

Abstract
The estimated incidence of mitochondrial diseases in humans is approximately 1:5000 to 1:10,000, whereas the molecular mechanisms for more than 50% of human mitochondrial disease cases still remain unclear. Here we report that mice lacking testicular nuclear receptor 4 (TR4−/−) suffered mitochondrial myopathy, and histological examination of TR4−/− soleus muscle revealed abnormal mitochondrial accumulation. In addition, increased serum lactate levels, decreased mitochondrial ATP production, and decreased electron transport chain complex I activity were found in TR4−/− mice. Restoration of TR4 into TR4−/− myoblasts rescued mitochondrial ATP generation capacity and complex I activity. Further real-time PCR quantification and promoter studies found TR4 could modulate complex I activity via transcriptionally regulating the complex I assembly factor NDUFAF1, and restoration of NDUFAF1 level in TR4−/− myoblasts increased mitochondrial ATP generation capacity and complex I activity. Together, these results suggest that TR4 plays vital roles in mitochondrial function, which may help us to better understand the pathogenesis of mitochondrial myopathy, and targeting TR4 via its ligands/activators may allow us to develop better therapeutic approaches.
The mitochondrion is the powerhouse of the cell. It generates the majority of ATP to support cellular function through the oxidative phosphorylation (OXPHOS) system, which consists of five multiprotein enzymatic complexes: complex I–V (1). Each individual complex is encoded by both the mitochondrial and nuclear genomes, except for complex II, which is exclusively nuclear encoded (2). Defects in the OXPHOS system in humans result in disastrous diseases with an estimated incidence of approximately 1:5000 to 1:10,000 (3).
Among all mitochondrial disorders, deficiency in complex I [reduced nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase; EC 1.6.5.3] is the most common cause. Composed of at least 45 different subunits, complex I is the largest and most intricate component in the mitochondria (4). Like cases in other mitochondrial diseases, the clinic presentations and genetic etiologies of complex I deficiency are highly diverse due to its genetic and biochemical complexity, which adds to the difficulties for proper diagnosis and treatment. Some typical clinical manifestations of complex I deficiency include progressive encephalomyopathy, leukodystrophy, Leigh syndrome, or mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (5–7). The identified genetic causes of complex I deficiency range from mutations in structure component encoding genes, such as NDUFS1 (8), NDUFV1 (5); to assembly factor encoding genes, such as B17.2L (9), C20orf7 (10); or various enzymes involved in production of electron carriers or maintenance of mitochondrial DNA integrity (11). Despite the improved characterization of its genetic defects in the past decade, the pathogenetic mechanisms of complex I deficiency are still not well understood.
Testicular nuclear receptor 4 (TR4) belongs to the nuclear receptor superfamily and is a key transcriptional regulator of many signaling pathways (12–14). Mice lacking TR4 (TR4KO, TR4−/−) display various phenotypes, including growth retardation, reduced fat mass, reduced fertility (15), and neurological abnormalities (16). TR4 is rhythmically expressed in skeletal muscles with the peak at Zeitgeber Time 12 (17), which is the time when mice start to be active, suggesting that TR4 is crucial for muscle function. Here we reported that TR4−/− mice developed mitochondrial myopathy with exercise intolerance, ragged red fibers in skeletal muscles, elevated serum lactate levels, impaired mitochondrial ATP generation capacity, and reduced complex I activity. We further demonstrated that TR4 regulates mitochondrial function through a complex I assembly factor NDUFAF1. Thus, our study provides a novel link between TR4 and mitochondrial function that may shed light on the pathogenesis and treatment of mitochondrial myopathy.
Results
Muscle weakness and impaired sustainable exercise capacity in TR4−/− mice
TR4−/− mice exhibited a variety of motor abnormalities, such as mild trembling, unsteady gait, and classical hind limb contractures upon tail suspension, suggesting that loss of TR4 might lead to abnormal motor function (16). To examine whether muscle weakness is one of the causes behind the abnormal motor function in TR4−/− mice, we carried out several tests. We first confirmed that TR4 expression was completely knocked out from skeletal muscle tissues in TR4−/− mice (Supplemental Fig. 1 published on The Endocrine Society's Journals Online website at http://mend.endojournals.org). Results from the rotarod test (18) showed the mean latency to fall in TR4−/− mice was significantly shorter than that found in littermate TR4+/+ mice. All TR4−/− mice fell off the rod within 20 sec, whereas TR4+/+ mice had no difficulty in hanging on for more than 90 sec (Fig. 1A).
Fig. 1.
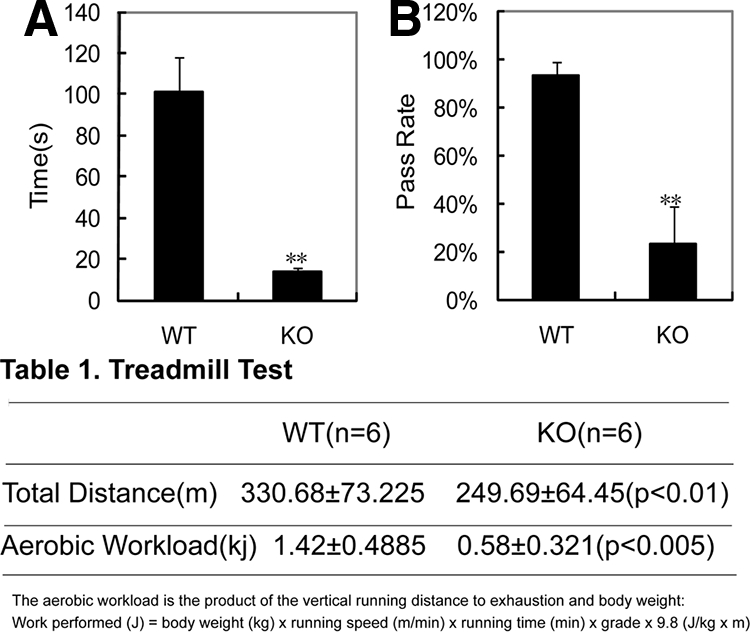
Muscle weakness and exercise intolerance in TR4−/ − mice. A, Determination of muscle strength by rotarod test. Four pairs of TR4−/− vs. TR4+/+ mice were tested, and the mean latency for TR4−/− mice to fall off the rod was significantly shorter than that for TR4+/+ mice. **, P < 0.01. B, Determination of muscle strength by whole-animal muscle strength and fatigability test. Three pairs of TR4−/− vs. TR4+/+ mice were used, and the percentage of passes over 15 trials of the test in a 3-min period in TR4−/ − mice was significantly reduced. **, P < 0.01. KO, Knockout; WT, wild type.
We then performed the whole animal strength and fatigability test (19) that tests the ability of the mice to pull themselves on top of a suspended rod by their forelimbs. The average success rate of 15 trials in a 3-min period, as an indication of whole-body strength, was less than 30% in TR4−/− mice as compared with 93% in TR4+/+ mice (Fig. 1B), indicating a potential muscle weakness in TR4−/− mice.
We also performed treadmill tests that were designed to determine the sustained exercise capacity (20), and a significant difference was found between TR4−/− and TR4+/+ mice with respect to the cumulative running distance and the total aerobic workload. TR4+/+ mice ran an average total distance of 330.68 ± 73.225 m with an average aerobic workload of 1.42 ± 0.4885 kJ, whereas TR4−/− mice ran an average total distance of 249.69 ± 64.45 m with an average aerobic workload of 0.58 ± 0.321 kJ (Table 1).
Table 1.
Treadmill test results in six pairs of TR4−/− and TR4+/+ mice
| WT (n = 6) | KO (n = 6) | |
|---|---|---|
| Total distance (m) | 330.68 ± 73.225 | 249.69 ± 64.45 (P < 0.01) |
| Aerobic workload (kJ) | 1.42 ± 0.4885 | 0.58 ± 0.321 (P < 0.005) |
The aerobic workload is the product of the vertical running distance to exhaustion and body weight: Work performed (J) = body weight (kg) × running speed (m/min) × running time (min) × grade × 9.8 (J/kg × m). WT, Wild type; KO, knockout.
Together, results of Fig. 1 and Table 1 from three different assays suggested that TR4−/− mice might have impaired sustainable exercise capacity with a likely muscle weakness.
Abnormal mitochondrial accumulation in the skeletal muscle tissues of TR4−/− mice
We then performed skeletal muscle histological examination to further validate the possible muscle deficiency in TR4−/− mice. First, we found that soleus muscle fibers in TR4−/− tissues showed a greater variety of sizes as compared with those in TR4+/+ tissues by hematoxylin and eosin (H&E) staining, in which we also found basophilic bands at the periphery of muscle fibers possibly due to the abnormal deposits of mitochondria only in TR4−/− tissues (Fig. 2, A and B). The abnormal mitochondrial deposits were confirmed by the moderate increase of the ragged red fibers in TR4−/− skeletal muscle samples (Fig. 2, C and D) by the modified Gomori trichrome staining. Ragged red fibers, the red staining in the peripheral rim of muscle fibers, consisting of abnormal subsarcolemmal aggregates of mitochondria, is a common feature in human mitochondrial disease patients (21). The succinate dehydrogenase (SDH) staining also revealed an increase of SDH reactivity in the same area of muscle fibers of TR4−/− mice (Fig. 2, E and F). The histological diagnosis of mitochondria abnormity in TR4−/− mice skeletal muscle was further supported by electron microscopy where we found subsarcolemmal aggregates of enlarged mitochondria with a high content of cristae in TR4−/− skeletal muscle (Fig. 2, G and H, and Supplemental Fig. 2).
Fig. 2.

Abnormal mitochondrial accumulation in TR4−/− mice muscle. Histological examination of frozen soleus muscle tissues from 1-yr-old TR4−/− and TR4+/+ mice (A–F). The cryostat sections were subjected to several staining procedures including H&E staining (A and B), Trichrome staining (C and D), and SDH staining (E and F). The typical ragged red fibers, which represent higher staining activities of mitochondria, are indicated by red arrows. The photos were taken under ×400 magnification. G and H, Electron microscopy (EM) examination of soleus muscle from 1-yr-old TR4−/− and TR4+/+ male mice. The abnormal subsarcolemmal aggregates of mitochondria in TR4−/− samples are indicated by red arrows. The soleus muscle tissues from the TR4−/− and TR4+/+ mice were fixed, embedded, and examined by electron microscopy. Examinations were carried out in three pairs of wild-type (WT) and TR4 knockout (KO) mice, and representative pictures are shown.
Together, muscle histological examination from Fig. 2 suggested that loss of TR4 might lead to mitochondrial myopathy that might explain the muscle weakness demonstrated in Fig. 1.
Increased serum lactate levels, decreased mitochondrial ATP production, and decreased electron transport chain complex I activity in TR4−/− mice
Defects of the oxidative phosphorylation in mitochondrial disorders usually lead to an accumulation of serum lactate (22, 23). We examined the resting serum lactate level in TR4−/− mice and their TR4+/+ littermates and found that TR4−/− mice displayed hyperlactatemia with 2.5-fold higher serum lactate levels as compared with TR4+/+ mice (Fig. 3A), supporting the above findings that TR4−/− mice may suffer mitochondrial myopathy.
Fig. 3.

Increased serum lactate level, decreased mitochondrial ATP production, and decreased electron transport chain complex I activity in TR4−/ − mice. A, Lactate content in the serum from seven pairs of TR4+/+ and TR4−/− mice was determined by measuring the oxidized DCPIP as described in Materials and Methods. TR4−/ − mice display elevated serum lactate levels as compared with TR4+/+ mice. B, Determination of ATP generation by a firefly luciferase-based assay. Mitochondria isolated from three pairs of TR4+/+ and TR4−/− skeletal muscle were subjected to ATP generation assay. TR4−/ − mice muscle exhibited markedly lower ATP production rate per gram mitochondrial protein compared with mitochondria from TR4+/+ mice. C, Determination of respiratory chain enzyme activity between TR4+/+ and TR4−/−. Mitochondria isolated from four pairs of TR4+/+ and TR4−/− skeletal muscle were applied to respiratory chain enzyme activity assays as described in Materials and Methods. Three independent experiments were carried out, and the mean ± sd was shown. **, P < 0.01. KO, Knockout; WT, wild type.
The defects of oxidative phosphorylation may lead to alteration of ATP production (24). We therefore compared ATP production capacity of the isolated mitochondria from TR4+/+ and TR4−/−skeletal muscle tissues. As shown in Fig. 3B, the mitochondrial ATP generation capacity declined 50% in tissues from TR4−/− mice as compared with TR4+/+ mice.
OXPHOS enzyme deficiency is a common cause for impaired ATP generation (25); therefore we examined the respiratory chain OXPHOS enzyme activities in isolated skeletal muscle mitochondria from both TR4+/+ and TR4−/− mice. Among all five complexes, we found complex I activity was reduced most significantly (near 45%) in skeletal muscles of TR4−/− mice (Fig. 3C) as compared with TR4+/+ mice. A mild reduction in complex V activity was also found in TR4−/− mice, whereas little change was seen in other enzyme complexes (Fig. 3C). Together, results from Fig. 3, A–C, indicated that the mitochondrial dysfunction in the skeletal muscle of TR4−/− mice could be due to the complex I or V deficiency. In addition to OXPHOS system, the proper function of tricarboxylic acid cycle (TCA cycle) is also essential for mitochondrial ATP generation. We also examined one of TCA enzymes, citrate synthase, in the skeletal muscle of TR4−/− mice and found no significant change in activity (data not shown). Further characterization in TCA activity will be necessary to fully understand the mitochondrial myopathy in TR4−/− mice.
Using primary myoblast cells to confirm the role of TR4 in mitochondrial function
We then established the in vitro culture of primary myoblast cells from TR4−/− and TR4+/+ mice to further investigate the role of TR4 in mitochondrial functions. As shown in Fig. 4, A and B, TR4−/− myoblast cells showed a decreased mitochondrial ATP generation capacity and a declined complex I activity as compared with TR4+/+ myoblast cells. More importantly, reconstitution of TR4 in TR4−/− myoblast cells by retroviral infection of pBabe-EGFP-TR4 improved both the mitochondrial ATP generation capacity and complex I activity to a level comparable to those of TR4+/+ cells (Fig. 4, A and B), whereas it did not change complex IV activity (Fig. 4).
Fig. 4.

Introduction of TR4 into TR4−/− myoblast cells partially restored mitochondrial ATP generation capacity and complex I activity. A, Examination of mitochondrial ATP generation capacity in wild-type (WT) myoblasts and TR4 knockout (KO) myoblasts infected with either pBabe-EGFP vector (KO+Vector) or pBabe-EGFP-TR4 (KO+TR4). B, Examination of complex I and IV activities by spectrometry assay in WT myoblasts and TR4KO myoblasts infected with either pBabe-EGFP vector (KO+Vector) or pBabe-EGFP-TR4 (KO+TR4). Three independent experiments were carried out, and the mean ± sd was shown. *, P < 0.05. **, P < 0.01.
Together, results from primary cultured myoblast cells in Fig. 4 strengthened our in vivo data from TR4−/− mice showing that TR4 might regulate mitochondrial function by modulating complex I activity.
Determination of complex I gene expressions in TR4−/− skeletal muscles
To dissect the molecular mechanisms through which TR4 modulates complex I functions, we next applied the real-time PCR quantification (Q-PCR) assay to compare the expression levels of complex I encoding genes between TR4−/− and TR4+/+ mice. Due to the relatively large numbers of the candidate genes, we started with the genes the mutation of which has been identified in human mitochondrial disease. We examined three groups of genes that could contribute to complex I activity: complex I structural components (Supplemental Fig. 3A), complex I assembly factors (Supplemental Fig. 3B), and mitochondrial DNA transcription factors (Supplemental Fig. 3C). Among all the genes we screened, we only found the mRNA level of NDUFAF1 (CIA30), one of the complex I assembly factors (26), was significantly decreased in TR4−/− muscles (Fig. 5A). We also found that the protein expression of NDUFAF1 was reduced in TR4−/− muscles (Fig. 5B).
Fig. 5.

Complex I assembly factor NDUFAF1 is a TR4 target gene. A, The mRNA expression level of complex I assembly factor NDUFAF1 was reduced in skeletal muscle tissues from TR4−/− mice. The mRNA was extracted from skeletal muscle tissues from TR4+/+ and TR4−/− mice and subsequently subjected to Q-PCR. B, The protein expression level of NDUFAF1 in skeletal muscle tissues from TR4+/+ and TR4−/− mice were detected by Western blot. Tublin level was used as an internal control. C, TR4 transactivates NDUFAF-1-Luc in a dose-dependent manner. Increasing amounts of pCMX-TR4 expression plasmid were cotransfected with pGL3-NDUFAF1-Luc reporter into CV-1 cells. After 48 h, cells were harvested and Luc activities were measured. D, Modulation of TR4 activity affects TR4-mediated Ndufaf1-luc activity. pGL3-NDUFAF1-Luc reporter (250 ng) and 750 ng of pCMX-TR4 expression plasmid or vector control were cotransfected into CV-1 cells. After 24 h, cells were treated with vehicle control or TR4 upstream inhibitors metformin or aminoimidazole carboxamide ribonucleotide (AICAR). After treatment for 24 h, cells were harvested and Luc activities were measured. E, TR4 binds to Ndufaf1 promoter by ChIP assay. The protein-DNA complex was pulled down using TR4-specific antibody from H1299 cells, and specific binding between TR4 and TR4 response elements in Ndufaf1 promoter was PCR amplified using primers that cover the regions approximately −1073 to −904 and −340 to −150 in Ndufaf1 promoter. PCR amplification using primers that cover the region approximately −2702 to −2579, which contains no TR4-binding site in Ndufaf1 promoter was used as a negative control. F, Determination of the mitochondrial ATP generation capacity in wild-type (WT) myoblasts, knockout (KO) myoblasts infected with either pWPI vector (KO+Vector), or pWPI-Ndufaf1 (KO+Ndufaf1). Mitochondria of myoblasts from different cells were extracted, and examined for their ATP generation capacity as described in Materials and Methods. G, Examination of complex I activity by spectrometry assays. Mitochondria of myoblasts from WT myoblasts, KO myoblasts infected with either pWPI vector (KO+Vector), or pWPI-Ndufaf1 (KO+Ndufaf1) were extracted and assayed for complex I activitiy. Shown were the mean ± sd. *, P < 0.05.
Complex I assembly factor NDUFAF1 is a TR4 target gene
The sequence analysis of human NDUFAF1 promoter revealed several putative TR4-hormone response elements. We then cloned human NDUFAF1 5′-promoter and constructed it into the pGL3-luciferase reporter plasmid. When we cotransfected the NDUFAF1-Luc with either the pCMX vector or increasing amounts of pCMX-TR4 into CV-1 cells, we found that TR4 activated NDUFAF1-Luc transcription up to 3.5-fold in a dose-dependent manner (Fig. 5C). We then applied a chromatin immunoprecipitation (ChIP) assay to test whether TR4 binds directly to the NDUFAF1 promoter and found that the NDUFAF1 promoter was recovered from immunoprecipitates using TR4 antibodies but not from immunoprecipitates using control IgG (Fig. 5D), indicating that TR4 directly binds to the 5′-promoter region of NDUFAF1. Together, results from Fig. 5, C and D, suggest that NDUFAF1 is a direct target gene of TR4.
Regulation of NDUFAF1 activity by modulating TR4 transactivation with AICAR and metformin
Because TR4 phosphorylation on S351 by AMP-activated protein kinase leads to suppression of TR4 transactivation (27), we then tested whether suppression of TR4 activity by AMP-activated protein kinase activators, AICAR and metformin, could affect NDUFAF1 transcription. As shown in Fig. 5E, both AICAR and metformin almost completely abolished TR4-induced NDUFAF1-Luc activity in CV-1 cells, whereas neither of them had significant effects on the basal level of NDUFAF1-Luc activity, indicating their effects on NDUFAF1 transcription are through modulation of TR4 transactivation. These results further demonstrate that TR4 modulates NDUFAF1 gene expression and suggest a possible way to regulate mitochondrial function by modulating TR4 activity via TR4 upstream signals.
Restoration of NDUFAF1 in TR4−/− primary myoblasts restored complex I activity
To prove that the reduction of NDUFAF1 is responsible for mitochondria defects found in TR4−/− mice, we restored NDUFAF1 level in TR4−/− primary mouse dermal fibroblasts and then examined the mitochondria function. We found when we reconstituted NDUFAF1 into TR4−/− cells by lentiviral infection of pWPI-NDUFAF1, both the mitochondrial ATP generation capacity and complex I activity increased to a comparable level of that in TR4+/+ cells (Fig. 5, F and G), implying that the TR4 mediates mitochondrial function through NDUFAF1.
Discussion
We first found interesting abnormalities in TR4−/− mice that resemble some pathological features observed in patients with mitochondrial OXPHOS disorders, including muscle weakness, exercise intolerance, ragged-red fibers in skeletal muscles, and hyperlactatemia (28). Although suggestive of a mitochondrial respiratory chain disease, these features alone are not sufficient enough to lead to an accurate diagnosis, because they are not very specific (29). The diagnosis of mitochondrial disorder in TR4−/− mice was further confirmed by the findings that both mitochondrial ATP production and respiratory chain complex I and V activity were decreased in TR4−/− skeletal muscle tissues.
Although it is still unclear whether other tissues, such as nervous system (16), also contribute to muscle weakness in TR4−/− mice, we are now sure that local defects do play roles here because we observed defects from in vitro culture of TR4−/− myoblasts, which was not under neuronal influence. The partial rescue of mitochondrial ATP generation capacity and complex I enzyme activity by reconstitution of TR4 into TR4−/− myoblast cells further supported our argument.
Interestingly, from electron microscopic examination of the soleus muscle from TR4−/− mice, we also found tubular aggregates (Supplemental Fig. 2) that have been found in muscle disorders in human and aged mice (30–32), and in some cases been linked to mitochondrial defects. It will be interesting in the future to dissect detailed mechanisms by which loss of TR4 may also lead to the tubular aggregates.
Other than abnormal mitochondrial accumulation and tubular aggregates, we also observed abnormal deposits of electron-dense granules that we suspected might be calcium in TR4−/− skeletal muscles (Supplemental Fig. 2), although further investigation is needed for the final identification of these particles. Calcium is an important modulator of skeletal muscle function (33) that contributes to the coordination of mitochondrial ATP production and local energy demand (34). Treatment of MERRF cells with drugs affecting calcium transport mostly restored both the agonist-dependent mitochondrial calcium uptake and the ensuing stimulation of ATP production (35). It will be interesting in the future to determine whether there is a linkage between mitochondrial calcium transport and TR4 function in the mitochondria.
Deficiencies in complex I is the most frequently encountered among all mitochondrial diseases. It has diverse clinical features ranging from single to multiple tissue involvement and has also been implicated in more common neurological diseases or impaired apoptotic signaling (4, 7, 36, 37). Clinical features associated with individual complex I deficiency also include encephalomyopathy, cardiomyopathy, cataracts, dementia, hepatopathy, and renal tubulopathy (38). We have not examined the impact of complex I deficiency in other organs, which remains an interesting issue for us to explore. The assembly process of complex I is poorly understood (39), although a number of recent studies have increased our understanding of the assembly of complex I in humans (9, 40). The only reported case of NDUFAF1 mutation in human harbors two heterozygous mutations in exon 3 of the NDUFAF1 gene, 1001A>C and 1140A>G (41). The NDUFAF1 level in this patient was decreased to approximately 10% of control values, and the level of steady-state complex I was also reduced to about 30% of control values. The activities of complexes II–IV were all normal. The patient displayed cardioencephalomyopathy, visual impairment, kyphoscoliosis, and mild intellectual handicaps. Interestingly, we did not observe any severe defects in the heart of TR4−/ − mice from histological examination or echocardiography (data not shown). The discrepancy might be explained by the fact that several other complex I structure genes also show severe decreases in this patient.
We also found reduced activity of the complex V in TR4−/− mice, although further investigation is needed to fully understand the molecular mechanism and the consequence of this defect in TR4−/− mice. Complex V deficiency in humans is mainly associated with the neurogenic weakness, ataxia, and retinitis pigmentosa syndrome (42), and Leigh syndrome (11). Are the skeletal muscle defects in TR4−/− mice the combinational results of both complex I and V defects? Further examination is needed to clarify this issue.
We previously found increased oxidative stress in TR4−/− mice (43). Considering that the election transport chain is the major site generating oxidative species in cells (44), the complex I defect we found here might account for the increased oxidative stress in TR4−/− mice. Consistent with our findings, patients with complex I defects also showed higher oxidative stress (45). The higher oxidative stress could in turn hamper complex I activity because mitochondria are susceptible to oxidative damage (46), thus forming a vicious circle in the function of complex I in TR4−/− mice.
In conclusion, our findings indicate that TR4 plays important roles in the regulation of mitochondrial function via complex I assembly factor NDUFAF1. The loss of TR4 results in defective skeletal muscle function due to impaired mitochondrial function. Considering that the genetic causes for more than 50% of human mitochondrial disease cases have not been identified, our finding suggests it might be necessary to also include TR4 in future genetic screening for human mitochondrial disorders. Targeting TR4 via small molecules to regulate mitochondrial function may provide a potential direction in the development of new therapeutic approaches for mitochondrial disorders.
Materials and Methods
Mice care and sample collection
Generation, breeding, and genotyping of TR4−/− mice were previously described (15). The original TR4−/− mice were of C57/BL6/129 mixed background. TR4−/−mice were backcrossed to C57/BL6 mice for five generations and used in this paper. Continuous backcross to C57/BL6 background over approximately six to seven generations caused embryonic lethality in TR4−/− mice. Unless specifically stated, male mice approximately 1 y old were used in this study. All blood and tissue samples were taken from mice under resting stage in the morning. All experimental protocols were approved by the University of Rochester University Committee on Animal Resources.
Cell culture, transfections, and luciferase assays
Primary myoblasts were isolated from 5-d-old pups. MEF cells were isolated from embryonic d 14.5 (E14.5) embryos after removal of the heads and all the internal organs, rinsed with PBS, added 5 ml of DMEM, and passed through a 22-gauge needle a few times to mince tissues. Cells were cultured in DMEM supplemented with 10% fetal bovine serum and 100 U ml−1 of penicillin-streptomycin mixture at 37 C in 5% CO2. CV1 cells were cultured in 24-well plates (BD Falcon, Bedford, MA), at a concentration of 105 cells per well, transfected with 1.2 μg DNA/well using SuperFect (QIAGEN, Chatsworth, CA). The internal control plasmid pRL-TK (Promega Corp., Madison, WI) was cotransfected in all transfection experiments. After 48 h transfection, the cells were harvested, and luciferase assays were performed using the Dual-Luciferase kit (Promega). For knockdown of TR4 in cells, we used retroviral delivery system (pRetro-H1G; Cellogenetics, Ijamsville, MD), with AACGGGAGAAACCAAGCAATT as the targeted sequence.
Plasmid constructs
The construction of plasmid pCMX and pCMX-TR4 has been described previously (47). pRL-TK (TK Renilla luciferase) was purchased from Promega. For the NDUFAF1 promoter luciferase (Luc) reporter, a total of 1001 bp human NDUFAF1 promoter sequence was amplified by PCR from human genomic DNA using Pfx DNA polymerase (Invitrogen, Carlsbad, CA) and put into pGL3 basic plasmid (Promega). The full-length NDUFAF1 cDNA was cloned from human WPMY-1 cells and constructed into pWPI vector.
Rotarod test
For determination of muscle weakness, a rotarod test was performed. Mice at the age of 3 months were put on an accelerating rotarod, and their ability to remain on the rotating rod was recorded. Each mouse was tested three times (18).
Strength and fatigability test
In brief, this test required the mice to pull themselves on top of a suspended rod. The measurement of muscle weakness was based on the mean percentage of passes over 15 trials of the test in a 3-min period. Fatigability was assessed as the average pass rate over time for each group of mice (19).
Treadmill test
Male wild-type (WT, TR4+/+) and TR4−/− mice were treadmill tested to measure their exercise capacity. All mice were familiarized to run on a motorized rodent treadmill with an electric grid at the rear of the treadmill (Exer-6M treadmill, Columbus Instruments, Columbus, OH) 2 d before completing an exercise performance test. For the performance test, the treadmill was started at a speed of 9 m/min with a 0° incline. After 9 min, the speed and incline were raised to 10 m/min and 5°, respectively, for the second stage of the test. Speed was then increased by 2.5 m/min every 3 min to a maximum of 40 m/min, and the incline progressively increased 5° every 9 min to a maximum of 15°. Exercise continued until exhaustion, defined as an inability to maintain running speed despite repeated contact with the electric grid (20). The exercise capacity for each mouse was evaluated by cumulative running distance and aerobic workload. The aerobic workload is the product of the vertical running distance to exhaustion and body weight: Work performed (J) = body weight (kg) × running speed (m/min) × running time (min) × grade × 9.8 (J/kg × m) (48).
Muscle histological staining
The fresh muscle specimen was mounted on cork using gum tragacanth with proper orientation. The specimen was frozen by immersion into the isopentane cooled to liquid nitrogen temperature. Tissues were sectioned using a cryostat for cross-sections. H&E stain, modified Gomori trichrome stain, SDH stain, and cytochrome c oxidase stain, were performed for each sample following protocols from http://neuromuscular.wustl.edu/lab/mbiopsy.htm#stain.
Electron microscopy
For electron-microscopic analysis, skeletal muscle tissues from TR4+/+ and TR4−/− mice were dissected in ice-cold fixative (4% glutaraldehyde in 0.1 m phosphate buffer;pH 7.4) and fixed in 2.5% glutaraldehyde for 1 h and then in 0.6% glutaraldehyde in PBS. Tissues were then postfixed in 1% OsO4 in PBS and embedded in Epon resin. Ultrathin sections were prepared and stained with uranyl/lead using standard methods. Sections (100 nm) were examined with a transmission electron microscope for mitochondrial abnormalities and accumulation of fusion vesicles on the subsarcolemmal membrane (49).
Plasma lactic acid determinations
Lactate content in plasma was determined by measuring the oxidized 2,6-dichlorophenol-indophenol (DCPIP), which is reduced by phenazine methosulfate, which in turn is reduced by NADH produced by lactate dehydrogenase, which oxidizes at the same time lactate to pyruvate. The plasma were analyzed in buffer S (10 mm KH2PO4, pH 7.8; 2 mm EDTA; 1 mg/ml BSA; 0.06 mm DCPIP; 0.5 mm phenazine methosulfate; 0.8 mm NAD+; 1.5 mm glutamate; 10 U/ml glutamate-pyruvate-transaminase, and 25 U/ml lactate dehydrogenase) and DCPIP oxidation was spectrophotometrically measured at 600 nm (50).
Isolation of mitochondria from skeletal muscle tissues
Mitochondria were isolated from skeletal muscle tissues using a MITOISO1 Mitochondria Isolation Kit (Sigma-Aldrich, St. Louis, MO) and following the manufacturer's instructions. In brief, the sample was quickly minced with scissors in isolation buffer (200 mm mannitol; 70 mm sucrose; 1 mm EGTA; in 10 mm HEPES, pH 7.5). After suspension in buffer with 0.25 mg/ml trypsin, the minced samples were homogenized. The homogenate was centrifuged at 600 × g for 5 min. The supernatant was recentrifuged at 11,000 × g for 10 min to obtain the final mitochondrial pellet. The pellet was suspended in 100 μl of storage buffer (250 mm sucrose, 1 mm ATP, 80 μm ADP, 5 mm sodium succinate, 2 mm K2HPO4, and 1 mm dithiothreitol in 10 mm HEPES, pH 7.5).
Mitochondrial ATP generation assay
Mitochondrial ATP generation rate was determined right after the isolation of mitochondria using a bioluminescence assay (Molecular Probes, Inc., Eugene, OR) and following the manufacturer's instructions. The luminescence change was calculated from the difference between the readings at 0 min and 1 min in a TD-20/20 luminometer (Turner Designs, Mountain View, CA), with the time of adding mitochondria as 0 min. The ATP values were calculated using an ATP standard curve. The mitochondrial protein concentrations were estimated using the Bradford assay (Bio-Rad Laboratories, Inc., Hercules, CA).
Enzymatic activities of the respiratory complexes
Enzymatic activities of the respiratory complexes were measured following published literatures (51–53). NADH-ubiquinone oxidoreductase activity was assayed in 50 mmol/liter potassium phosphate buffer (pH 7.7), 200 μmol/liter NADH, 2 mmol/liter KCN, and 90 μmol/liter decylubiquinone. Oxidation of NADH was monitored at 340 nm with a mmol/liter extinction coefficient of 6.2 for NADH. After 4 min, 1.0 μl 1 mmol/liter rotenone was added, and the absorbance was measured again at 30-sec intervals for 4 min. Succinate ubiquinone oxidoreductase activity was assayed in 80 μm DCPIP, 4 μm rotenone, 0.2 mm ATP, 10 mm succinate, and 80 μm decylubiquinone. The decrease in absorbance resulting from the reduction of DCPIP was monitored at 600 nm. Ubiquinol cytochrome c reductase was assayed in 80 mm decylubiquinol, 240 μm KCN, 4 μm rotenone, 200 μm ATP, and 40 μm oxidized cytochrome c. The increase in absorbance resulting from the reduction of cytochrome c was monitored at 550 nm. Complex V activity (ATP hydrolysis) can be measured spectrophotometrically by a coupled assay using lactate dehydrogenase and pyruvate kinase as the coupling enzyme. Reaction was started by adding mitochondria into 200 μl 50 mm Tris, pH 8.0; 5 mg/ml BSA; 20 mm MgCl2; 50 mm KCl; 15 μm carbonyl cyanide m-chlorophenyl hydrazone; 5 μm antimycin A; 10 mm phosphoenol pyruvate; 2.5 mm ATP; 4 U of lactate dehydrogenase and pyruvate kinase; and 1 mm NADH. Oligomycin (3 μm) was added and the reaction was followed for an additional 3 min to distinguish the ATPase activity coupled to the respiratory chain, after which the increase in absorbance at 340 nm resulting from NADH oxidation was determined. Citrate synthase activity was measured as described previously (54). Each measurement was performed three times.
ChIP assay
ChIP assays were performed in H1299 cells as previously described (55). Immunoprecipitations were performed at 4 C overnight, with 2 μg monoclonal antibody no. 15, which were generated using NH2 termini of TR4 as an antigen (AndroScience, San Diego, CA).
Real-time PCR quantification (Q-PCR) of gene expression
For RT-PCR and Q-PCR analysis of the mRNA expression of TR4 and mitochondrial disease-related genes, total RNA was isolated from TR4+/+ and TR4−/− mouse skeletal muscles, and from primary myoblast cultures using TRIzol reagent (Invitrogen). Q-PCR amplifications of reverse-transcribed first-strand DNA samples were performed using the iCycler iQ PCR cycler (Bio-Rad Laboratories). The relative abundance of target mRNA was quantified relative to the control GAPDH gene expression from the same reaction.
Statistical analysis
The data are presented as mean ± sd. Student's t test was used for comparisons between experimental groups. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Shey-Shing Sheu, University of Rochester (Rochester, NY) for his helpful discussion.
This work was supported by a George Whipple Professorship endowment, National Institutes of Health Grant CA127548, American Cancer Society RSG-06-123, and Taiwan Department of Health Clinical Trial and Research Center of Excellence grant DOH99-TD-B-111-004 (China Medical University, Taichung, Taiwan).
S.C. is retired from the Norris ALS Neuromuscular Research Institute.
Disclosure Summary: The authors declare no conflict of interest.
NURSA Molecule Pages†:
Nuclear Receptors: TR4.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AICAR
- Aminoimidazole carboxamide ribonucleotide
- ChIP
- chromatin immunoprecipitation
- DCPIP
- dichlorophenol-indophenol
- H&E
- hematoxylin and eosin
- NADH
- reduced nicotinamide adenine dinucleotide
- OXPHOS
- oxidative phosphorylation
- Q-PCR
- real-time PCR quantification
- SDH
- succinate dehydrogenase
- TCA
- tricarboxylic acid
- TR4
- testicular nuclear receptor 4.
References
- 1. Saraste M. 1999. Oxidative Phosphorylation at the fin de siècle. Science 283:1488–1493 [DOI] [PubMed] [Google Scholar]
- 2. Larsson NG, Clayton DA. 1995. Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 29:151–178 [DOI] [PubMed] [Google Scholar]
- 3. Thorburn DR. 2004. Mitochondrial disorders: Prevalence, myths and advances. J Inherited Metab Dis 27:349–362 [DOI] [PubMed] [Google Scholar]
- 4. Janssen RJ, Nijtmans LG, van den Heuvel LP, Smeitink JA. 2006. Mitochondrial complex I: structure, function and pathology. J Inherited Metab Dis 29:499–515 [DOI] [PubMed] [Google Scholar]
- 5. Schuelke M, Smeitink J, Mariman E, Loeffen J, Plecko B, Trijbels F, Stöckler-Ipsiroglu S, van den Heuvel L. 1999. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet 21:260–261 [DOI] [PubMed] [Google Scholar]
- 6. Procaccio V, Mousson B, Beugnot R, Duborjal H, Feillet F, Putet G, Pignot-Paintrand I, Lombès A, De Coo R, Smeets H, Lunardi J, Issartel JP. 1999. Nuclear DNA origin of mitochondrial complex I deficiency in fatal infantile lactic acidosis evidenced by transnuclear complementation of cultured fibroblasts. J Clin Invest 104:83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smeitink J, van den Heuvel L. 1999. Human mitochondrial complex I in health and disease. Am J Hum Genet 64:1505–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bénit P, Chretien D, Kadhom N, de Lonlay-Debeney P, Cormier-Daire V, Cabral A, Peudenier S, Rustin P, Munnich A, Rötig A. 2001. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet 68:1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ogilvie I, Kennaway NG, Shoubridge EA. 2005. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest 115:2784–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugiana C, Pagliarini DJ, McKenzie M, Kirby DM, Salemi R, Abu-Amero KK, Dahl HH, Hutchison WM, Vascotto KA, Smith SM, Newbold RF, Christodoulou J, Calvo S, Mootha VK, Ryan MT, Thorburn DR. 2008. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am J Hum Genet 83:468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DiMauro S, Schon EA. 2003. Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656–2668 [DOI] [PubMed] [Google Scholar]
- 12. Tanabe O, Shen Y, Liu Q, Campbell AD, Kuroha T, Yamamoto M, Engel JD. 2007. The TR2 and TR4 orphan nuclear receptors repress Gata1 transcription. Genes Dev 21:2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Young WJ, Smith SM, Chang C. 1997. Induction of the intronic enhancer of the human ciliary neurotrophic factor receptor (CNTFRα) gene by the TR4 orphan receptor. A member of steroid receptor superfamily. J Biol Chem 272:3109–3116 [DOI] [PubMed] [Google Scholar]
- 14. Lee YF, Shyr CR, Thin TH, Lin WJ, Chang C. 1999. Convergence of two repressors through heterodimer formation of androgen receptor and testicular orphan receptor-4: a unique signaling pathway in the steroid receptor superfamily. Proc Natl Acad Sci USA 96:14724–14729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins LL, Lee YF, Heinlein CA, Liu NC, Chen YT, Shyr CR, Meshul CK, Uno H, Platt KA, Chang C. 2004. Growth retardation and abnormal maternal behavior in mice lacking testicular orphan nuclear receptor 4. Proc Natl Acad Sci USA 101:15058–15063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen YT, Collins LL, Uno H, Chang C. 2005. Deficits in motor coordination with aberrant cerebellar development in mice lacking testicular orphan nuclear receptor 4. Mol Cell Biol 25:2722–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM. 2006. Nuclear receptor expression links the circadian clock to metabolism. Cell 126:801–810 [DOI] [PubMed] [Google Scholar]
- 18. Grohmann K, Rossoll W, Kobsar I, Holtmann B, Jablonka S, Wessig C, Stoltenburg-Didinger G, Fischer U, Hübner C, Martini R, Sendtner M. 2004. Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1). Hum Mol Genet 13:2031–2042 [DOI] [PubMed] [Google Scholar]
- 19. Joya JE, Kee AJ, Nair-Shalliker V, Ghoddusi M, Nguyen MA, Luther P, Hardeman EC. 2004. Muscle weakness in a mouse model of nemaline myopathy can be reversed with exercise and reveals a novel myofiber repair mechanism. Hum Mol Genet 13:2633–2645 [DOI] [PubMed] [Google Scholar]
- 20. Massett MP, Berk BC. 2005. Strain-dependent differences in responses to exercise training in inbred and hybrid mice. Am J Physiol Regul Integr Comp Physiol 288:R1006–R1013 [DOI] [PubMed] [Google Scholar]
- 21. Reichmann H, Vogler L, Seibel P. 1996. Ragged red or ragged blue fibers. Eur Neurol 36:98–102 [DOI] [PubMed] [Google Scholar]
- 22. Hoppel CL, Kerr DS, Dahms B, Roessmann U. 1987. Deficiency of the reduced nicotinamide adenine dinucleotide dehydrogenase component of complex I of mitochondrial electron transport. Fatal infantile lactic acidosis and hypermetabolism with skeletal-cardiac myopathy and encephalopathy. J Clin Invest 80:71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trijbels JM, Scholte HR, Ruitenbeek W, Sengers RC, Janssen AJ, Busch HF. 1993. Problems with the biochemical diagnosis in mitochondrial (encephalo-)myopathies. Eur J Pediatr 152:178–184 [DOI] [PubMed] [Google Scholar]
- 24. Vojtísková A, Jesina P, Kalous M, Kaplanová V, Houstek J, Tesarová M, Fornůsková D, Zeman J, Dubot A, Godinot C. 2004. Mitochondrial membrane potential and ATP production in primary disorders of ATP synthase. Toxicol Mech Methods 14:7–11 [DOI] [PubMed] [Google Scholar]
- 25. McKenzie M, Liolitsa D, Akinshina N, Campanella M, Sisodiya S, Hargreaves I, Nirmalananthan N, Sweeney MG, Abou-Sleiman PM, Wood NW, Hanna MG, Duchen MR. 2007. Mitochondrial ND5 gene variation associated with encephalomyopathy and mitochondrial ATP consumption. J Biol Chem 282:36845–36852 [DOI] [PubMed] [Google Scholar]
- 26. Janssen R, Smeitink J, Smeets R, van Den Heuvel L. 2002. CIA30 complex I assembly factor: a candidate for human complex I deficiency? Hum Genet 110:264–270 [DOI] [PubMed] [Google Scholar]
- 27. Kim ES, Liu NC, Yu IC I-Chen, Lin HY, Lee YF, Sparks JD, Chen LM, Chang C. 2011. Metformin Inhibits Nuclear Receptor TR4-Mediated Hepatic Stearoyl-Coenzyme A Desaturase 1 Gene Expression With Altered Insulin Sensitivity. Diabetes 60:1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zeviani M, Di Donato S. 2004. Mitochondrial disorders. Brain 127:2153–2172 [DOI] [PubMed] [Google Scholar]
- 29. Hui J, Kirby DM, Thorburn DR, Boneh A. 2006. Decreased activities of mitochondrial respiratory chain complexes in non-mitochondrial respiratory chain diseases. Dev Med Child Neurol 48:132–136 [DOI] [PubMed] [Google Scholar]
- 30. Schubert W, Sotgia F, Cohen AW, Capozza F, Bonuccelli G, Bruno C, Minetti C, Bonilla E, Dimauro S, Lisanti MP. 2007. Caveolin-1(−/−)- and caveolin-2(−/−)-deficient mice both display numerous skeletal muscle abnormalities, with tubular aggregate formation. Am J Pathol 170:316–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luan X, Chen B, Liu Y, Zheng R, Zhang W, Yuan Y. 2009. Tubular aggregates in paralysis periodica paramyotonica with T704M mutation of SCN4A. Neuropathology 29:579–584 [DOI] [PubMed] [Google Scholar]
- 32. Chou SM. 1967. Myxovirus-like structures in a case of human chronic polymyositis. Science 158:1453–1455 [DOI] [PubMed] [Google Scholar]
- 33. Endo M. 2009. Calcium-induced calcium release in skeletal muscle. Physiol Rev 89:1153–1176 [DOI] [PubMed] [Google Scholar]
- 34. Valsecchi F, Esseling JJ, Koopman WJ, Willems PH. 2009. Calcium and ATP handling in human NADH:ubiquinone oxidoreductase deficiency. Biochim Biophys Acta 1792:1130–1137 [DOI] [PubMed] [Google Scholar]
- 35. Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. 1999. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med 5:951–954 [DOI] [PubMed] [Google Scholar]
- 36. Parker WD, Jr, Parks JK, Swerdlow RH. 2008. Complex I deficiency in Parkinson's disease frontal cortex. Brain Res 1189:215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Esteitie N, Hinttala R, Wibom R, Nilsson H, Hance N, Naess K, Teär-Fahnehjelm K, von Döbeln U, Majamaa K, Larsson NG. 2005. Secondary metabolic effects in complex I deficiency. Ann Neurol 58:544–552 [DOI] [PubMed] [Google Scholar]
- 38. Vogel H. 2001. Mitochondrial myopathies and the role of the pathologist in the molecular era. J Neuropathol Exp Neurol 60:217–227 [DOI] [PubMed] [Google Scholar]
- 39. Lazarou M, Thorburn DR, Ryan MT, McKenzie M. 2008. Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta 1793:78–88 Corrected Proof [DOI] [PubMed] [Google Scholar]
- 40. Saada A, Edvardson S, Rapoport M, Shaag A, Amry K, Miller C, Lorberboum-Galski H, Elpeleg O. 2008. C6ORF66 is an assembly factor of mitochondrial complex I. Am J Hum Genet 82:32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dunning CJ, McKenzie M, Sugiana C, Lazarou M, Silke J, Connelly A, Fletcher JM, Kirby DM, Thorburn DR, Ryan MT. 2007. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J 26:3227–3237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parfait B, de Lonlay P, von Kleist-Retzow JC, Cormier-Daire V, Chrétien D, Rötig A, Rabier D, Saudubray JM, Rustin P, Munnich A. 1999. The neurogenic weakness, ataxia and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia. Eur J Pediatr 158:55–58 [DOI] [PubMed] [Google Scholar]
- 43. Li G, Lee YF, Liu S, Cai Y, Xie S, Liu NC, Bao BY, Chen Z, Chang C. 2008. Oxidative stress stimulates testicular orphan receptor 4 through forkhead transcription factor forkhead box O3a. Endocrinology 149:3490–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hirst J, King MS, Pryde KR. 2008. The production of reactive oxygen species by complex I. Biochem Soc Trans 36:976–980 [DOI] [PubMed] [Google Scholar]
- 45. Verkaart S, Koopman WJ, van Emst-de Vries SE, Nijtmans LG, van den Heuvel LW, Smeitink JA, Willems PH. 2007. Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim Biophys Acta 1772:373–381 [DOI] [PubMed] [Google Scholar]
- 46. Iuso A, Scacco S, Piccoli C, Bellomo F, Petruzzella V, Trentadue R, Minuto M, Ripoli M, Capitanio N, Zeviani M, Papa S. 2006. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J Biol Chem 281:10374–10380 [DOI] [PubMed] [Google Scholar]
- 47. Kim E, Yang Z, Liu NC, Chang C. 2005. Induction of apolipoprotein E expression by TR4 orphan nuclear receptor via 5′ proximal promoter region. Biochem Biophys Res Commun 328:85–90 [DOI] [PubMed] [Google Scholar]
- 48. Kinugawa S, Wang Z, Kaminski PM, Wolin MS, Edwards JG, Kaley G, Hintze TH. 2005. Limited exercise capacity in heterozygous manganese superoxide dismutase gene-knockout mice: roles of superoxide anion and nitric oxide. Circulation 111:1480–1486 [DOI] [PubMed] [Google Scholar]
- 49. Martin-Touaux E, Puech JP, Château D, Emiliani C, Kremer EJ, Raben N, Tancini B, Orlacchio A, Kahn A, Poenaru L. 2002. Muscle as a putative producer of acid α-glucosidase for glycogenosis type II gene therapy. Hum Mol Genet 11:1637–1645 [DOI] [PubMed] [Google Scholar]
- 50. Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schägger H, Rustin P, Kroemer G. 2004. AIF deficiency compromises oxidative phosphorylation. EMBO J 23:4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ. 2007. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 53:729–734 [DOI] [PubMed] [Google Scholar]
- 52. de Wit LE, Sluiter W. 2009. Chapter 9 Reliable assay for measuring complex I activity in human blood lymphocytes and skin fibroblasts. Methods Enzymol 456:169–181 [DOI] [PubMed] [Google Scholar]
- 53. Barrientos A, Fontanesi F, Diaz F. 2009. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet Chapter 19:Unit19 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trounce IA, Kim YL, Jun AS, Wallace DC. 1996. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 264:484–509 [DOI] [PubMed] [Google Scholar]
- 55. Liu NC, Lin WJ, Kim E, Collins LL, Lin HY, Yu IC, Sparks JD, Chen LM, Lee YF, Chang C. 2007. Loss of TR4 orphan nuclear receptor reduces phosphoenolpyruvate carboxykinase-mediated gluconeogenesis. Diabetes 56:2901–2909 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.