

Abstract
Surfactant protein D (SP-D) is a constituent of the innate immune system that plays a role in the host defense against lung pathogens and in modulating inflammatory responses. While SP-D has been detected in extrapulmonary tissues, little is known about its expression and function in the vasculature. Immunostaining of human coronary artery tissue sections demonstrated immunoreactive SP-D protein in smooth muscle cells (SMCs) and endothelial cells. SP-D was also detected in isolated human coronary artery SMCs (HCASMCs) by PCR and immunoblot analysis. Treatment of HCASMCs with endotoxin (LPS) stimulated the release of IL-8, a proinflammatory cytokine. This release was inhibited >70% by recombinant SP-D. Overexpression of SP-D by adenoviral-mediated gene transfer in HCASMCs inhibited both LPS-and TNF-α-induced IL-8 release. Overexpression of SP-D also enhanced uptake of Chlamydia pneumoniae elementary bodies into HCASMCs while attenuating IL-8 production induced by bacterial exposure. Both LPS and TNF-α increased SP-D mRNA levels by five to eightfold in HCASMCs, suggesting that inflammatory mediators upregulate the expression of SP-D. In conclusion, SP-D is expressed in human coronary arteries and functions as an anti-inflammatory protein in HCASMCs. SP-D may also participate in the host defense against pathogens that invade the vascular wall.
Keywords: inflammation, Chlamydia pneumoniae, innate immunity, interleukin-8
Chronic low levels of inflammation caused by agents such as endotoxin (LPS) have been implicated in the development of atherosclerosis in experimental animal models and in humans (14, 37). Monocytes/macrophages respond vigorously to stimuli of innate immunity and in this way contribute to lesion formation in atherosclerosis. We have reported that human vascular smooth muscle cells (SMCs) are also highly sensitive to endotoxin, suggesting a potential role for SMC innate immune signaling in atherosclerosis (30). Moreover, human vascular SMCs express CD14, a component of the innate immune system that serves as a signal transducer for LPS as well as other inflammatory mediators (4, 8, 35, 39).
The discovery of CD14 expression in SMCs (35) prompted us to search for other components of the innate immune system in SMCs that might also regulate inflammation and host defense. Surfactant protein A (SP-A) and D (SP-D), along with mannose-binding protein, belong to the collectin family of proteins, which are characterized by a type IV collagen-like domain and a carboxy terminal Ca2+-dependent lectin domain (9). SPs can modify the response to innate immune stimuli and can bind to LPS present on the surface of many bacteria as well as to CD14 (7, 19, 31, 32). In the lung, SPs not only enhance the phagocytic function of macrophages and neutrophils and modulate inflammation but also have direct antimicrobial effects (23, 32). SPs also enhance the phagocytosis of bacterial pathogens such as Chlamydia, which could potentially be pertinent to cardiovascular disease (26). Although SPs were initially described as lung proteins, several investigators have reported that SP-D is also present in extrapulmonary tissues (2, 34). Notably, Leth-Larsen and co-workers (20) have recently reported that SP-D is present in the walls of blood vessels in the female reproductive tract.
In the present study, we examined the expression of SP-D in human coronary arteries and, in particular, in human coronary artery SMCs (HCASMCs). We examined the capacity of SP-D to modulate inflammation as well as the role of inflammation in regulating SP-D gene expression in SMCs. Based on our findings, we propose that SP-D is produced by vascular SMCs in response to inflammatory stimuli and that it may function to modulate the host inflammatory response and pathogen defense in the vascular wall.
MATERIALS AND METHODS
Preparation and culture of vascular cells
HCASMCs, human coronary artery endothelial cells (HCAECs), human aortic SMCs (HASMCs), and human aortic endothelial cells (HAECs) were isolated using a modification of a previously described method (41). Human aortas and coronary arteries removed at the time of heart transplantation surgery were obtained from the operating room at the University of Iowa Hospitals and Clinics according to a protocol approved by the University of Iowa Human Subjects Office. Fat and adventitia were thoroughly dissected from the vessel. The vessel was then cut longitudinally and incubated in ~40 ml PBS containing collagenase (type IV, 2 mg/ml, Worthington, Lakewood, NJ) for 20 min. The luminal surface was scraped gently, and the tissue was washed repeatedly in HBSS to remove endothelial cells. The arterial smooth muscle was then minced and incubated in 10 ml DMEM containing 2 mg/ml type IV collagenase, 0.4 mg/ml soybean trypsin inhibitor (Worthington), 0.5 mg/ml elastase (Worthington), and 1 mg/ml BSA (Sigma, St. Louis, MO) at 37°C with vigorous periodic mixing (every 15 min) for up to 2 h. Undigested tissue pieces were removed, and SMCs were collected by centrifugation. Cells were cultured in medium formulated for SMC [smooth muscle growth medium (SmGM), Cell Applications, San Diego, CA]. Cells from passages 4–9 were used in these experiments. Cells were treated with either 1–5 ng/ml TNF-α (Sigma) or 1–5 ng/ml rough LPS from Esherichia coli (List Biological Laboratories, Campbell, CA) for periods ranging up to 24 h, as previously described (35).
Immunolocalization of SP-D protein
Paraffin sections of human coronary arteries obtained from four individuals were immunostained for SP-D using a Vectastain Elite kit (Vector Laboratories, Burlingame, CA) and a rabbit polyclonal antibody directed against human SP-D (1:1,000, Chemicon, Temecula, CA). In addition, sections of formalin-fixed, paraffin-embedded cardiovascular tissues (7 μm thick) were obtained from the Department of Pathology at the University of Iowa and immunostained for SP-D. Nonspecific binding was blocked by incubating the sections with 2% normal goat serum for 30 min at room temperature. Sections were rinsed in PBS and then incubated for 1 h at 23°C with rabbit SP-D antiserum, nonimmune rabbit IgG, or PBS alone. Sections were washed twice in PBS followed by an incubation for 30 min with a biotinylated secondary antibody. After being rinsed in PBS, sections were incubated for 45 min in avidin-peroxidase reagent, rinsed again, and incubated in diaminobenzidine (0.7 mg/ml, Sigma) for 1–3 min. Sections were counterstained with hematoxylin, dehydrated, and mounted with coverslips.
Immunoblot analysis
Cardiovascular tissues and cells were homogenized in sterile water containing 1 mM PMSF; the homogenate was centrifuged at 600 g for 10 min at 4°C. The supernatant was collected, and the protein concentration was measured using the Bradford method (6). Homogenate proteins (50 μg) were separated by one-dimensional gel electrophoresis on a 10% polyacrylamide-SDS minigel (Bio-Rad, Hercules, CA), and proteins were subsequently transferred to nitrocellulose membranes (Bio-Rad) electrophoretically. Nonspecific binding sites were blocked by incubating the membranes in Blotto (7% nonfat dry milk in 20 mM Tris buffer containing 0.1% Tween 20) overnight at 4°C. Membranes were then incubated with antibodies for SP-D (1:1,000 in Blotto) for 1 h at room temperature with gentle agitation. After three 15-min rinses in 0.1% Tris-NaCl with 0.1% Tween 20 buffer, membranes were incubated in a secondary antibody conjugated to horseradish peroxidase (Cappel, Irvine, CA, diluted 1:10,000 in Blotto) for 45 min at room temperature. After three 15-min washes in Tris-buffered saline, membranes were incubated in ECL solution (Amersham, Piscataway, NJ) and exposed to film. The relative amount of immunoreactive protein in each band was quantitated by densitometry using Quantity One software (Bio-Rad).
IL-8 assay
IL-8 released into the culture medium was measured by ELISA using matched antibodies from R&D Systems (Minneapolis, MN), as previously described (10). To evaluate the effects of SP-D on IL-8 release, HCASMC cultures were incubated with 0.36 μg/ml of recombinant rat SP-D protein (a kind gift from Dr. Scott Ferguson, Dept. of Internal Medicine, University of Iowa, Iowa City, Iowa).
RT and PCR for SP-D
The medium was removed from confluent cultures of HCASMCs, and cells were washed twice with PBS before total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA). Two micrograms of total RNA from each sample was then reverse transcribed using an SP-D oligomer, 5′-TCA GAA CTC GCA GAC CA-3′, as a primer (21). An aliquot of the RT reaction was used for SP-D PCR (forward primer: 5′-ATG TTG CTT CTC TGA GG-3′ and reverse primer: 5′-TCA GAA CTC GCA GAC CAC AAG-3′) (21). PCR conditions were as follows: 35 cycles, denaturation at 94°C for 30 s, annealing at 62°C for 1 min, and extension at 72°C for 2 min. The PCR product was analyzed on a 1% agarose gel, and the 431-bp SP-D band was visualized with UV illumination.
Real-time PCR
Cultured HCASMCs were washed two times in PBS. Total RNA was isolated as described above, resuspended in water, and quantitated by absorbance at 260 nm. Two micrograms of total RNA from each sample was reversed transcribed using oligo-dT primers. The resulting cDNAs were diluted (1:50–1:100), and equal amounts were aliquoted in replicates for real-time PCR analysis using a Stratogene Mx 3000P instrument. Primers and FAM-labeled probes for human SP-D, 18S rRNA, and a Universal Taqman master mix for the reaction were purchased from Applied Biosystems (ABI Systems, Foster City, CA). SP-D mRNA was assayed by the comparative quantitation method; calculated differences in mRNA expression were determined according to the manufacturer’s User Bulletin 2 (10/2001, ABI Systems).
SP-D adenovirus
An adenovirus expressing the human SP-D gene (AdSP-D) was constructed using a SP-D cDNA plasmid obtained from the American Type Culture Collection (no. 6644024). Briefly, the plasmid was cloned into an E1 shuttle (pVQAdCMV) and then linearized. This plasmid, along with a linearized RAPAd backbone plasmid, was transfected into HEK-293 cells (3). The resulting virus particles were isolated by two rounds of CsCl gradient centrifugation, dialyzed against 3% sucrose in PBS, and then stored at −80°C until use. HCASMCs were infected in six-well tissue culture plates with AdSP-D [0–100 multiplicity of infection (MOI)] or 100 MOI of a control virus, AdLacZ (Ad5CMVLacZ, University of Iowa Gene Therapy Vector Core Facility), when the cells were at ~75% confluence. To infect the cells, the virus was added into 1 ml of the culture medium (SmGM) containing 1.5% FBS plus 250 molecules of poly-L-lysine/virus particle. The cells and adenovirus were incubated at 37°C for 4 h, and an additional 1 ml of SmGM containing 5% FBS was then added. The incubation was continued for 24–48 h before the experimental protocols were performed. At the end of the experiments, media were frozen for the subsequent determination of IL-8 by ELISA and SP-D protein by immunoblot analysis. Cells were washed and frozen at −80°C for subsequent immunoblot analysis for SP-D. In separate experiments, confocal microscopy was used to localize the SP-D protein in AdSP-D-infected cells.
Chlamydia pneumoniae infection
Following adenoviral infection, HCASMCs were incubated with C. pneumoniae elementary bodies (2 MOI, a gift from Dr. Scott Boitano, University of Arizona, Tuscon, AZ) in 2 ml SmGM + 5% FBS for 4 h at 37°C, after which the culture medium was removed and replaced with SmGM + 5% FBS. Cells were cultured for an additional 3 days, and the medium was removed and assayed for IL-8. After the cells had been washed and extracted with lysis buffer, immunoblot analysis was performed to determine the amount of C. pneumoniae major outer membrane protein (MOMP; antibody from Fitzgerald Industries, Cambridge, MA) as an index of bacterial content. Intracellular inclusions in HCASMCs containing viable C. pneumoniae were quantified using a bioassay and the Pathfinder Chlamydia Culture Confirmation Test (Bio-Rad) as previously described (27). Fifteen photomicrographs (×40) of random fields from each treatment group were scored blindly by two investigators to determine the number of cells infected with C. pneumoniae per field in two separate experiments.
Statistical analyses
All data are expressed as means ± SE. Differences between mean values of two groups were analyzed by Student’s t-tests. Differences between mean values of multiple groups were analyzed by one-way ANOVA with a Newman-Keul post hoc analysis. Probability values of 0.05 or less were considered to be statistically significant.
RESULTS
Detection of SP-D in coronary arteries and vascular cells
SP-D was detected by immunostaining in tissue sections of coronary arteries obtained from four donor hearts. SP-D protein was present in the cytoplasm of both endothelial cells and vascular SMCs (Fig. 1, A and C). As a positive control, human fetal lung explants were immunostained for SP-D; as expected, staining was restricted to alveolar type II epithelial cells (Fig. 1E). Negative controls included omission of the primary antibody (Fig. 1, B and D) and staining with nonimmune IgG (Fig. 1F).
Fig. 1.
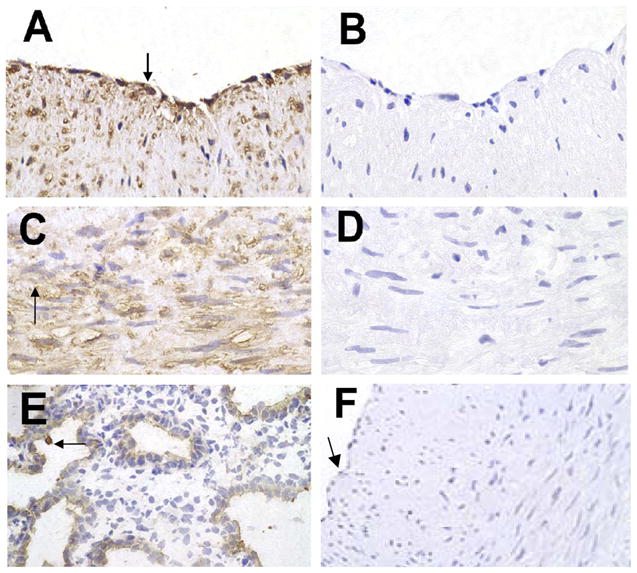
Surfactant protein D (SP-D) in human coronary arteries (HCAs). Paraffin sections of HCAs were immunostained for SP-D and then counterstained with hematoxylin. Positive staining was detected in endothelial cells (ECs; A, arrow) and medial smooth muscle cells (SMCs; C, arrow). B and D: staining controls in which PBS was used in place of the primary antibody. E: positive control of human fetal lung explant tissue immunostained for SP-D protein; the arrow points to type II ECs. F: control stained with nonimmune IgG; the arrow points to the EC layer. Images are representative of sections from 4 different specimens (all images were photographed at ×40 and enlarged similarly).
RT-PCR analysis of total RNA isolated from primary cultures of HCAECs, HCASMCs, HASMCs, and HAECs revealed the presence of SP-D mRNA in each of these cell types (Fig. 2A). In addition, SP-A mRNA was detected in HCASMCs (data not shown). SP-D protein was detected in HCAECs and HCASMCs, migrating as a single immunoreactive band at ~43 kDa, the same molecular weight as a SP-D standard (Fig. 2B).
Fig. 2.
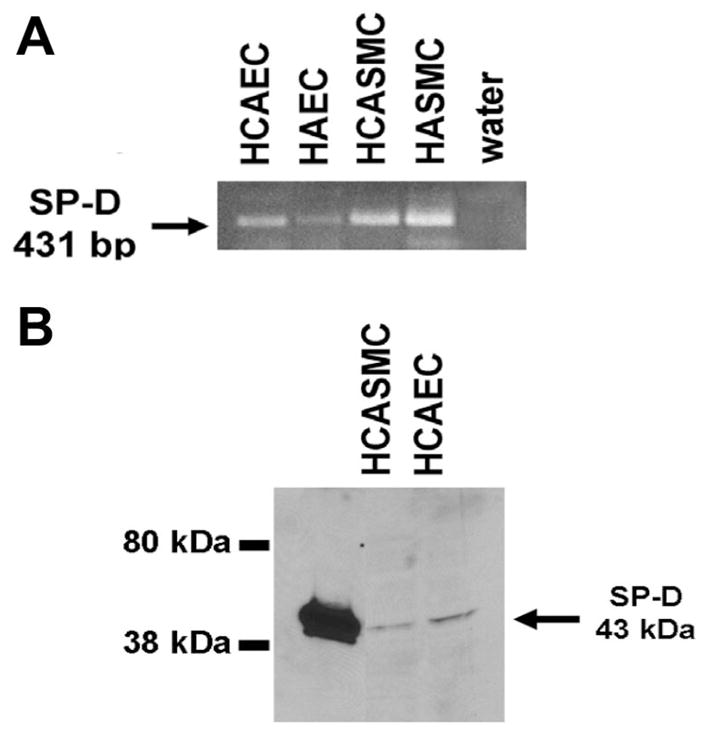
SP-D mRNA in ECs and vascular SMCs. Total RNA and protein from cultured vascular cells were analyzed for SP-D mRNA and protein using RT-PCR (A) and Western blot analysis (B), respectively. SP-D mRNA (431-bp RT-PCR product) was detected in HCAECs, human aortic ECs (HAECs), HCASMCs, and human aortic SMCs (HASMCs) (A). SP-D protein was detected in HCAECs and HCASMCs (B). Lane 1 contains a SP-D standard, which migrates at ~43 kDa.
Effect of SP-D on IL-8 release in HCASMCs
The biological activity of SP-D in the vascular system has not been investigated. However, in the lung, SP-D binds to bacterial pathogens, facilitates their interaction with alveolar macrophages, and suppresses inflammatory responses (28, 32). We therefore investigated the ability of exogenous SP-D protein to suppress LPS-induced inflammation in HCASMCs. Treatment of HCASMCs with 1 ng/ml LPS induced a ~10-fold increase in the production of the proinflammatory cytokine IL-8 during a 24-h incubation (Fig. 3). Coincubation of HCASMCs with 0.36 μg/ml rat SP-D abolished the LPS-induced IL-8 release, consistent with a potent anti-inflammatory effect of SP-D on these cells. SP-D added alone had no effect on IL-8 release by cells (Fig. 3). Preliminary experiments indicated that the application of SP-A also inhibited LPS-induced IL-8 release by HCASMCs (data not shown).
Fig. 3.

SP-D inhibits LPS-induced IL-8 release in cultured HCASMCs. Cells were treated for 24 h in medium containing either vehicle (control) or 1 ng/mL LPS with or without 0.36 μg/ml recombinant rat SP-D. Media were removed, and IL-8 was quantified by ELISA. Responses were normalized to control values (i.e., cells not treated with LPS). Data are representative of 2 experiments with 3 samples/data point. *P < 0.05 from control.
Overexpression of SP-D
Purified SP-D can contain contaminants such as LPS (26). Therefore, an adenovirus containing the human SP-D gene (AdSP-D) was constructed to overexpress SP-D protein. Infection of HCASMCs with AdSP-D resulted in a MOI-dependent increase in SP-D protein detected in the culture medium within 2 days after adenoviral infection (Fig. 4, A and B). Infection with 50 –100 MOI resulted in as much as a fivefold increase in the levels of SP-D protein released into the medium compared with control levels (Fig. 4B). In addition, the amount of SP-D protein present within HCASMCs increased approximately fourfold after infection with AdSP-D (Fig. 4B). To examine the cellular distribution of SP-D protein, immunofluorescence confocal microscopy was performed. As shown in Fig. 4C, SP-D protein was detected throughout the cytoplasm in HCASMCs after adenoviral-mediated overexpression of SP-D.
Fig. 4.

Infection of HCASMCs with AdSP-D increases the production of SP-D protein. Cultured HCASMCs were infected with 0–100 multiplicity of infection (MOI) of AdSP-D as described in MATERIALS AND METHODS. A: SP-D protein expression in media and cells following adenoviral gene transfer. After 48 h, culture media (lanes 1–6) and cell extracts (lanes 7 and 8) were immunoblotted for SP-D protein, which was quantitated by densitometry and expressed as relative optical density (OD; B). Two immunoreactive bands were detected in the media; the upper band was produced by cross-reactivity with a protein in the serum-containing culture medium. The lower band corresponds to authentic human SP-D protein. The data in B are representative of 2 experiments; levels of SP-D are expressed relative to control. C: uniform cellular distribution of SP-D protein in the cytoplasm of HCASMCs, as detected by confocal microscopy.
Effects of overexpression of SP-D on LPS-induced IL-8 release
The biological activity of SP-D protein produced by AdSP-D-infected cells was evaluated by treating transfected HCASMCs with LPS and then measuring IL-8 release into the medium. Transfection with 100 MOI of AdSP-D, the highest titer employed, did not affect basal IL-8 release from HCASMCs during the 24-h incubation period (Fig. 5A). Treatment with LPS (2 ng/ml) produced an increase in IL-8 release that was significantly inhibited in a MOI-dependent fashion in cells infected with AdSP-D (Fig. 5A). Incubation of HCASMCs with another inflammatory mediator, 2 ng/ml TNF-α, increased the release of IL-8 by approximately fivefold over control levels (Fig. 5B). In cell cultures infected with increasing MOI of AdSP-D for 2 days before a 24-h challenge with TNF-α, IL-8 secretion was significantly diminished at all levels of AdSP-D infection (Fig. 5B).
Fig. 5.

Overexpression of SP-D by adenoviral-mediated gene transfer inhibits IL-8 release induced by LPS (A) and TNF-α (B). HCASMCs were infected with 0–100 MOI of AdSP-D. Two days later, cells were treated with either vehicle (control), 2 ng/ml LPS, or 2 ng/ml TNF-α for 24 h, after which the release of IL-8 was measured by ELISA. Experiments were performed in triplicate, and 3 separate experiments were conducted. +P < 0.05 compared with control; *P < 0.05 compared with LPS alone.
Inflammatory stimuli affect SP-D mRNA
Having shown that SP-D is expressed in vascular cells and modulates LPS- and TNF-α-induced cytokine production in HCASMCs, we next investigated whether the level of SP-D mRNA in cells is regulated by proinflammatory stimuli. HCASMCs were exposed to either 2 ng/ml LPS or 2 ng/ml TNF-α for 2–24 h, and levels of SP-D mRNA were quantified by real-time PCR. LPS treatment rapidly increased SP-D mRNA levels in HCASMCs, with a maximum increase (~10-fold) observed by 12 h (Fig. 6). By 24 h after LPS application, SP-D levels had declined substantially. TNF-α likewise increased SP-D mRNA levels by nearly 10-fold. However, the stimulation of SP-D mRNA by TNF-α was more gradual in onset than was observed with LPS and continued to rise throughout the course of the experiment (Fig. 6).
Fig. 6.

SP-D mRNA levels are increased in HCASMCs by LPS and TNF-α. HCASMCs were treated with either LPS (2 ng/ml, solid line) or TNF-α (2 ng/ml, dashed line) for 2–24 h. SP-D mRNA levels were assessed by real-time PCR. Data were normalized to the level of SP-D mRNA in control (untreated) cells harvested at time 0. Data represent the average increase in the relative amount of SP-D mRNA in 2 experiments with duplicate samples collected at each time point.
Effects of overexpression of SP-D on responses to C. pneumoniae in HCASMCs
To determine whether SP-D can modulate the inflammatory response produced by an LPS-bearing pathogen, C. pneumoniae, HCASMCs were infected with 0–100 MOI AdSP-D to produce elevated levels of SP-D in cells and culture medium. Two days later, HCASMCs were exposed to C. pneumoniae elementary bodies (2 MOI). In the presence of increasing amounts of SP-D, there was a concentration-dependent increase in the levels of intracellular C. pneumoniae MOMP in HCASMCs as detected by Western blot analysis (Fig. 7A). We also evaluated cell cultures for the presence of viable Chlamydia intracellular inclusions (an index of bacterial infection). Infection in the presence of AdSP-D resulted in an ~40% reduction in the number of viable Chlamydia intracellular inclusions compared with noninfected control cells or infection with AdLacZ (average number of infected cells in 15 photomicrographs/treatment: control, 15.9 ± 0.9 cells/field; 100 MOI AdLacZ, 15.2 ± 1.2 cells/field; and 100 MOI AdSP-D, 9.2 ± 0.6 cells/field). Moreover, the inflammatory response to C. pneumoniae by HCASMCs (indicated by IL-8 release into the culture medium) was progressively reduced with increasing SP-D levels (Fig. 7B). Together, these results suggest that SP-D in SMC participates both in the host defense and modulation of the inflammatory response to invading pathogens.
Fig. 7.

Effect of overexpression of SP-D on the uptake of Chlamydia pneumoniae (A) and release of IL-8 (B) by HCASMCs. Forty-eight hours after infection with AdSP-D at the indicated MOI or with AdLacZ at 100 MOI, HCASMCs were exposed to C. pneumoniae elementary bodies (2 MOI) for 3 days. The relative amount of C. pneumoniae present in HCASMCs was then estimated by immunoblot analysis for C. pneumoniae major outer membrane protein (MOMP; quantified by densitometry and expressed as relative OD; A). Cell extracts from 3 replicate cultures were combined for each condition. The control group consisted of cells that were not infected with an adenoviral vector or with Chalmydia. In B, IL-8 release into the media from the experiment described above was determined by ELISA. Media from 3 replicate cultures were assayed. *P < 0.05 compared with control; +P < 0.05 compared with 0 MOI AdSP-D.
DISCUSSION
Innate immune system proteins, such as Toll-like receptors and CD14, have been detected in both endothelial cells and vascular SMCs (35). We now report that human vascular SMCs express SP-D, a member of the collectin family of proteins that modulates innate inflammation in the lung. SP-D inhibited IL-8 released from HCASMCs in response to LPS and TNF-α. In addition, LPS and TNF-α upregulated the expression of SP-D mRNA in HCASMCs. SP-D also facilitated the uptake of C. pneumoniae elementary bodies by HCASMCs while modulating the host cell inflammatory response. Collectively, these data suggest that SP-D may play a role in regulating inflammatory processes and innate host defense in vascular SMCs.
SPs are synthesized as glycoprotein monomers that initially form trimers covalently linked by disulfide bonds in their collagen domains; the trimers then aggregate in groups of six (SP-A) or four (SP-D) to form the mature, functional collectin molecules (9). All of the collectins are involved in innate host defense (16). Although first described in lung surfactant (29), SPs (particularly SP-D) are widely distributed in other mammalian tissues (2, 22, 34), including human vascular endothelial cells (20). However, to our knowledge, SP-D expression has not been examined in atherosclerosis-prone blood vessels such as coronary arteries. It is conceivable that some of the SP-D protein detected in arteries was taken up from serum. However, SP-D mRNA and protein were also detected in cultured human vascular SMCs and endothelial cells, indicating that SP-D is endogenously produced by these cells. Likewise, SP-A mRNA was detected in HCASMCs. Moreover, we found that HCASMCs respond to an inflammatory challenge with LPS or TNF-α by increasing the expression of SP-D. These results suggest that SP-D gene expression in HCASMCs may be regulated via proinflammatory signaling pathways, as has been demonstrated in the lung in a variety of injury models, including LPS administration (9).
In addition to demonstrating that HCASMCs express SP-D, we found that the application of exogenous SP-D, or the overexpression of endogenous SP-D, significantly decreased the IL-8 release induced by either LPS or TNF-α. IL-8 is an 8.4-kDa cytokine with potent proinflammatory activity that is believed to play an important role in monocyte chemotaxis and atherosclerotic lesion formation (12). TNF-α is an inflammatory cytokine that stimulates IL-8 production through a different family of receptors than LPS; however, both LPS and TNF-α share some common signaling intermediates and act via a common transcription factor, NF-κB (25, 36). Thus, our results suggest that SP-D can modulate inflammatory responses to mediators that may contribute to the initiation and/or progression of atherosclerosis.
The precise mechanisms whereby SP-D exerts its anti-inflammatory effects in SMCs remain to be determined. Protection against LPS-mediated inflammatory signaling could potentially be mediated by SP-D binding to LPS and/or CD14, as has been demonstrated in the lung (7). Thus, we speculate that SP-D may physically block LPS binding to CD14 in vascular SMCs. However, such a mechanism could not explain the inhibitory effects of SP-D on TNF-α signaling, which does not involve CD14. There is also evidence that SP-D binds to one or more cellular receptors in pulmonary macrophages, including glycoprotein 340 (15), signal inhibitory regulatory peptide-α, and/or a calreticulin/CD91 complex (11). The latter two receptors have been postulated to initiate a signal transduction cascade that inhibits inflammation. Thus, SP-D may act as part of a feedback mechanism to repress inflammatory signaling pathways in vascular cells.
Indolent bacterial infections may be a local source of LPS and other inflammatory mediators such as heat shock proteins that stimulate smooth muscle inflammatory mechanisms in atherosclerotic lesions. Although a causative association between atherosclerosis and C. pneumoniae has not been established, infection of atherosclerotic lesions by C. pneumoniae has been reported (18). When C. pneumoniae were incubated with HCASMCs in culture, we detected a robust increase in IL-8 production that corresponded to the level of bacterial exposure. Overexpression of SP-D in HCASMCs reduced this inflammatory response to Chlamydia infection in the same way that SP-D reduced LPS- and TNF-α-stimulated IL-8 release. Interestingly, with increasing levels of SP-D in the culture medium, there was an increase in intracellular C. pneumoniae MOMP (Fig. 7A), suggesting an increase in the uptake of Chlamydia by HCASMCs. While overexpression of SP-D increased C. pneumoniae MOMP within HCASMCs, the number of viable intracellular C. pneumoniae inclusions (an index of active bacterial infection) was reduced, suggesting that SP-D not only facilitates the uptake of C. pneumoniae elementary bodies by HCASMCs but may also direct the elementary bodies to a phagocytic cellular pathway that results in killing of the Chlamydia. Thus, our data are suggestive of two mechanisms whereby overexpression of SP-D may modulate responses to C. pneumoniae in HCASMCs: 1) repression of the inflammatory response and 2) promotion of the phagocytosis of C. pneumoniae by HCASMCs.
The involvement of SP-D in the pathogenesis of atherosclerosis may be complex. For example, deletion of SP-D has been reported to increase the levels of reactive oxygen species in lung macrophages, thereby increasing NF-κB activity and matrix metalloproteinase production (40). This observation suggests that SP-D may play a direct role in regulating macrophage activation, perhaps independently of innate immune signaling. Conversely, binding of SP-D to vascular cells can potentially augment proinflammatory responses, consistent with a dual-function mechanism that has been proposed in macrophages (38). In the latter study, the authors suggested that the physical presentation of the SP-D molecule to the cell may determine which of the cell surface receptors interact with the SP-D and, thus, whether the signal to the NF-κB pathway is either enhanced or repressed.
SP-D knockout mice have been generated and exhibit chronic pulmonary inflammation leading to emphysema and other lung maladies (38). These mice were also used to evaluate the effects of SP-D gene deletion on atherosclerosis in vivo (33). In this study, deletion of SP-D was reported to reduce atherosclerotic lesion formation in mice fed a high-fat diet, in conjunction with an improvement in plasma lipid profile and a reduction of plasma TNF-α levels. To our knowledge, the effects of overexpression of SP-D on atherosclerosis in mice have not been reported. Our data in human vascular cells suggest that overexpression of SP-D could potentially modulate innate immune responses elicited by LPS, a proatherogenic factor (14, 37). However, it is important to point out the species differences between mice and humans with regard to the innate immune system (for a review, see Ref. 24). For example, humans are extremely responsive to endotoxin, whereas mice are far less sensitive, requiring much higher doses of endotoxin to elicit a response (5). These differences have been attributed to structural differences in both Toll-like receptor 4 and an adaptor protein, MD-2, in the two species (1, 13, 17). Thus, it is difficult to predict whether overexpression of SP-D would sufficiently suppress innate immune activation in mice to attenuate atherosclerosis. In addition, it is not known whether endogenous SP-D expression is modulated in the cardiovascular system during the development of atherosclerosis in mice or humans. Further studies are needed to ascertain the role of SP-D in regulating atherosclerosis as well as other aspects of cardiovascular pathophysiology, such as ischemic myocardial injury and repair.
In summary, the data presented in this study provide the first evidence that SP-D is expressed in human vascular SMCs. Moreover, we propose that SP-D may function to modulate inflammation and host defense in the vasculature, which may impact the development or progression of cardiovascular disease.
Acknowledgments
GRANTS
This work was supported by National Institutes of Health Grants HL-070860, HL-076684, and HL-62984 (to N. L. Weintraub), HL-50050 (to J. M. Snyder), Training Grant Award 2-T32-HL-07638-16 (to R. E. Oberley-Deegan), and Diabetes and Endocrine Research Center Grant DK-25295 and by a Merit Review Grant from the Department of Veteran’s Affairs (to N. L. Weintraub).
References
- 1.Akashi S, Nagai Y, Ogata H, Oikawa M, Fukase K, Kusumoto S, Kawasaki K, Nishijima M, Hayashi S, Kimoto M, Miyake K. Human MD-2 confers on mouse Toll-like receptor 4 species-specific lipopolysaccharide recognition. Int Immunol. 2001;13:1595–1599. doi: 10.1093/intimm/13.12.1595. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama J, Hoffman A, Brown C, Allen L, Edmondson J, Poulain F, Hawgood S. Tissue distribution of surfactant proteins A and D in the mouse. J Histochem Cytochem. 2002;50:993–996. doi: 10.1177/002215540205000713. [DOI] [PubMed] [Google Scholar]
- 3.Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 2000;7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- 4.Antal-Szalmas P. Evaluation of CD14 in host defense. Eur J Clin Invest. 2000;30:167–179. doi: 10.1046/j.1365-2362.2000.00610.x. [DOI] [PubMed] [Google Scholar]
- 5.Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 6.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Chaby R, Garcia-Verdugo I, Espinassous Q, Augusto LA. Interactions between LPS and lung surfactant proteins. J Endotoxin Res. 2005;11:181–185. doi: 10.1179/096805105X37358. [DOI] [PubMed] [Google Scholar]
- 8.Choi HC, Lee KY. CD14 glycoprotein expressed in vascular smooth muscle cells. J Pharm Sci. 2004;95:65–70. doi: 10.1254/jphs.95.65. [DOI] [PubMed] [Google Scholar]
- 9.Crouch EC. Surfactant protein-D and pulmonary host defense. Respir Res. 2000;1:93–108. doi: 10.1186/rr19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll LL, Britigan BE. Pseudomonas pyocyanin increases interleukin-8 expression by human airway epithelial cells. Infect Immun. 1998;66:5777–5784. doi: 10.1128/iai.66.12.5777-5784.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 2003;115:13–23. doi: 10.1016/s0092-8674(03)00758-x. [DOI] [PubMed] [Google Scholar]
- 12.Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Hing HA, Gimbrone MAJ, Luster AD, Luscinskas FW, Rosenzweig A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 13.Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat Immun. 2002;3:354–359. doi: 10.1038/ni777. [DOI] [PubMed] [Google Scholar]
- 14.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 15.Holmskov U, Mollenhauer J, Madsen J, Vitved L, Gronlund J, Tornoe I, Kliem A, Reid KB, Poustka A, Skjodt K. Cloning of gp-340, a putative opsonin receptor for lung surfactant protein D. Proc Natl Acad Sci USA. 1999;96:10794–10799. doi: 10.1073/pnas.96.19.10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003;21:547–578. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki K, Gomi K, Nishijima M. Cutting edge: Gln22 of mouse MD-2 is essential for species-specific lipopolysaccharide mimetic action of taxol. J Immunol. 2001;166:11–14. doi: 10.4049/jimmunol.166.1.11. [DOI] [PubMed] [Google Scholar]
- 18.Kol A, Bourcier T, Lichtman AH, Libby P. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J Clin Invest. 1999;103:571–577. doi: 10.1172/JCI5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuan SF, Rust K, Crouch E. Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J Clin Invest. 1992;90:97–106. doi: 10.1172/JCI115861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leth-Larsen R, Floridon C, Nielsen O, Holmskov U. Surfactant protein D in the female genital tract. Mol Hum Reprod. 2004;10:149–154. doi: 10.1093/molehr/gah022. [DOI] [PubMed] [Google Scholar]
- 21.Lu J, Willis AC, Reid KB. Purification, characterization and cDNA cloning of human lung surfactant protein D. Biochem J. 1992;284:795–802. doi: 10.1042/bj2840795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madsen J, Tornoe I, Nielsen O, Koch C, Steinhilber W, Holmskov U. Expression and localization of lung surfactant protein A in human tissues. Am J Respir Cell Mol Biol. 2003;29:591–597. doi: 10.1165/rcmb.2002-0274OC. [DOI] [PubMed] [Google Scholar]
- 23.McCormack FX, Gibbons R, Ward SR, Kuzmenko A, Wu H, Deepe GS., Jr Macrophage-independent fungicidal action of the pulmonary collectins. J Biol Chem. 2003;278:36250–36256. doi: 10.1074/jbc.M303086200. [DOI] [PubMed] [Google Scholar]
- 24.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 25.Muller JM, Ziegler-Heitbrock HW, Baeuerle PA. Nuclear factor kappa B, a mediator of lipopolysaccharide effects. Immunobiology. 1993;187:233–256. doi: 10.1016/S0171-2985(11)80342-6. [DOI] [PubMed] [Google Scholar]
- 26.Oberley RE, Ault KA, Neff TL, Khubchandani KR, Crouch EC, Snyder JM. Surfactant proteins A and D enhance the phagocytosis of Chlamydia into THP-1 cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L296–L306. doi: 10.1152/ajplung.00440.2003. [DOI] [PubMed] [Google Scholar]
- 27.Oberley RE, Goss KL, Ault KA, Crouch EC, Snyder JM. Surfactant protein D is present in human female reproductive tract and inhibits Chlamydia trachomatis infection. Mol Human Reprod. 2004;10:861–870. doi: 10.1093/molehr/gah117. [DOI] [PubMed] [Google Scholar]
- 28.O’Riordan DM, Standing JE, Kwon KY, Chang D, Crouch EC, Limper AH. Surfactant protein D interacts with Pneumocystis carinii and mediates organism adherence to alveolar macrophages. J Clin Invest. 1995;95:2699–2710. doi: 10.1172/JCI117972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persson A, Chang D, Rust K, Moxley M, Longmore W, Crouch E. Purification and biochemical characterization of CP4 (SP-D), a collagenous surfactant-associated protein. Biochemistry. 1989;28:6361–6367. doi: 10.1021/bi00441a031. [DOI] [PubMed] [Google Scholar]
- 30.Rice JB, Stoll LL, Li WG, Denning GM, Weydert J, Charipar E, Richenbacher WE, Miller FJ, Jr, Weintraub NL. Low-level endotoxin induces potent inflammatory activation of human blood vessels: inhibition by statins. Arterioscler Thromb Vasc Biol. 2003;23:1576–1582. doi: 10.1161/01.ATV.0000081741.38087.F9. [DOI] [PubMed] [Google Scholar]
- 31.Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem. 2000;275:22442–22451. doi: 10.1074/jbc.M001107200. [DOI] [PubMed] [Google Scholar]
- 32.Sano H, Kuroki Y. The lung collectins, SP-A and SP-D, modulate pulmonary innate immunity. Mol Immunol. 2005;42:279–287. doi: 10.1016/j.molimm.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 33.Sorensen GL, Madsen J, Kejling K, Tornoe I, Nielsen O, Townsend P, Poulain F, Nielsen CH, Reid KBM, Hawgood S, Falk E, Holmskov U. Surfactant protein D is proatherogenic in mice. Am J Physiol Heart Circ Physiol. 2006;290:H2286–H2294. doi: 10.1152/ajpheart.01105.2005. [DOI] [PubMed] [Google Scholar]
- 34.Stahlman MT, Gray ME, Hull WM, Whitsett JA. Immunolocalization of surfactant protein-D (SP-D) in human fetal, newborn, and adult tissues. J Histochem Cytochem. 2002;50:651–660. doi: 10.1177/002215540205000506. [DOI] [PubMed] [Google Scholar]
- 35.Stoll LL, Denning GM, Li WG, Rice JB, Harrelson AL, Romig SA, Gunnlaugsson ST, Miller FJ, Jr, Weintraub NL. Regulation of endotoxin-induced proinflammatory activation in human coronary artery cells: expression of functional membrane-bound CD14 by human coronary artery smooth muscle cells. J Immunol. 2004;173:1336–1343. doi: 10.4049/jimmunol.173.2.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Wiedermann CJ, Keith S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck study. J Am Coll Cardiol. 1999;34:1975–1981. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 38.Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, Ross GF, Fisher JH, Korfhagen TR, Whittsett JA. Increased metalloproteinase activity, oxidant production and emphysema in surfactant D gene-inactivated mice. Proc Natl Acad Sci USA. 2000;97:5972–5977. doi: 10.1073/pnas.100448997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Coriolan D, Murthy V, Schultz K, Golenbock DT, Beasley D. Proinflammatory phenotype of vascular smooth muscle cells: role of efficient Toll-like receptor 4 signaling. Am J Physiol Heart Circ Physiol. 2005;289:H1069–H1076. doi: 10.1152/ajpheart.00143.2005. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol. 2001;166:7514–7519. doi: 10.4049/jimmunol.166.12.7514. [DOI] [PubMed] [Google Scholar]
- 41.Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Ionic currents in rat pulmonary and mesenteric arterial myocytes in primary culture and subculture. Am J Physiol Lung Cell Mol Physiol. 1993;264:L107–L115. doi: 10.1152/ajplung.1993.264.2.L107. [DOI] [PubMed] [Google Scholar]