

Abstract
To determine the reproducibility and quantitation of the amplicon sequencing-based detection approach for analyzing microbial community structure, a total of 24 microbial communities from a long-term global change experimental site were examined. Genomic DNA obtained from each community was used to amplify 16S rRNA genes with two or three barcode tags as technical replicates in the presence of a small quantity (0.1% wt/wt) of genomic DNA from Shewanella oneidensis MR-1 as the control. The technical reproducibility of the amplicon sequencing-based detection approach is quite low, with an average operational taxonomic unit (OTU) overlap of 17.2%±2.3% between two technical replicates, and 8.2%±2.3% among three technical replicates, which is most likely due to problems associated with random sampling processes. Such variations in technical replicates could have substantial effects on estimating β-diversity but less on α-diversity. A high variation was also observed in the control across different samples (for example, 66.7-fold for the forward primer), suggesting that the amplicon sequencing-based detection approach could not be quantitative. In addition, various strategies were examined to improve the comparability of amplicon sequencing data, such as increasing biological replicates, and removing singleton sequences and less-representative OTUs across biological replicates. Finally, as expected, various statistical analyses with preprocessed experimental data revealed clear differences in the composition and structure of microbial communities between warming and non-warming, or between clipping and non-clipping. Taken together, these results suggest that amplicon sequencing-based detection is useful in analyzing microbial community structure even though it is not reproducible and quantitative. However, great caution should be taken in experimental design and data interpretation when the amplicon sequencing-based detection approach is used for quantitative analysis of the β-diversity of microbial communities.
Keywords: metagenomics, amplicon sequencing, pyrosequencing, reproducibility, quantitation, random sampling
Introduction
Soil microbial communities are among the most complex, diverse and important assemblages in the biosphere. It is estimated that 1 g of forest soil contains 4 × 107 prokaryotic cells, whereas 1 g of grassland soil has 2 × 109 prokaryotic cells (Daniel, 2005). DNA reassociation kinetic studies have estimated that 1 g of soil contains approximately 2000–18 000 genomes (Torsvik et al., 1996, 1998; Torsvik and Ovreas, 2002), suggesting that prokaryotic diversity in soil is extremely high. Owing to such high diversity, soil microbial communities are among the most difficult to characterize both phenotypically and genetically. Although introduction of molecular biology approaches have greatly advanced microbial community studies (Zhou et al., 2004), our understanding of the extent of microbial diversity in soil environments is still very limited, especially in contrast to plants and animals. In addition, due to the uncultivated status of the majority of microorganisms (>99%) in nature, understanding their genetic properties, metabolic characteristics, biochemical functions and ecological functions is even more difficult.
Large-scale genome sequencing and associated genomic technologies have revolutionized the study of microbial communities. Recently, several new high-throughput sequencing approaches such as 454 pyrosequencing, Illumina (Illumina, San Diego, CA, USA) and SOLiD (Applied Biosystems, Foster City, CA, USA) have been developed (Margulies et al., 2005; Hamady et al., 2008; Smith et al., 2008; Schulte et al., 2010). At present, the 454 pyrosequencing technology has been widely used to reveal microbial diversity and ecology in different natural environments, such as the deep sea (Sogin et al., 2006; Huber et al., 2007), extreme hydrogeological conditions (Edwards et al., 2006) and soil (Leininger et al., 2006; Roesch et al., 2007). In addition, multiplexed high-throughput pyrosequencing of individual genes (for example, 16S rRNA) by tagging or bar coding with short nucleotides (also called pyrotag sequencing (Kunin et al., 2010)) (Huse et al., 2007; Parameswaran et al., 2007; Roesch et al., 2007; Hamady et al., 2008) has been developed to process many samples simultaneously and has been widely used in microbial community studies (Youssef et al., 2009; Cheung et al., 2010; Chun et al., 2010; He et al., 2010a, 2010b; Koopman et al., 2010; Schutte et al., 2010; Teixeira et al., 2010; Uroz et al., 2010). A similar strategy has also been used for sequencing functional genes (Iwai et al., 2010). Thus, as PCR-amplified products (amplicons) are used for sequencing, this approach is generally referred to as amplicon sequencing. Various studies have clearly demonstrated that amplicon sequencing is powerful for studying microbial community diversity and activity.
Some technical problems inherent to amplicon sequencing have recently been observed, such as overestimating the rare biosphere because of pyrosequencing errors (Quince et al., 2008, 2009; Kunin et al., 2010) and overestimating gene and taxon abundance because of inherent, systematic artifacts (Gomez-Alvarez et al., 2009). Amplicon size and primer pair also seem to have a great influence on estimating microbial community richness and evenness (Engelbrektson et al., 2010). However, it is unclear whether amplicon sequencing-based detection is reproducible and quantitative. In this study, we have examined these important issues by analyzing the composition and structure of 24 microbial communities from a field site in Oklahoma (USA) for a long-term study of global climate change. Genomic DNA obtained from each community was used to amplify 16S rRNA genes with two or three bar-coded tags as technical replicates in the presence of a small quantity (0.1% wt/wt) of Shewanella oneidensis MR-1 genomic DNA as the control. In this study, technical replicates mean that the same genomic DNA obtained from a community is amplified multiple times and subsequently sequenced. Our results indicated that the reproducibility among technical replicates and quantitative capability were quite low for amplicon sequencing-based detection. Various strategies for improving data comparability are discussed. Although experimental data are generated by the pyrosequencing approach in this study, the conclusions should be applicable to other sequencing approaches as well.
Materials and methods
Site description and sampling
Soil samples used in this study were obtained from the KFFL (Kessler Farm Field Laboratory) at the Great Plain Apiaries in McClain County (OK, USA; 34°58′54′′N, 97°31′14′′W) and were part of a long-term experiment using a paired nested design with warming as the main factor and clipping as a secondary factor (Luo et al., 2001). There were six replicates for each of the following treatments: (1) clipped, warmed, (2) unclipped, warmed, (3) clipped control and (4) unclipped control. The warming plots have been continually warmed for ∼2 °C since November 1999. A total of 24 soil samples were taken from a 0–15 cm layer in April 2007. Each sample was composited from four soil cores (2 cm diameter × 15 cm deep) after being sieved (2 mm). All samples were stored at −80 °C.
DNA extraction
Soil DNA was extracted by freeze-grinding mechanical lysis as described previously (Zhou et al., 1996) and was purified using a low melting agarose gel, followed by phenol extraction. DNA quality was assessed based on the absorbance ratios of 260/280 nm and 260/230 nm using NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), whereas DNA concentration was quantified by PicoGreen (Promega, Sunnyvale, CA, USA) (Ahn et al., 1996) using FLUOstar Optima (BMG Labtech, Jena, Germany).
Sample tagging and PCR amplicon preparations for sequencing
On the basis of the V4-V5 hypervariable regions of bacterial 16S rRNAs (Escherichia coli positions 515–907), the PCR primers, F515: GTGCCAGCMGCCGCGG and R907: CCGTCAATTCMTTTRAGTTT were selected. Both primers covered >98% of the 16S gene sequences in the ribosomal database (Cole et al., 2007, 2009). To pool multiple samples for one run of 454 sequencing, a sample tagging approach was used (Binladen et al., 2007; Hamady et al., 2008). Each tag (6 mer) was added to the 5′-end of both forward and reverse primers (Supplementary Table S1), synthesized by Invitrogen (Carlsbad, CA, USA) and then used for the generation of PCR amplicons. To evaluate the reproducibility of sequencing results, a total of 60 sequencing tags were used: 12 samples were amplified with 3 tagged primers (n=36) and 12 with 2 tagged primers (n=24). Pyrosequencing data for four samples were excluded from data analysis because of some errors in primers used.
The amplification mix contained 10 Units of Pfu polymerase (BioVision, Mountain View, CA, USA), 5 μl Pfu reaction buffer, 200 μ dNTPs (Amersham, Piscataway, NJ, USA) and a 0.2 μ concentration of each primer in a volume of 50 μl. Genomic DNA (10 ng) was added to each amplification mix. Cycling conditions were an initial denaturation at 94 °C for 3 min, 25 cycles of 95 °C for 30 s, 58 °C for 60 s, 72 °C for 60 s and a final 2-min extension at 72 °C. To obtain enough PCR products for sequencing, multiple (5–10) 100-μl reactions were used for each sample. The products from each tagged primer were pooled and purified by agarose gel electrophoresis. The amplified PCR products were recovered and then quantitated using PicoGreen (Ahn et al., 1996). Finally, amplicons of all samples were pooled in an equimolar concentration for 454 pyrosequencing.
454 Pyrosequencing
Pyrosequencing was carried out at the Genome Sequencing Center of the University of Oklahoma. In brief, fragments in the amplicon libraries were repaired and ligated to the 454 sequencing adapters, and the resulting products were bound to beads under conditions that favor one fragment per bead. The beads were emulsified in a PCR mixture in oil, and PCR amplification occurred in each droplet, generating millions of copies of a unique DNA template. After breaking the emulsion, DNA strands were denatured, and beads carrying single-stranded DNA clones were deposited into wells on a PicoTiter Plate (454 Life Sciences, Branford, CT, USA) for pyrosequencing (Margulies et al., 2005) on a FLX 454 system. For this study, we recovered both forward and reverse sequencing reads of the 24 samples with an average length of ∼240 bp. All pyrosequencing reads were initially processed using the RDP (Ribosomal Database Project) pyrosequencing pipeline (http://pyro.cme.msu.edu/) (Cole et al., 2009).
Pyrosequencing data preprocessing
To minimize the effects of random sequencing errors, we eliminated low-quality sequences as follows: (1) sequences that did not perfectly match the PCR primer at the beginning of a read; (2) sequences with non-assigned tags; (3) short sequence reads (<200 bp) after the proximal PCR primer if they terminated before reaching the distal primer and (4) sequences that contained more than one undetermined nucleotide (N). Only the first 240 bp after the proximal PCR primer of each sequence was included as the quality of sequences degrades beyond this point. After removing low-quality sequences, the raw sequences were sorted and distinguished by unique sample tags and each sample had two or three unique tags as replicates. The tags and primers were then trimmed for each replicate.
On the basis of sequences obtained, there is ∼120 bp overlap between the sequences from the forward and reverse primers. To identify sequences that potentially originated from the same 16S rRNA gene molecule, sequences from forward and reverse primers were compared. If the number of overlapped nucleotides was >120 bp and their sequence identity was >97%, these two forward and reverse sequences were considered to originate from the same DNA molecule. If one sequence from the forward (or reverse) primer was >97% identical to multiple sequences, the one with the highest identity from the reverse (forward) primer was considered to be its corresponding counterpart. The two matched sequences were then combined to generate a longer sequence (356 nt). All combined sequences and individual sequences from both the forward and the reverse primers were then pooled as a full set of sequences of each tag.
All sequences from the 24 samples were aligned by RDP Infernal Aligner, a fast secondary-structure aware aligner (Ribosomal Database Project, Michigan State University, East Lansing, MI, USA) (Nawrocki and Eddy, 2007) and then complete linkage clustering was used to define operational taxonomic units (OTUs) within a 0.03 difference (Stackebrandt and Goebel, 1994). As 97% identify is used as the cutoff and singleton sequence reads were removed, each OTU contains more than two sequence reads from at least two samples. The sequence reads within an OTU were partitioned into individual samples based on the original sample-tagging information. In addition, sequences were then assigned to a taxonomy by the RDP classifier (Wang et al., 2007) with a confidence cutoff of 0.8. The lineage of each OTU was summarized with all phylogenetic information.
As the sequence numbers of individual OTUs obtained varied significantly among different samples, the relative proportions of sequence numbers were used for subsequent analysis. The RA (relative abundance) was calculated as:
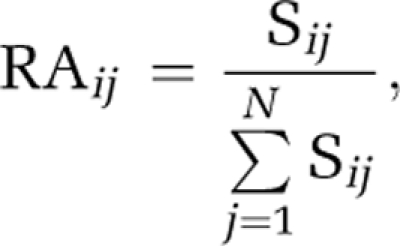 |
where Sij is the number of sequencing reads of the jth OTU (1 to n) in the ith sample (1 to m).
Statistical analysis
The Mothur program (University of Michigan Health System, Ann Arbor, MI, USA) (Patrick Schloss, http://www.mothur.org/) was used for rarefaction analysis at different levels based on individual tags, samples, treatments and the whole data set. Rarefaction curves were created by plotting the results of random sequence re-sampling from OTUs. The rarefaction curves were fitted with the model y=a(1−e−bx), and maximum OTUs were estimated as predicted diversity based on the fitted rarefaction curve. OTU predictions were also obtained using the Chao1 method (Chao, 1987). Both Sørensen similarity (Ss) and Bray-Curtis similarity (BCs) were calculated between any pair of two samples, and the complement (Sd) of Sørensen similarity (1−Ss) and the complement (BCd) of Bray-Curtis similarity (1−BCs) were used to measure β-diversity of microbial communities among different samples. One-way ANOVA (analysis of variance) was used to compare the β-diversity at technical (tag) replicate and biological replicate levels, and that between control and the treatments (warming and control). The Duncan multiple range test was used to determine statistical significance of the differences between microbial communities at different levels.
Ordination analyses were further performed using PC-ORD (McCune and Mefford, 1999) and confirmed by CANOCO 4.5 (Biometris—Plant Research International, Wageningen, The Netherlands). Detrended correspondence analysis was used to determine the overall phylogenetic composition changes in microbial communities. Detrended correspondence analysis is an ordination technique that uses detrending to remove the arch effect, in which data points are organized in a horseshoe-like shape, in correspondence analysis (Hill and Gauch, 1980).
Data sets of microbial communities generated by different analytical methods were used to examine whether warming or clipping had significant effects on soil microbial communities. Typically, it is difficult for all data sets to meet the assumptions (for example, normality, equal variances, independence) of parametric statistics. Thus, in this study, three different complementary non-parametric analyses for multivariate data were used: analysis of similarity (ANOSIM) (Clarke, 1993), non-parametric multivariate ANOVA (Adonis) using distance matrices (Anderson, 2001) and multiresponse permutation procedure (MRPP). We used the Bray-Curtis similarity index to calculate the distance matrix for ANOSIM, Adonis and MRPP analyses. MRPP is a non-parametric procedure that does not depend on assumptions, such as normally distributed data or homogeneous variances. Rather, it depends on the internal variability of data (Mielke and Berry, 2001; McCune and Grace, 2002). All three methods are based on dissimilarities among samples and their rank order to calculate test statistics. The Monte Carlo permutation was used to test the significance of statistics. All three procedures (ANOSIM, Adonis and MRPP) were performed using the Vegan package (version 1.15-1, Department of Statistics, Iowa State University, Ames, IA, USA) in R v. 2.8.1 (R Development Core Team, 2006).
Results
OTU overlaps among technical replicates
The V4-V5 regions of 16S rRNA genes were amplified and sequenced with one pyrosequencing run, and a total of 52.2 Mbp sequences with 213 329 reads were obtained (see Supplementary text for overview of sequencing statistics). A total of 115 741 sequence reads were obtained after preprocessing. An average of 1121±390 OTUs were obtained for each tag based on the combined samples. Rarefaction analysis indicated that the maximum number of OTUs in this warming site is ∼17 370, and that the diversity of the abundant populations in these communities was recovered in this study (Supplementary text).
As community DNA from the same sample was amplified with two or three tagged primers as technical replicates, followed by sequencing, theoretically, a maximum of 100% overlap of OTUs detected among these technical replicates should have been expected if all populations in these communities were sampled. However, in reality, the expected percentage of overlap will be lower because of undersampling, random sampling and the complexity of microbial communities. Surprisingly, the overlap among these technical replicates was quite low (Supplementary Table S2). The average OTU overlap among the samples with two technical replicates for the forward primer was 15.5%±2.5% (Supplementary Figure S1A), ranging from 10.9% to 19.2% (Supplementary Table S2A) and 18.8%±2.1% (Supplementary Figure S1B) for the reverse primer with the range of 13.9–21.4% (Supplementary Table S2B). The difference between forward and reverse primers was significant (P=0.003 based on Mann–Whitney U-test). The average OTU overlap among the samples with three technical replicates was 7.4%±2.1% (Supplementary Figure S2A) in the range of 4.1–11.0% (Supplementary Table S2A) for the forward primer, and 9.1%±2.5% (Supplementary Figure S2B) in the range of 3.5–12% for the reverse primer (Supplementary Table S2B). The difference between forward and reverse primers was also significant (P=0.05 based on Mann–Whitney U-test). These results suggest that variations in technical replicates for the amplicon sequencing-based detection approach are quite high, which are most likely due to problems associated with random sampling processes.
The OTU overlap among technical replicates was increased significantly by removing singleton sequences. After removing singleton sequences, the OTU overlap among the samples with two technical replicates ranged from 15.1–30.2% (Supplementary Table S2A), with an average of 23.3%±4.3% (Supplementary Figure S1C, P<0.001 based on Mann–Whitney U-test) for the forward primer, and from 23.8% to 32.2% (Supplementary Table S2B) with an average of 26.6%±2.1% (Figure 1a, P<0.001) for the reserve primer. For samples with three technical replicates, they ranged from 6.2% to 16.6% (Supplementary Table S2A), with an average of 11.7%±3.3% (Supplementary Figure S2C, P=0.009) for the forward primer, and from 5.8% to 17.7% (Supplementary Table S2B) with an average of 13.3%±3.3% (Figure 1b, P=0.005) for the reverse primer. In addition, when biological replicates are available, one could remove the less-representative OTUs for a reliable diversity comparison across different samples. The percentages of OTUs overlap were significantly increased among technical replicates (Supplementary Table S2A–C) by removing the less-representative OTUs across all biological replicates within a treatment. In addition, to determine whether more abundant OTUs are reproducibly detected among technical replicates, OTUs were ranked based on their RA in each sample against their frequencies detected among the technical replicates. Interestingly, no consistent patterns were observed between the RA of the OTUs and their reproducibility among technical replicates (data not shown).
Figure 1.

The average overlap of OTUs of microbial communities sequenced with two or three pyrotags. (a) Reverse primer sequences with two tags, singleton sequences removed. (b) Reverse primer sequences with three tags, singleton sequences removed.
Effects of variations in technical replicates on diversity estimations
To determine how such variations in technical replicates affect the estimations of microbial local diversity (α-diversity), the Shannon–Weaver index was calculated at levels of the technical replicates, biological replicates and treatments. Three-way ANOVA indicates that gene richness, functional gene diversity and evenness were all significantly different among technical replicates (P<0.05) (Supplementary Table S3), but most of them, if not all, were not significantly different at the biological replicate or treatment level. These results suggest that variations in technical replicates may not affect the estimation of α-diversity at biological replicate and/or treatment levels.
To understand whether variations in technical replicates affect the comparison of different microbial communities (that is, β-diversity), two popular similarity metrics, Sørensen's incidence-based and Bray-Curtis's abundance-based methods, are used. Their complements are used as metrics of β-diversity, which are widely used in many studies and range from 0 (when all OTUs/individuals are shared between two communities) to 1 (when no OTUs/individuals are shared). β-Diversity was calculated for each pair of microbial communities at the levels of technical replicates, biological replicates and treatments based on the combined OTU data from both forward and reverse primers (Table 1). The average β-diversities at the technical replicate level were 0.756±0.042 based on the Sørensen index and 0.763±0.049 based on the Bray-Curtis index without removing singletons (Table 1). These results indicated that the background noise in estimating β-diversity is quite high. However, in this case, one-way ANOVA revealed that the average β-diversity at both biological replicate and treatment levels based on both the Sørensen and the Bray-Curtis indices were all still significantly (P<0.05) higher than those at the technical replicate level (Table 1). As expected, removing singletons significantly (P<0.05) decreased β-diversity estimations at all three levels, but had more impact on the β-diversity estimation based on the Bray-Curtis index (Table 1).
Table 1. One-way ANOVA and Duncan grouping method to assess β-diversity at the levels of technical replicates, biological replicates and treatments based on the combined OTUs from both forward and reverse primers.
| Data sizea |
With singletons |
Singletons removed |
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
Sørensen |
Bray-Curtis |
Sørensen |
Bray-Curtis |
||||||
| β-Diversityb | Significancec | β-Diversityb | Significancec | β-Diversityb | Significancec | β-Diversityb | Significancec | ||
| Technical replicate | 42 | 0.756 | c | 0.763 | c | 0.734 | c | 0.475 | b |
| Biological replicate | 322 | 0.814 | b | 0.866 | a | 0.798 | b | 0.533 | a |
| Warming | 196 | 0.83 | a | 0.854 | b | 0.815 | a | 0.526 | a |
| Clipping | 196 | 0.834 | a | 0.853 | b | 0.82 | a | 0.525 | a |
Abbreviation: ANOVA, analysis of variance.
Data sizes (n) are the number of data points of the pairwise comparisons within the technical replicates, biological replicates or treatments.
We calculate two popular β-diversity dissimilarity measurements, Sørensen and Bray-Curtis, in which Sørensen dissimilarity is based on OTUs richness and Bray-Curtis dissimilarity takes OTUs abundance into account.
Significance at [pr(>F)]<0.05, using Duncan grouping method is shown. a, b and c represent the significance of β-diversity differences between technical replicates, biological replicates and treatments. a marks the highest β-diversity, the one less than the highest but not significant is still marked with a, then the ones significantly lower than the highest is marked with b or c.
Quantitation of the amplicon sequencing
To determine whether amplicon sequencing-based detection is quantitative, 10 pg gDNA from S. oneidensis MR-1 was spiked into PCR reactions containing 10 ng community DNA of each sample, and then PCR amplified and pyrosequenced. MR-1 has nine copies of the 16S rRNA gene, with 100% identity among them in the sequenced regions. Thus, only a single OTU from MR-1 would be obtained. Theoretically, 0.1% (wt/wt) sequences from the MR-1 genome would be detected in all of these samples. However, our experimental results indicated that the percentages of MR-1 OTUs detected varied substantially among different soil samples, ranging from 0.06% to 5.34% for the forward primer and 0.0% to 4.42% for the reverse primer. The difference between the maximum and minimum percentages of detected MR-1 sequences is ∼66.7-fold for the forward primer. In contrast, no MR-1 sequence was detected in some tags for the reverse primer, whereas the maximum percentage of detected MR-1 sequences was 4.42% (Supplementary Table S4).
Improving data comparability
Various methods were used to improve data comparability, including increasing biological replicates, and removing singleton sequences and less-representative OTUs across biological replicates (see Supplementary text). Among three statistical methods (MRPP, ANOSIM and Adonis), which were used to compare the differences of microbial community structure between warming and non-warming or clipping and non-clipping, Adonis seems to be the most sensitive (Tables 2 and 3, Supplementary Tables S5 and S6). Thus, only the results from Adonis are presented. The significance value (P) is presented in these tables and smaller P-values indicate better resolution. The following generalizations can be drawn. First, removing singleton sequences yielded better resolution (Tables 2 and 3). Removing less-representative OTUs is another option based on the results of biological replicates. Considerable differences in P-values were obtained by removing less-representative OTUs (one or two) across samples (Tables 2 and 3). In addition, generally only one-strand DNA sequences were obtained by amplicon sequencing, and hence, approximately half of the sequencing data are not used. In this study, both primers for PCR amplification were tagged, pyrosequenced and then assigned to OTUs. Thus, using sequencing reads from both forward and reverse primers leads to increased sequencing efforts. As expected, considerably better resolution was obtained using the combined sequencing data sets from both primers (Tables 2 and 3).
Table 2. Effects of data preprocessing on the significance (P-values) in resolving the differences of microbial communities between warming and unwarminga.
| Primers |
Not removing singletons |
Removing singletons |
||||
|---|---|---|---|---|---|---|
| Cut 0b | Cut 1c | Cut 2d | Cut 0b | Cut 1c | Cut 2d | |
| Forward | 0.55 | 0.05 | 0.03 | 0.44 | 0.07 | 0.02 |
| Reverse | 0.51 | 0.08 | 0.04 | 0.19 | 0.13 | 0.03 |
| Forward and reverse | 0.63 | 0.02 | 0.03 | 0.04 | 0.03 | <0.01 |
Abbreviations: ANOSIM, analysis of similarity; MRPP, multiresponse permutation procedure.
The values in the table are probability values (P), indicating significance with the Adonis method. Three statistical methods (MRPP, ANOSIM and Adonis) were used. However, Adonis is more sensitive; thus, only results using this method are presented.
No OTUs were removed.
OTUs present only in 1 of the 14 tag sequence data sets of a treatment were removed.
OTUs present in 1 or 2 of the 14 tag sequence data sets of a treatment were removed.
Table 3. Effects of data preprocessing on the significance (P-values) in resolving the differences of microbial communities between clipping and unclippinga.
| Primers |
Not removing singletons |
Removing singletons |
||||
|---|---|---|---|---|---|---|
| Cut 0b | Cut 1c | Cut 2d | Cut 0b | Cut 1c | Cut 2d | |
| Forward | 0.6 | 0.1 | 0.03 | 0.59 | 0.07 | 0.04 |
| Reverse | 0.62 | 0.18 | 0.04 | 0.16 | 0.16 | 0.09 |
| Forward and reverse | 0.58 | 0.02 | <0.01 | 0.06 | 0.03 | <0.01 |
Abbreviations: ANOSIM, analysis of similarity; MRPP, multiresponse permutation procedure.
The values in the table are probability values (P), indicating significance with the Adonis method. Three statistical methods (MRPP, ANOSIM and Adonis) were used. However, Adonis is more sensitive; thus, only results using this method are presented.
No OTUs were removed.
OTUs present only in 1 of the 14 tag sequence data sets of a treatment were removed.
OTUs present only in 1 or 2 of the 14 tag sequence data sets of a treatment were removed.
Despite several findings for improving data comparability, the real question is whether the effects of warming and/or clipping on microbial communities can be detected based on such amplicon sequencing data. Detrended correspondence analysis showed that samples from warming and/or clipping plots clustered together and were well separated from control plots (Figure 2), suggesting that microbial community composition and structure were markedly different between warming and unwarming or between clipping and unclipping. To examine whether these observed differences are statistically significant, three complementary non-parametric multivariate statistical tests (ANOISM, Adonis and MRPP) were performed. The phylogenetic community structure based on the 16S rRNA gene was also significantly different between warming and unwarming, and between clipping and unclipping when more than five biological replicates were used, according to at least one of the three methods (Table 4). It is clear that increasing biological replicates provide better resolution in differentiating the treatment effects of warming or clipping. Taken together, these results indicated that the phylogenetic composition and structure of microbial communities under experimental warming or clipping were significantly different from those under control conditions.
Figure 2.

Detrended correspondence analysis (DCA) of 454 pyrosequencing data showing that warming and clipping significantly affected soil microbial community composition and structure. The relative abundance of all detected OTUs at 97% similarity cutoff in at least 4 of 14 tagged sequence data sets for each treatment were used for DCA analysis. Red dots are for unclipped, unwarmed control (UC), blue dots are for unwarmed, clipped (CC), dark purple dots are for unclipped, warmed (UW) and dark yellow dots are for clipped, warmed (CW). The effects of warming and clipping on soil microbial community composition and structure appeared to be well separated by the second axis.
Table 4. Significance tests of the effects of warming or clipping on the overall microbial community structure with three different statistical approaches.
| Data sets | Adonisa |
ANOSIMb |
MRPPc | |||
|---|---|---|---|---|---|---|
| F | P-valued | R | P-valued | δ | P-valued | |
| Warming vs unwarming | ||||||
| Six replicates | 2.607 | <0.01 | 0.519 | <0.001 | 33.43 | 0.025 |
| Five replicates | 2.419 | <0.01 | 0.5 | 0.009 | 33.12 | 0.052 |
| Four replicates | 1.960 | 0.04 | 0.427 | 0.059 | 32.02 | 0.107 |
| Three replicates | 1.797 | 0.06 | 0.482 | 0.179 | 30.82 | 0.220 |
| Clipping vs unclipping | ||||||
| Six replicates | 2.243 | <0.01 | 0.406 | 0.014 | 32.76 | 0.072 |
| Five replicates | 1.966 | 0.05 | 0.396 | 0.037 | 33.18 | 0.188 |
| Four replicates | 1.637 | 0.06 | 0.344 | 0.087 | 32 | 0.307 |
| Three replicates | 1.549 | 0.15 | 0.407 | 0.213 | 28.55 | 0.304 |
Abbreviations: ANOSIM, analysis of similarity; MRPP, multiresponse permutation procedure.
All three tests are non-parametric multivariate analyses based on dissimilarities among samples.
Permutational multivariate analysis of variance using distance matrices. Significance tests were performed by F-tests based on sequential sums of squares from permutations of the raw data.
ANOSIM: Statistic R is based on the difference of mean ranks between groups and within groups. The significance of observed R is assessed by permuting the grouping vector to obtain the empirical distribution of R under the null model.
MRPP: Statistic δ is the overall weighted mean of within-group means of the pairwise dissimilarities among sampling units. The significance test is the fraction of permuted deltas that are less than the observed delta;
P-value of corresponding significance test.
The values in bold indicate the significant difference between the treatments (warming and clipping) and the control (at the levels of P<0.01, or P<0.05) by the tests.
Discussion
With the recent development and application of metagenomic technologies such as large-scale high-throughput sequencing (Sogin et al., 2006; Huber et al., 2007; Hamady et al., 2008), PhyloChip and GeoChip (He et al., 2007, 2008, 2010a, 2010b; Zhou et al., 2008; Zhou, 2009; Vaishampayan et al., 2010), spatial and temporal information on microbial community structure and functional activities can be rapidly obtained at a community-wide level. Metagenomic technologies allow scientists to address research questions, which could not be approached previously. High-throughput metagenomic technologies have greatly advanced microbiologists' capabilities for analyzing microbial communities in the environment and significantly shaped the field of microbial ecology. However, great caution is required when using high-throughput sequencing technologies for characterizing microbial community composition and structure as demonstrated in this study and in several others (Huse et al., 2007; Gomez-Alvarez et al., 2009; Engelbrektson et al., 2010; Kunin et al., 2010).
Despite several reports on the rigorous analyses of pyrosequencing error rates of 16S rRNA genes (Huse et al., 2007; Kunin et al., 2010), overestimation of rare biosphere communities (Huse et al., 2010; Kunin et al., 2010) and gene/taxon abundance (Gomez-Alvarez et al., 2009), as well as PCR amplification conditions on estimating species richness and evenness (Engelbrektson et al., 2010), little is known about the technical reproducibility and quantitation of amplicon sequencing. Our results in this study demonstrated that the reproducibility of the detected OTUs among these technical replicates is quite low, with an average of 13.1%±1.5% OTU overlap for the two tags and 5.9%±1.6% OTU overlap for the three tags. Such low percentages of OTU overlap among technical replicates raise a concern of comparing the β-diversity of microbial communities across different samples for amplicon sequencing. Inherent high variations in technical replicates could be a significant problem for comparative studies (for example, β-diversity), but it could be less problematic for diversity discovery studies (for example, α-diversity). To make meaningful comparisons across different samples, variations among different samples must be larger than variations in technical replicates. Otherwise, it will be difficult to draw confident biological conclusions.
The high variations among technical replicates is most likely due to sampling artifacts associated with random sampling (Zhou et al., 2008), as well as the performance of the technology per se. Many steps in the pyrotag-based sequencing analysis are associated with random sampling, including PCR amplification of target genes, ligation of amplified PCR products to sequencing adaptors for generating single-stranded DNA bound beads, emulsion and immobilization of beads, as well as bead deposition into wells on a PicoTiter Plate (454 Life Sciences). Given the vast complexity of a soil microbial community, the random sampling process could have more dramatic influence on estimating the difference of microbial communities across different samples (that is, β-diversity) (Zhou et al., 2008). However, further theoretical analysis is required to support such speculation. In addition, based on rarefaction analysis, <10% of the microbial populations/genomes were sampled in each sampling event (that is, herein equivalent to sequencing a community with a tag). If microbial population abundance in the community is evenly distributed, theoretical values of 1% and 0.1% overlap would be expected for two and three sampling events, respectively. However, the actual percentages of OTU overlap were higher than the expected values (Figure 1), which could be explained by the facts that microbial populations could be not evenly distributed and dominant populations have a higher probability of being re-sampled. In addition, although this study addressed the random sampling problem of pyrosequencing, such problems should also exist in other similar sequencing technologies, such as Illumina and SOLiD sequencing platforms.
In general, only sequences from one strand are used for analysis with pryotag sequencing. Therefore, about half of the available information is not used (Acosta-Martinez et al., 2008; Hollister et al., 2010). As demonstrated in this study, combining sequences from both forward and reverse primers alleviates the problem of random sampling because it doubles the sampling effort. As demonstrated in this study, the statistical resolution is greatly improved when sequences from both ends are used for determining OTUs. To combine them together, an appropriate length of overlap of sequences from both ends is required. The 16S primers used in this study (F515, R907) seem to be an appropriate choice because they are highly conserved and their derived sequences have an appropriate length of overlap (123 bp) with the FLEX pyrosequencing technology. Various studies have demonstrated that the average length (240 bp) of DNA fragments generated by these primers should be sufficient to determine statistically significant differences among microbial genomes that reflect the most likely biology occurring in the environment (Edwards et al., 2006; Sogin et al., 2006).
Quantitative capability is a critical issue when sequencing-based technology is used for environmental and ecological studies. Using the control DNA spiked into PCR reactions, substantial variations in the control sequences were observed among the 56 tag sequencing, despite that various strategies were used to alleviate the amplification biases on quantitation, including combining several amplifications together and using less cycle numbers to avoid PCR product saturation. Therefore, amplicon sequencing is not quantitative. This observation is consistent with previous pyrotag sequencing studies (Engelbrektson et al., 2010) and the general consensus that conventional PCR amplification is not quantitative (Suzuki and Giovannoni, 1996; Qiu et al., 2001). Therefore, great caution is required when using the amplicon sequencing approach to make quantitative inferences of microbial community diversity in comparative studies.
One way to improve reproducibility and quantitation is to use biological replicates. As demonstrated in this study, increasing biological replicates greatly helps to differentiate the effects of warming or clipping on microbial communities and allows for a more rigorous statistical analysis, which is important for comparative studies. Notably, microarray-based technologies have significant advantages in minimizing/eliminating the sampling artifacts associated with random sampling processes (Zhou et al., 2008); combining high-throughput sequencing with array-based technologies could provide robust measurements for comparative studies.
As demonstrated in this study, variations in technical replicates could lead to overestimating β-diversity of microbial communities. As such effects vary in experiments, depending on the complexity of microbial communities examined and sampling efforts, it is important to assess the effects of random sampling processes on estimating β-diversity for later appropriate data interpretation. Thus, we recommend that technical replicates (for example, more than three for each biological sample) should be considered in individual experiments. These technical replicates will allow us to assess the background noise level in β-diversity estimation. With the most recent advanced sequencing technologies (such as Illumina HiSeq 2000), having several technical replicates for each biological sample should be not difficult to achieve. In addition, the three methods (MRPP, ANOSIM and Adonis) used in this study should be useful to assess the statistical significance of microbial community diversities under different conditions.
In conclusion, amplicon sequencing of the rRNA gene and other functional genes is rapidly becoming the method of choice for profiling microbial communities and has generated great insights into microbial community diversity. Although the amplicon sequencing-based detection approach is powerful, it suffers from several inherent drawbacks such as high sequencing errors and artifacts as demonstrated by several previous studies. This study revealed that the amplicon sequencing-based detection approach is not reproducible and quantitative with soil microbial communities, which was most likely caused by artifacts associated with random sampling processes. Such variations in technical replicates could have substantial effects on estimating β-diversity, but less on α-diversity. Therefore, great caution should be taken when amplicon sequencing-based detection is used for making quantitative inferences about the β-diversity of microbial communities. In addition, various strategies can be used to improve the comparability of amplicon sequencing data, including removing singleton sequences, adding sequences together from technical replicates, combining the sequences from both primers, removing less frequently encountered OTUs across biological replicates and increasing biological replicates. Consistent with many previous studies, application of the amplicon sequencing-based detection approach to analyze microbial communities from a long-term global change experiment revealed clear differences in the composition and structure of microbial communities between warming and non-warming or between clipping and non-clipping. These results suggest that if used appropriately, amplicon sequencing-based detection is useful for analyzing microbial community structure despite the fact that it has several inherent drawbacks, such as poor reproducibility and quantitation.
Acknowledgments
We thank Dr Fares Najar and Dr Bruce Roe at the University of Oklahoma for providing sequencing services, and Qiong Wang and James Cole at the Michigan State University for helping process pyrosequencing data. This work has been supported, through contract DE-AC02-05CH11231 (as part of ENIGMA, a Scientific Focus Area) and contract DE-SC0004601, by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, Genomics: GTL Foundational Science, the United States Department of Agriculture (Project 2007-35319-18305) through NSF-USDA Microbial Observatories Program, Oklahoma Bioenergy Center (OBC) and The Fundamental Research Foundation of Tsinghua University.
Footnotes
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Acosta-Martinez V, Dowd S, Sun Y, Allen V. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Biochem. 2008;40:2762–2770. [Google Scholar]
- Ahn SJ, Costa J, Emanuel JR. PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res. 1996;24:2623–2625. doi: 10.1093/nar/24.13.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- Binladen J, Gilbert MTP, Bollback JP, Panitz F, Bendixen C, Nielsen R, et al. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PloS ONE. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A. Estimating the population-size for capture recapture data with unequal catchability. Biometrics. 1987;43:783–791. [PubMed] [Google Scholar]
- Cheung MK, Au CH, Chu KH, Kwan HS, Wong CK. Composition and genetic diversity of picoeukaryotes in subtropical coastal waters as revealed by 454 pyrosequencing. ISME J. 2010;4:1053–1059. doi: 10.1038/ismej.2010.26. [DOI] [PubMed] [Google Scholar]
- Chun J, Kim KY, Lee JH, Choi Y. The analysis of oral microbial communities of wild-type and toll-like receptor 2-deficient mice using a 454 GS FLX Titanium pyrosequencer. BMC Microbiol. 2010;10:101. doi: 10.1186/1471-2180-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke KR. Nonparametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–143. [Google Scholar]
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The Ribosomal Database Project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–D172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R. The metagenomics of soil. Nat Rev Microbiol. 2005;3:470–478. doi: 10.1038/nrmicro1160. [DOI] [PubMed] [Google Scholar]
- Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM, et al. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrektson A, Kunin V, Wrighton KC, Zvenigorodsky N, Chen F, Ochman H, et al. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010;4:642–647. doi: 10.1038/ismej.2009.153. [DOI] [PubMed] [Google Scholar]
- Gomez-Alvarez V, Teal TK, Schmidt TM. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3:1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Deng Y, Van Nostrand JD, Tu QC, Xu MY, Hemme CL, et al. GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J. 2010a;4:1167–1179. doi: 10.1038/ismej.2010.46. [DOI] [PubMed] [Google Scholar]
- He Z, Gentry TJ, Schadt CW, Wu LY, Liebich J, Chong SC, et al. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 2007;1:67–77. doi: 10.1038/ismej.2007.2. [DOI] [PubMed] [Google Scholar]
- He Z, Xu MY, Deng Y, Kang SH, Kellogg L, Wu LY, et al. Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2. Ecol Lett. 2010b;13:564–575. doi: 10.1111/j.1461-0248.2010.01453.x. [DOI] [PubMed] [Google Scholar]
- He ZL, Van Nostrand JD, Wu LY, Zhou JZ. Development and application of functional gene arrays for microbial community analysis. Transactions of Nonferrous Metals Society of China. 2008;18:1319–1327. [Google Scholar]
- Hill MO, Gauch HG. Detrended correspondence-analysis – an improved ordination technique. Vegetation. 1980;42:47–58. [Google Scholar]
- Hollister EB, Engledow AS, Hammett AJM, Provin TL, Wilkinson HH, Gentry TJ. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010;4:829–838. doi: 10.1038/ismej.2010.3. [DOI] [PubMed] [Google Scholar]
- Huber JA, Mark Welch D, Morrison HG, Huse SM, Neal PR, Butterfield DA, et al. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- Huse SM, Huber JA, Morrison HG, Sogin ML, Mark Welch D. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai S, Chai BL, Sul WJ, Cole JR, Hashsham SA, Tiedje JM. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 2010;4:279–285. doi: 10.1038/ismej.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman MM, Fuselier DM, Hird S, Carstens BC. The carnivorous pale pitcher plant harbors diverse, distinct, and time-dependent bacterial communities. Appl Environ Microbiol. 2010;76:1851–1860. doi: 10.1128/AEM.02440-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, et al. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- Luo YQ, Wan SQ, Hui DF, Wallace LL. Acclimatization of soil respiration to warming in a tall grass prairie. Nature. 2001;413:622–625. doi: 10.1038/35098065. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune B, Grace JB. Analysis of Ecological Communities. MJM Software Design: Gleneden Beach, OR; 2002. [Google Scholar]
- McCune B, Mefford MJ.1999PC-ORD: Multivariate Analysis of Ecological DataVersion 4. User's guideMJM Software Design: Gleneden Beach, OR [Google Scholar]
- Mielke PW, Berry KJ. Permutation Methods: A Distance Function Approach. Springer Series in Statistics. Springer: New York, NY; 2001. [Google Scholar]
- Nawrocki EP, Eddy SR. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput Biol. 2007;3:540–554. doi: 10.1371/journal.pcbi.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M, et al. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 2007;35:e130. doi: 10.1093/nar/gkm760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu XY, Wu LY, Huang HS, McDonel PE, Palumbo AV, Tiedje JM, et al. Evaluation of PCR-generated chimeras: mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl Environ Microbiol. 2001;67:880–887. doi: 10.1128/AEM.67.2.880-887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Curtis TP, Sloan WT. The rational exploration of microbial diversity. ISME J. 2008;2:997–1006. doi: 10.1038/ismej.2008.69. [DOI] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods. 2009;6:639–U627. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria; 2006. [Google Scholar]
- Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD, et al. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1:283–290. doi: 10.1038/ismej.2007.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JH, Marschall T, Martin M, Rosenstiel P, Mestdagh P, Schlierf S, et al. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res. 2010;38:5919–5928. doi: 10.1093/nar/gkq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte UME, Abdo Z, Foster J, Ravel J, Bunge J, Solheim B, et al. Bacterial diversity in a glacier foreland of the high Arctic. Mol Ecol. 2010;19:54–66. doi: 10.1111/j.1365-294X.2009.04479.x. [DOI] [PubMed] [Google Scholar]
- Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, Woolf B, et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18:1638–1642. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, et al. Microbial diversity in the deep sea and the underexplored ‘rare biosphere'. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackebrandt E, Goebel BM. A place for DNA-DNA reassociation and 16S ribosomal-RNA sequence-analysis in the present species definition in bacteriology. Int J Syst Microbiol. 1994;44:846–849. [Google Scholar]
- Suzuki MT, Giovannoni SJ. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol. 1996;62:625–630. doi: 10.1128/aem.62.2.625-630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira L, Peixoto RS, Cury JC, Sul WJ, Pellizari VH, Tiedje J, et al. Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J. 2010;4:989–1001. doi: 10.1038/ismej.2010.35. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Daae FL, Sandaa RA, Ovreas L. Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol. 1998;64:53–62. doi: 10.1016/s0168-1656(98)00103-5. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Ovreas L. Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol. 2002;5:240–245. doi: 10.1016/s1369-5274(02)00324-7. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Sorheim R, Goksoyr J. Total bacterial diversity in soil and sediment communities—a review. J Ind Microbiol. 1996;17:170–178. [Google Scholar]
- Uroz S, Buee M, Murat C, Frey-Klett P, Martin F. Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Reports. 2010;2:281–288. doi: 10.1111/j.1758-2229.2009.00117.x. [DOI] [PubMed] [Google Scholar]
- Vaishampayan P, Osman S, Andersen G, Venkateswaran K. High-density 16S microarray and clone library-based microbial community composition of the phoenix spacecraft assembly clean room. Astrobiology. 2010;10:499–508. doi: 10.1089/ast.2009.0443. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef N, Sheik CS, Krumholz LR, Najar FZ, Roe BA, Elshahed MS. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl Environ Microbiol. 2009;75:5227–5236. doi: 10.1128/AEM.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JZ. Predictive microbial ecology. Microb Biotechnol. 2009;2:154–156. doi: 10.1111/j.1751-7915.2009.00090_21.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JZ, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62:316–322. doi: 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JZ, Kang S, Schadt CW, Garten CT. Spatial scaling of functional gene diversity across various microbial taxa. Proc Natl Acad Sci USA. 2008;105:7768–7773. doi: 10.1073/pnas.0709016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JZ, Thompson DK, Xu Y, Tiedje JM. Microbial Functional Geomics. John Wiley & Sons: Hoboken, NJ; 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.