attenuated podocyte apoptosis in Akita mice at 4–5 wk of age. As shown in Table 2, over this time period mice develop hyperglycemia that precedes an increase in urinary albumin excretion. For the experiments, 4-wk-old Akita mice and age-matched controls were treated for 1 wk with FK506 after which apoptosis was assessed in 5-wk-old animals. Urine collections (24 h) were obtained the day before beginning FK506 treatment and at the end of the treatment period. As shown in Fig. 6B, apoptosis was significantly increased in 5-wk-old Akita mice compared with controls, and this apoptotic response was attenuated by treatment with FK506. Consistent with the results of other investigators (9), the absolute number of apoptotic podocytes was low, which likely reflects the dynamic aspects of this apoptotic process in which apoptotic podocytes are rapidly cleared from the glomerulus and, in turn, only a small number of apoptotic cells can be detected at a single time point. As shown in Fig. 6C, urinary albumin excretion increased significantly in vehicle-treated Akita mice at 5 wk of age compared with the 4-wk time point. FK506 significantly decreased albuminuria in 5-wk-old Akita mice compared with age-matched vehicle-treated controls. Serum glucose levels were not affected by FK506 treatment at the 5-wk time point [690 ± 44 mg/dl (vehicle) vs. 709 ± 45 mg/dl (FK506); P = 0.81), and FK506 had no significant effect of albuminuria in 5-wk-old control mice [39 ± 4.2 μg/24 h (vehicle) vs. 49 ± 7.7 μg/24 h (FK506); P = 0.50]. Lastly, to determine whether the average level of hyperglycemia was similar in Akita mice treated with FK506 or vehicle, we measured serum fructosamine levels, which reflect the average glucose concentration over the prior 2–3 wk (40). We found that serum fructosamine was elevated in Akita mice compared with controls at the 5-wk time point [155 ± 13 mg/dl μ m (controls) vs. 246 ± 23 mg/dl μm (Akita); P < 0.025]. Moreover, there was no significant difference in fructosamine levels in FK506-treated Akita mice (249 ± 25 μm) compared with Akita mice treated with vehicle (239 ± 56 μm; P = 0.88).
Fig. 6.

Podocyte apoptosis is blocked by FK506 in Akita mice. In panel A (left panel), we assessed podocyte apoptosis in tissue sections by TUNEL using a commercially available kit (rhodamine tag). In the middle panel, tissue sections were also stained with an antibody to WT1, which specifically stains podocyte nuclei (fluorescein tag). Podocyte number was quantitated by counting WT1-positive nuclei (middle panel) per glomerular profile, and apoptotic podocytes were quantitated by counting TUNEL-stained nuclei that colocalized with WT1 (right panel). For the studies shown in panel B, 4-wk-old Akita mice were treated with FK506 for 1 wk, and then kidneys were harvested for measurement of podocyte apoptosis by IHC. As shown in panel B, podocyte apoptosis was significantly increased in Akita mice compared with control animals at 4–5 wk of age, and treatment with FK506 significantly decreased the number of apoptotic podocytes in Akita mice. Panel C shows urinary albumin excretion in Akita mice treated with either vehicle or FK506. For the studies, urine was collected the day before treatment with FK506 and at the end of the treatment period. Urinary albumin excretion increased significantly in Akita mice treated with vehicle between 4 and 5 wk of age. Treatment with FK506 significantly reduced albuminuria in Akita mice at the 5-wk time point. Four to 10 mice were studied in each group. *, P < 0.05 vs. age matched controls; †, P < 0.01 vs. Akita mice treated with vehicle; **, P < 0.01 vs. 4-wk-old Akita mice; ƒ, P < 0.025 vs. 5-wk-old Akita mice treated with vehicle.
Table 2.
Serum glucose levels and albuminuria in Akita mice
| Glucose (mg/dl) |
Albuminuria (μg/24 h) |
|||
|---|---|---|---|---|
| Control | Akita | Control | Akita | |
| 3 wk | 183 ± 11 | 255 ± 16 | 17 ± 1.8 | 20 ± 7.9 |
| 4 wk | 230 ± 21 | 513 ± 33a | 50 ± 10 | 45 ± 7.8 |
| 5 wk | 190 ± 41 | 695 ± 32a | 51 ± 9.4 | 281 ± 47a |
, P < 0.001 vs. age-matched control.
Discussion
The beneficial effects of CN inhibitors in glomerular disease have long been attributed to inhibition of immune responses (41). The present findings, together with recent studies (42), suggest that CN has important actions in glomerular disease processes that are independent of its immunomodulatory effects. In this regard, we found that: 1) Gq-dependent CN activation promotes apoptosis of glomerular podocytes in vitro by mechanisms that require NFAT-dependent gene induction, and 2) Pharmacological CN inhibition attenuates podocyte apoptosis in a mouse model of type 1 diabetes mellitus. Because, it is likely, that multiple Gq-coupled GPCR contribute to disease progression in glomerular diseases (16), a strategy that inhibits a final common, injury-promoting pathway activated by these GPCR systems may provide more comprehensive protection from glomerular injury. The observation that FK506 blocks podocyte apoptosis in vivo suggests that treatment with CN inhibitors may be one strategy for targeting a final common signaling cascade activated by Gq-coupled GPCR, which, in turn, may have beneficial effects in glomerular disease processes.
The VIVIT peptide blocks CN-dependent dephosphorylation of NFAT by preventing the protein-protein interaction between CN and NFAT substrate (32). Although recent studies suggest that VIVIT inhibits the protein-protein interaction between CN and at least one additional CN substrate (43), the selectivity of VIVIT for the NFAT signaling pathways is significantly enhanced compared with the CN inhibitors FK506 and cyclosporine A (32). The observation that VIVIT inhibits both Gq(+)- and CN(+)-induced podocyte apoptosis is, therefore, consistent with the notion that CN/NFAT-dependent gene induction plays an important role in the apoptotic response induced by Gq activation. Whereas a large number of genes are regulated by CN/NFAT signaling (44), COX2 is potently induced by CN activation through NFAT-responsive sequences in its promoters (21, 28). Moreover, up-regulation of COX2 in podocytes contributes to podocyte damage in several glomerular diseases including diabetic kidney disease (45). Whereas the mechanisms of COX2-mediated injury are likely multifactorial, the enzymatic activity of COX2 generates ROS (36) as well as prostaglandins (46). The present studies found that both ROS and PGE2 generation contributed to Gq-dependent apoptosis in cultured podocytes. With regard to PGE2, the mechanism appears to be autocrine activation of the Gq-coupled EP1 receptor, which would further potentiate Gq activation. Whereas additional studies will be necessary to determine the role of COX2 in mediating podocyte apoptosis in diabetes mellitus, it is possible that therapies targeting either COX2 enzymatic activity or its enzymatic products might be an alternative strategy for preventing podocyte apoptosis in DN. Indeed, diabetic kidney disease in rodent models is attenuated by either COX2 inhibitors (45), EP1 receptor blockers (17), or strategies to inhibit ROS generation (9).
Although we found that CN inhibition attenuated apoptosis in diabetic Akita mice, Chen and co-workers (47) did not detect apoptosis after induction of a constitutively active NFAT2 in podocytes in nondiabetic mice. The lack of an apoptotic effect in the study by Chen and co-workers (47) may be dependent on the cellular context. For example, overexpression of COX2 in podocytes using a transgenic strategy does not promote podocyte injury in unmanipulated mice but kidney damage is more severe after treatment with the podocyte toxins puromycin aminonucleoside or adriamycin (48). Moreover, we found that targeting a constitutively active Gq construct (21) to podocytes in vivo did not cause a kidney phenotype, although expression of the same construct in cultured podocytes caused podocyte apoptosis in the present study. Given that multiple signaling cascades modulate the apoptotic response, cellular outcome is likely dependent on differential activation of the signaling pathways that promote either cell survival or apoptosis.
The present studies, taken together with published reports (11, 12), suggest that CN inhibition is promising strategy for slowing the progression of diabetic kidney disease. Unfortunately, current pharmacological CN inhibitors have the potential for nephrotoxicity (49), which may limit the usefulness of these drugs as therapeutic agents in glomerular diseases. Pharmacological CN inhibitors, however, block CN phosphatase activity and, as a result, modulate multiple signaling pathways including dephosphorylation of proapoptotic protein BAD (BCL2-associated agonist of cell death) (31) and activation of prosurvival nuclear factorκB signaling cascade (32, 50). In contrast, the protein inhibitor VIVIT prevents activation of NFAT transcription factors by CN without affecting CN phosphatase activity (32). As a result, VIVIT blocks NFAT-dependent gene induction without inhibiting other signaling cascades (32). Some investigators have suggested that selective inhibition of CN-dependent NFAT activation may have less toxicity than currently available pharmacological CN inhibitors (51). Indeed, unlike the pharmacological CN inhibitors cyclosporine A (CyA) and FK506, VIVIT does not decrease insulin secretion even at fully immunosuppressive dosages (51). Although additional studies will be necessary to determine the nephrotoxic potential of VIVIT, it is possible that this agent will have a more favorable nephrotoxic profile than CyA or FK506, which may provide the impetus for the development of more selective agents that block CN-mediated NFAT activation without affecting dephosphorylation of other CN substrates.
Although the CN inhibitor FK506 (20) blocked Gq-dependent apoptosis by mechanisms that are mediated, in part, by COX2, we previously found (21) that FK506 potently enhanced COX2 expression both in vitro and in vivo. Increased eicosanoid production, perhaps related to up-regulation of COX2, may play a role in nephrotoxicity induced by FK506 (49). Despite enhancing COX2 expression, however, FK506 blocked apoptosis induced by either Gq(+) or CN(+) in cultured podocytes as well as reduced podocyte apoptosis in vivo. These data are consistent with the notion that CN may be a final common pathway that promotes podocyte apoptosis after Gq activation. In support of this hypothesis, the apoptotic effect of COX2 was dependent on autocrine activation of the Gq-coupled EP1 receptor by PGE2, which would tend to further stimulate CN activity. The other product of COX2 enzymatic activity, ROS, contributed to the apoptotic effect and has also been suggested to promote increases in intracellular calcium and CN activation (12). These data indicate that CN may play a central role in podocyte apoptosis induced by Gq-coupled GPCR in podocytes.
Figure 7 summarizes the proposed signaling pathways mediating Gq-dependent apoptosis in glomerular podocytes. We found that stimulation of CN activity by both Gq-coupled receptors and Gq(+) promotes podocyte apoptosis and that this apoptotic effect is mimicked by CN(+). The apoptotic response induced by both Gq(+) and CN(+) required CN-dependent NFAT activation and, in turn, NFAT-dependent gene induction. Moreover, the CN/NFAT-responsive gene COX2 is up-regulated by CN activation and contributes to the apoptotic response, in part, by autocrine activation of the Gq-coupled EP1 receptor as well as ROS generation. Finally, the findings appear relevant to the in vivo situation because treatment with a pharmacological CN inhibitor attenuated podocyte apoptosis in a genetic model of type 1 diabetes mellitus. In addition to modulating immune function and cytoskeletal dynamics (42), the beneficial effects of CN inhibitors in glomerular diseases may be mediated, at least in part, by inhibiting podocyte apoptosis.
Fig. 7.
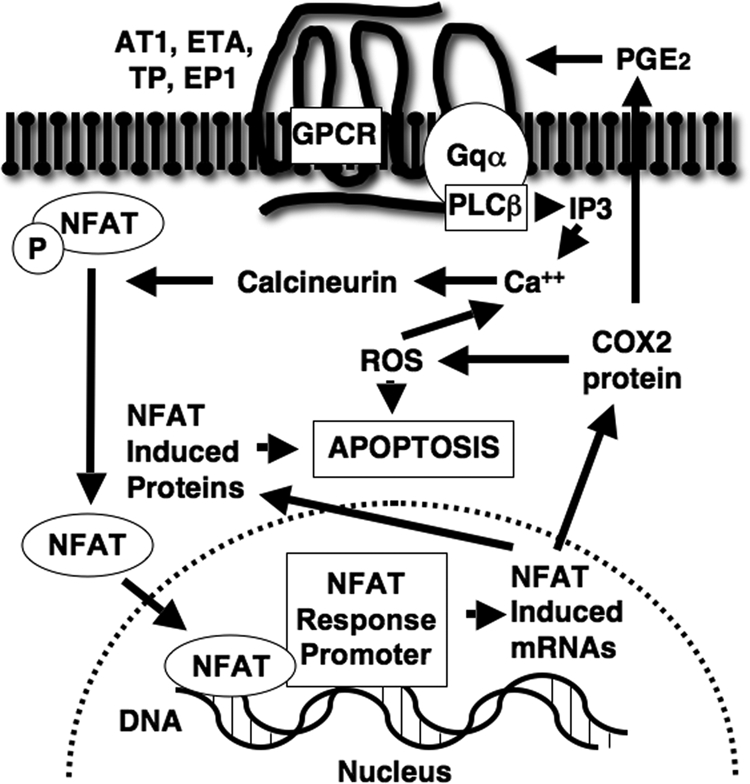
Mechanisms of Gq-dependent apoptosis. Podocytes express numerous GPCR implicated in the pathogenesis of DN including receptors for ANGII (AT1), TP, E-series prostaglandins (EP1), and ET-1 (ETA). Receptor activation stimulates phospholipase C (PLC)β via Gq α-subunits and generates inositol phosphate (IP), which mobilizes calcium from intracellular stores and activates CN. Activation of CN dephosphorylates NFAT family members and permits their translocation to the nucleus. Nuclear localization of NFAT isoforms induces transcription of genes such as COX2 as well as other NFAT-responsive genes that likely contribute to the apoptotic response. COX2 enzymatic activity generates both PGE2 (21) and ROS (36). PGE2 synergizes with other Gq-coupled receptor systems to further activate CN through the Gq-coupled EP1 receptor. ROS are also important mediators of podocyte apoptosis (9) and may further enhance intracellular calcium levels and, in turn, potentiate CN activation (11, 12).
Materials and Methods
Materials
Reagents were obtained from the following suppliers: 1) Tempol, ET-1, and ANGII were obtained from Sigma-Aldrich (St. Louis, MO); 2) the cell-permeable peptide inhibitor of CN VIVIT was obtained from Calbiochem (La Jolla, CA); and 3) FK506 for injection was obtained from the pharmacy at Duke University Medical Center (Durham, NC). CAY10404 and SC51089 were obtained from Cayman Chemical (Ann Arbor, MI).
Culture of SV40-transformed mouse glomerular epithelial cells
The immortalized mouse podocyte cell line was a gift of Dr. Paul E. Klotman (Mount Sinai Medical Center, New York, NY) (21). The cells were derived from animals bred with the H-2Kb-tsA58 Immortomice (Charles River Laboratories, Wilmington, MA). Cell lines were established that expressed the podocyte-specific markers WT1, synaptopodin, and podocalyxin (21). For the studies, podocytes were maintained in culture and differentiated as previously described (21). Briefly, to permit immortalized growth, cells were grown at 33 C in medium supplemented with 100 units/ml γ-interferon to induce the H-2Kb promoter driving synthesis of the temperature-sensitive (tsA58) SV-40 T antigen (permissive conditions). For differentiation, cells were grown at 37 C in medium lacking γ-interferon, resulting in degradation of the T antigen (nonpermissive conditions). Cells were discarded after a maximum of 10 passages.
Animal experiments
FVB/NJ Akita mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred at Duke Medical Center. Mice were treated with FK506 (10 mg/kg/d) or vehicle by twice daily sc injection starting at 4 wk of age. After 1 wk, mice were killed and kidneys were harvested for examination by IHC as described below. All animal care conformed to National Institutes of Health guidelines and was approved by the IACUC at Duke University Medical Center. Blood glucose levels were measured using an AlphaTRAK testing system (Abbott Laboratories, Inc., Abbott Park, IL).
Measurement of serum fructosamine levels
Serum fructosamine was measured by the reduction of nitroblue tetrazolium (40) using reagents and standards from Catachem (Bridgeport, CT).
DNA constructs
A constitutively active form of the Gq α-subunit (GqQ>L) was obtained from Dr. J. Silvio Gutkind at the National Institutes of Dental Health (Bethesda, MD) (24). In this construct, glutamine at position 209 is mutated to leucine (24). This region of the Gq α-subunit is involved in binding and hydrolysis of GTP. Dr. Paul Rosenberg, at Duke University Medical Center, provided the human MCIP1 (27) construct and a constitutively active form of the CN catalytic subunit (33). The CN construct lacks functional calcium binding- and autoinhibitory domains in the catalytic subunit (33). The peptide Gq inhibitor was obtained from Dr. Robert Lefkowitz at our institution (30). This construct codes for the carboxyl terminus of the Gq α-subunit, which interacts with the intracellular domains of Gq-coupled receptors and, in turn, prevents their interaction with endogenous Gq α-subunits (30). To create TAT proteins, the constructs were cloned in the pTAT-HA vector (23) in frame with the TAT protein sequence and a sequence coding for the HA epitope.
Treatment of cultured podocytes with TAT proteins
Protein transduction was used to introduce TAT proteins into cultured podocytes by tagging the proteins with the HIV TAT sequence using methods adapted from Becker-Hapak et al. (23) as previously described (52). Briefly, podocytes were plated in six-well tissue culture clusters (Evergreen Scientific, Los Angeles, CA) and then differentiated for 5–7 d as described (21) before treatment overnight with 100–200 nm of TAT proteins in serum free medium in the presence or absence of the indicated agents as previously described (52). Pilot experiments suggested that this concentration of TAT protein optimized uptake of the constructs by podocytes with maximal uptake after 10–15 min. After the overnight incubation, cells were harvested, and apoptotic cells were identified by either TUNEL or annexin V staining using a kits from Roche (Mannheim, Germany) and BD Pharmingen (San Diego, CA), respectively, according to the directions of the manufacturer. Quantitation of the apoptotic cells was performed by flow cytometric analysis at the Duke Comprehensive Cancer facility. For the annexin V studies, apoptotic podocytes were differentiated from necrotic cells by counting cells that stained with both the annexin V antibody and 7-amino-actinomycin D. Results were similar with both methods (TUNEL and annexin V staining), and the results were combined for the statistical analyses.
Luciferase reporter assays
Podocytes were transfected in 12-well tissue culture clusters (Evergreen Scientific, Los Angeles, CA) after which luciferase reporter assays were performed as previously described (21). Briefly, podocytes were transfected with the reporter construct pNFAT-luc (0.5 μg per well, Stratagene) or its control vector (0.5μg per well, Stratagene) using Lipofectamine (Invitrogen) according to the directions of the manufacturer. The promoter of this reporter construct contains direct repeats of the transcription recognition sequences for NFAT transcription factors driving expression of the Photinus pyralis (firefly) luciferase gene. To control for transfection efficiency, cells were also transfected with pRL-TK (0.25 μg per well, Promega Corp., Madison, WI) in which the thymidine kinase promoter drives expression of Renilla luciferase. The cells were harvested 2 d after transfection using the Promega Dual Luciferase Reporter Assay System, and both firefly and Renilla luciferase intensity were measured with a luminometer (MGM Instruments, Hamden, CT) according to the directions of the manufacturer (Promega). To correct for transfection efficiency, the firefly luciferase values were divided by the Renilla luciferase values after subtracting the background light intensity. Data are expressed as a percentage of the response in vehicle (DMEM)-treated control cells.
Immunoblotting
Immunoblotting was performed using methods adapted from previous studies (21, 52) and the following antibodies: 1) mouse monoclonal antibodies to NFAT2 (sc-7294) and NFAT4 (sc-8405) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); 2) a mouse monoclonal antibody to actin (Chemicon International, Temecula, CA); 3) a mouse monoclonal antibody to histone 3 (Abcam, Cambridge, MA); and 4) a rabbit monoclonal antibody to the HA antigen (UpState Biotechnology, Inc., Lake Placid, NY). To assess protein-loading immunoblots were stripped according to the directions of the manufacturer, and immunoblotting was performed using mouse monoclonal antibodies to either β-actin or histone 3. The immunoblots were converted into a digital format using an Epson Perfection scanner 1670 (Seiko Epsom Corp., Nagano, Japan) and then analyzed using ScanAnalysis 2.5 software (Biosoft, Ferguson, MO). Densitometric data were normalized by dividing the NFAT signals by the signals for either actin (cytosolic fractions) or histone 3 (nuclear fractions).
IHC
IHC of mouse kidney cortex was performed using methods adapted from previous studies (21). The WT1 antibody was added at a 1:400 dilution, the NFAT2 antibody was added at a concentration of 1:100, and TUNEL labeling was performed using an ApoTag Red In Situ Apoptosis kit from Millipore Corp. (Bedford, MA) according to the directions of the manufacturer.
For IHC of cultured podocytes, cells were grown on glass coverslips and then fixed in acetone before evaluation by IHC using the NFAT2 antibody at a 1:400 dilution, a fluorescein-labeled secondary antibody, and methods adapted from previous studies (52). Nuclei of cultured podocytes were stained with 0.5 μg/ml 4′,6-diamidino-2-phenylindole.
Quantitation of NFAT2 nuclear localization by IHC
To quantitate nuclear localization of NFAT2 in vivo, tissue sections were stained for WT1 and NFAT2 as described above and then examined blinded to genotype. Using Adobe Photoshop CS, the number of pixels in digital images of the glomerular area was determined after defining the glomerular area using the lasoo tool and using the histogram function. Similarly, the number of NFAT2-stained pixels that colocalized with WT1 was determined by selecting yellow pixels in the glomerular area using the select color tool and the histogram function. The number of pixels in the NFAT2-WT1-stained area (yellow area) was then divided by the number of pixels in the glomerular area to determine the percentage of NFAT-stained pixels that colocalized with WT1 within each glomerulus. In all experiments, digital images were obtained using the same exposure duration, and the areas were quantitated without altering the brightness or contrast levels. For each mouse kidney examined, the percent NFAT2-stained pixels that colocalized with WT1-stained areas was determined by averaging results from 10 different digital pictures. Ten glomeruli were assessed in tissue sections from each mouse, and the average of the values was used for the statistical analysis.
Statistical analysis
Data are presented as the sem. For comparison of continuous variables between two groups, statistical significance was assessed by a t test using the InStat computer program (GraphPad Sofware, Inc., San Diego, CA). For comparisons between more than two groups, statistical analysis was performed using an ANOVA followed by a Bonferonni multiple comparisons posttest using the InStat program.
Acknowledgments
These studies were supported by National Institutes of Health Grants RO1-DK075688 (to R.F.S.) and 3R01DK075688-01A2S1 (to R.F.S.) from the National Institute of Diabetes, Digestive and Kidney Diseases.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ANGII
- Angiotensin II
- AT1
- receptor for ANGII
- CN
- calcineurin
- CN(+)
- constitutively active CN tagged with the TAT protein sequence
- CN(−)
- constitutively active CN lacking the TAT protein sequence
- CN1
- regulator of calcineurin 1
- COX2
- cyclooxygenase 2
- CyA
- cyclosporine A
- DMSO
- dimethylsulfoxide
- DN
- diabetic nephropathy
- EP1
- type 1 E-series prostaglandin receptor
- ETA
- type A endothelin receptor
- ET-1
- endothelin-1
- Gq
- Gq α-subunits
- Gq(+)
- constitutively active Gq tagged with the TAT protein sequence
- Gq(−)
- constitutively active Gq lacking the TAT protein sequence
- Gqi
- Gq inhibitor peptide
- Gqi(+)
- Gqi tagged with the TAT protein sequence
- Gqi(−)
- Gqi lacking the TAT protein sequence
- GPCR
- G protein-coupled receptor
- HA
- hemagglutinin
- IHC
- immunohistochemistry
- MCIP1
- myocyte-enriched CN-interacting protein 1
- MCIP(+)
- MCIP1 tagged with the TAT protein sequence
- MCIP(−)
- MCIP1 lacking the TAT protein sequence
- NFAT
- nuclear factor of activated T cells
- PGE2
- prostaglandin E2
- ROS
- reactive oxygen species
- TAT
- transactivator of transcription
- TP
- thromboxane receptor
- TUNEL
- terminal deoxynucleotide transferase-mediated dUTP nick end labeling
- WT1
- Wilms tumor antigen 1.
References
- 1. U.S. Renal Data System, USRDS 2010. Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: NIH, National Institute of Diabetes and Digestive and Kidney Diseases [Google Scholar]
- 2. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. 2001. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345:861–869 [DOI] [PubMed] [Google Scholar]
- 3. Wolf G, Chen S, Ziyadeh FN. 2005. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54:1626–1634 [DOI] [PubMed] [Google Scholar]
- 4. Meyer TW, Bennett PH, Nelson RG. 1999. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia 42:1341–1344 [DOI] [PubMed] [Google Scholar]
- 5. White KE, Bilous RW, Marshall SM, El Nahas M, Remuzzi G, Piras G, De Cosmo S, Viberti G. 2002. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 51:3083–3089 [DOI] [PubMed] [Google Scholar]
- 6. Dalla Vestra M, Masiero A, Roiter AM, Saller A, Crepaldi G, Fioretto P. 2003. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes 52:1031–1035 [DOI] [PubMed] [Google Scholar]
- 7. Wiggins RC. 2007. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71:1205–1214 [DOI] [PubMed] [Google Scholar]
- 8. Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P. 2009. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20:322–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Susztak K, Raff AC, Schiffer M, Böttinger EP. 2006. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55:225–233 [PubMed] [Google Scholar]
- 10. Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, Giannoni M, Rimoldi L, Lauria F, Miji M, Deferrari G, Garibotto G. 2007. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int 72:1262–1272 [DOI] [PubMed] [Google Scholar]
- 11. Gooch JL, Barnes JL, Garcia S, Abboud HE. 2003. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol 284:F144–F154 [DOI] [PubMed] [Google Scholar]
- 12. Gooch JL, Gorin Y, Zhang BX, Abboud HE. 2004. Involvement of calcineurin in transforming growth factor-β-mediated regulation of extracellular matrix accumulation. J Biol Chem 279:15561–15570 [DOI] [PubMed] [Google Scholar]
- 13. Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Böttinger EP. 2001. Apoptosis in podocytes induced by TGF-β and Smad7. J Clin Invest 108:807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding G, Reddy K, Kapasi AA, Franki N, Gibbons N, Kasinath BS, Singhal PC. 2002. Angiotensin II induces apoptosis in rat glomerular epithelial cells. Am J Physiol Renal Physiol 283:F173–F180 [DOI] [PubMed] [Google Scholar]
- 15. Jia J, Ding G, Zhu J, Chen C, Liang W, Franki N, Singhal PC. 2008. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol 28:500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Winn MP, Daskalakis N, Spurney RF, Middleton JP. 2006. Unexpected role of TRPC6 channel in familial nephrotic syndrome: does it have clinical implications? J Am Soc Nephrol 17:378–387 [DOI] [PubMed] [Google Scholar]
- 17. Makino H, Tanaka I, Mukoyama M, Sugawara A, Mori K, Muro S, Suganami T, Yahata K, Ishibashi R, Ohuchida S, Maruyama T, Narumiya S, Nakao K. 2002. Prevention of diabetic nephropathy in rats by prostaglandin E receptor EP1-selective antagonist. J Am Soc Nephrol 13:1757–1765 [DOI] [PubMed] [Google Scholar]
- 18. Wenzel RR, Littke T, Kuranoff S, Jürgens C, Bruck H, Ritz E, Philipp T, Mitchell A. 2009. Avosentan reduces albumin excretion in diabetics with macroalbuminuria. J Am Soc Nephrol 20:655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kontessis PS, Jones SL, Barrow SE, Stratton PD, Alessandrini P, De Cosmo S, Ritter JM, Viberti GC. 1993. Effect of selective inhibition of thromboxane synthesis on renal function in diabetic nephropathy. J Lab Clin Med 121:415–423 [PubMed] [Google Scholar]
- 20. Rao A, Luo C, Hogan PG. 1997. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15:707–747 [DOI] [PubMed] [Google Scholar]
- 21. Wang L, Flannery PJ, Rosenberg PB, Fields TA, Spurney RF. 2008. Gq-dependent signaling upregulates COX2 in glomerular podocytes. J Am Soc Nephrol 19:2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asai M, Iwasaki Y, Yoshida M, Mutsuga-Nakayama N, Arima H, Ito M, Takano K, Oiso Y. 2004. Nuclear factor of activated T cells (NFAT) is involved in the depolarization-induced activation of growth hormone-releasing hormone gene transcription in vitro. Mol Endocrinol 18:3011–3019 [DOI] [PubMed] [Google Scholar]
- 23. Becker-Hapak M, McAllister SS, Dowdy SF. 2001. TAT-mediated protein transduction into mammalian cells. Methods 24:247–256 [DOI] [PubMed] [Google Scholar]
- 24. Kalinec G, Nazarali AJ, Hermouet S, Xu N, Gutkind JS. 1992. Mutated α subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol Cell Biol 12:4687–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boss V, Abbott KL, Wang XF, Pavlath GK, Murphy TJ. 1998. The cyclosporin A-sensitive nuclear factor of activated T cells (NFAT) proteins are expressed in vascular smooth muscle cells. Differential localization of NFAT isoforms and induction of NFAT-mediated transcription by phospholipase C-coupled cell surface receptors. J Biol Chem 273:19664–19671 [DOI] [PubMed] [Google Scholar]
- 26. Ruff VA, Leach KL. 1995. Direct demonstration of NFATp dephosphorylation and nuclear localization in activated HT-2 cells using a specific NFATp polyclonal antibody. J Biol Chem 270:22602–22607 [DOI] [PubMed] [Google Scholar]
- 27. Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. 2001. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 98:3328–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. 2006. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 116:3114–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ortmann J, Amann K, Brandes RP, Kretzler M, Münter K, Parekh N, Traupe T, Lange M, Lattmann T, Barton M. 2004. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension 44:974–981 [DOI] [PubMed] [Google Scholar]
- 30. Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. 1998. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280:574–577 [DOI] [PubMed] [Google Scholar]
- 31. Shibasaki F, Hallin U, Uchino H. 2002. Calcineurin as a multifunctional regulator. J Biochem 131:1–15 [DOI] [PubMed] [Google Scholar]
- 32. Aramburu J, Yaffe MB, López-Rodriguez C, Cantley LC, Hogan PG, Rao A. 1999. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285:2129–2133 [DOI] [PubMed] [Google Scholar]
- 33. O'Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O'Neill EA. 1992. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature 357:692–694 [DOI] [PubMed] [Google Scholar]
- 34. Iñiguez MA, Martinez-Martinez S, Punzón C, Redondo JM, Fresno M. 2000. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem 275:23627–23635 [DOI] [PubMed] [Google Scholar]
- 35. Parashar B, Latha Shankar S, O'Guin K, Butler J, Vikram B, Shafit-Zagardo B. 2005. Inhibition of human neuroblastoma cell growth by CAY10404, a highly selective Cox-2 inhibitor. J Neurooncol 71:141–148 [DOI] [PubMed] [Google Scholar]
- 36. Lee SH, Williams MV, Dubois RN, Blair IA. 2005. Cyclooxygenase-2-mediated DNA damage. J Biol Chem 280:28337–28346 [DOI] [PubMed] [Google Scholar]
- 37. Wilcox CS, Pearlman A. 2008. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev 60:418–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsuo M, Yoshida N, Zaitsu M, Ishii K, Hamasaki Y. 2004. Inhibition of human glioma cell growth by a PHS-2 inhibitor, NS398, and a prostaglandin E receptor subtype EP1-selective antagonist, SC51089. J Neurooncol 66:285–292 [DOI] [PubMed] [Google Scholar]
- 39. Serfling E, Chuvpilo S, Liu J, Höfer T, Palmetshofer A. 2006. NFATc1 autoregulation: a crucial step for cell-fate determination. Trends Immunol 27:461–469 [DOI] [PubMed] [Google Scholar]
- 40. Armbruster DA. 1987. Fructosamine: structure, analysis, and clinical usefulness. Clin Chem 33:2153–2163 [PubMed] [Google Scholar]
- 41. Mathieson PW. 2008. Proteinuria and immunity–an overstated relationship? N Engl J Med 359:2492–2494 [DOI] [PubMed] [Google Scholar]
- 42. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. 2008. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14:931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Czirják G, Enyedi P. 2006. Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K+ channel, TRESK. J Biol Chem 281:14677–14682 [DOI] [PubMed] [Google Scholar]
- 44. Hogan PG, Chen L, Nardone J, Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205–2232 [DOI] [PubMed] [Google Scholar]
- 45. Cheng HF, Wang CJ, Moeckel GW, Zhang MZ, McKanna JA, Harris RC. 2002. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Int 62:929–939 [DOI] [PubMed] [Google Scholar]
- 46. FitzGerald GA, Patrono C. 2001. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med 345:433–442 [DOI] [PubMed] [Google Scholar]
- 47. Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, Liapis H, Miner JH, Chen F. 2010. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol 21:1657–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng H, Wang S, Jo YI, Hao CM, Zhang M, Fan X, Kennedy C, Breyer MD, Moeckel GW, Harris RC. 2007. Overexpression of cyclooxygenase-2 predisposes to podocyte injury. J Am Soc Nephrol 18:551–559 [DOI] [PubMed] [Google Scholar]
- 49. Gaston RS. 2009. Chronic calcineurin inhibitor nephrotoxicity: reflections on an evolving paradigm. Clin J Am Soc Nephrol 4:2029–2034 [DOI] [PubMed] [Google Scholar]
- 50. Lee JI, Burckart GJ. 1998. Nuclear factor κ B: important transcription factor and therapeutic target. J Clin Pharmacol 38:981–993 [DOI] [PubMed] [Google Scholar]
- 51. Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, Tanaka N, Matsui H. 2004. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med 10:305–309 [DOI] [PubMed] [Google Scholar]
- 52. Wang L, Gesty-Palmer D, Fields TA, Spurney RF. 2009. Inhibition of WNT signaling by G protein-coupled receptor (GPCR) kinase 2 (GRK2). Mol Endocrinol 23:1455–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]