

Abstract
The engineering of drug-encapsulated targeted nanoparticles (NPs) has the potential to revolutionize drug therapy. A major challenge for the smooth translation of targeted NPs to the clinic has been developing methods for the prediction and optimization of the NP surface composition, especially when targeting ligands (TL) of different chemical properties are involved in the NP self-assembly process. Here we investigated the self-assembly and properties of two different targeted NPs decorated with two widely used TLs that have different water solubilities, and developed methods to characterize and optimize NP surface composition. We synthesized two different biofunctional polymers composed of poly(lactide-co-glycolide)-b-polyethyleneglycol-RGD (PLGA-PEG-RGD, high water solubility TL) and PLGA-PEG-Folate (low water solubility TL). Targeted NPs with different ligand densities were prepared by mixing TL-conjugated polymers with non-conjugated PLGA-PEG at different ratios through nanoprecipitation. The NP surface composition was quantified and the results revealed two distinct nanoparticle assembly behaviors: for the case of PLGA-PEG-RGD, nearly all RGD molecules conjugated to the polymer were found to be on the surface of the NPs. In contrast, only ~20% of the folate from PLGA-PEG-Folate was present on the NP surface while the rest remained presumably buried in the PLGA NP core due to hydrophobic interactions of PLGA and folate. Finally, in vitro phagocytosis and cell targeting of NPs was investigated, from which a window of NP formulations exhibiting minimum uptake by macrophages and maximum uptake by targeted cells was determined. These results underscore the impact the ligand chemical properties have on the targeting capabilities of self-assembled targeted nanoparticles and provide an engineering strategy for improving their targeting specificity.
Keywords: Nanoparticles, Targeting Ligand, Surface Ligand Density, RGD, Folate
1. Introduction
Targeted nanoparticles (NPs), which have the capability of encapsulating different therapeutic agents and preferentially delivering them to specific tissues and cells, hold the promise of revolutionizing the treatment of many diseases including cancer, cardiovascular diseases, and immunological disorders [1–4]. With respect to cancer, NPs able to encapsulate chemotherapeutic drugs and directly deliver them to tumor cells can reduce systemic cytotoxicity caused by these agents and improve their efficacy [5]. Among the different classes of NPs, polymeric nanoparticles have gained much attention due to several attractive properties, including biodegradability and biocompatibility of materials, favorable synthesis conditions, self-assembly in aqueous solution, and requirement of relatively little post-processing and purification work [6]. A central challenge, however, has been the development of methods to reproducibly synthesize targeted NPs with well-characterized and optimized surface compositions. Although there are examples showing the synthesis of targeted polymeric NPs with different targeting ligands such as the glycoprotein trasnferrin [7], HER-2 antibody [8], RGD peptide [9], and folic acid [10], it is common to see characterization and optimization of the surface ligand density underemphasized. Due to the delicate balance between the amount of targeting ligand to promote effective targeting and the protective shield (commonly conferred by poly(ethylene glycol), PEG) to avoid immune detection [11], methods that enable the prediction and characterization of NP surface composition and the control of NP surface density become essential for the clinical translation of these vehicles.
Conventional methods of synthesizing polymeric targeted NPs involve use of amphiphilic block copolymers that self-assemble into drug-encapsulated NP in aqueous solution, followed by chemical conjugation of the targeting ligands to the surface of the NP [12]. Post-synthesis NP surface modification often requires the addition of excess reactant to drive the chemical reaction, and thus makes it difficult to adjust the density of TL on NP surface in a meaningfully reproducible manner. Furthermore, the post-synthesis conjugation of TL to drug encapsulated NPs is often done under aqueous condition during which there is an uncontrolled release of the drug from NPs primarily through diffusion. Consequently, the post-synthesis particle-processing methods offer limited ability to precisely engineer the NP surface properties in a robust and reproducible manner, and the targeted NPs produced by such methods may have significant batch-to-batch variations in their biophysicochemical properties. Our group recently developed a pre-functionalized biopolymer composed of hydrophobic poly(lactide–co–glycolide) (PLGA) that makes up the NP core, PEG that forms a corona-like ‘stealth’ shell, and A10 2-fluoropyrimidine RNA aptamer (Apt), which binds to the prostate-specific membrane antigen (PSMA) on the surface of prostate cancer (PCa) cells [13]. This functionalized polymer possessed all required components for a targeted NP and enabled single step self-assembly of targeted functional particles, simplifying the optimization and the potential manufacturing of the NPs [14].
Extension of this single-step self-assembly technology to other types of targeting molecules, such as peptides and small molecules with distinct solubilities and modes of action, is desired along with methods that enable the characterization and optimization of these ligands in vitro before embarking on in vivo studies. Likewise, it is of interest to understand the effect of some of the chemical properties of targeting ligands on the properties and performance of self-assembled NPs. Currently, two of the most frequently employed targeting ligands are RGD peptide, which has high water solubility (>50 mg/mL at neutral pH), and folic acid, which has a relatively poor water solubility (0.0016 mg/mL at neutral pH) [15]. RGD is a small peptide that preferentially binds to integrin-ανβ3 receptor, which is often over-expressed in endothelial cells and tumor neovasculature [16]. There are reports showing preparation of NPs encapsulating drugs and genes in which the RGD ligand enables preferential targeting of angiogenic tumor blood vessels, thus producing an anticancer response [17]. Folic acid is a small molecule that binds to the folate receptor, which is over-expressed in some cancer cells including breast, lung, kidney, ovary, brain, among others [18]. Although there are a few examples of single-step preparation of NPs using a polymer functionalized with either RGD or folic acid [9, 19], previous studies lack investigation of the effects of varying ligand densities on the NP biophysichochemical properties together with characterization and optimization of the NP surface composition.
This work presents a comparison of the surface composition and targeting specificity of self-assembled RGD- and Folate-targeted NPs – two widely used TLs with different solubility profiles. Two different biofunctional polymers composed of PLGA-PEG-RGD (high water solubility TL) and PLGA-PEG-Folate (low water solubility TL) were synthesized, and targeted NPs with different ligand densities were prepared by controlling the ratios of TL-conjugated polymers with non-conjugated PLGA-PEG lacking the TL in a nanoprecipitation process (otherwise known as solvent displacement method) [20, 21]. The surface ligand densities of these NPs were quantified and compared to the theoretical ligand densities estimated from their initial concentrations. Finally, in vitro binding and uptake of these NPs against macrophages, HUVEC cells (for NP-RGD), and KB cells (for NP-Folate) was investigated to determine an optimum formulation window where NPs exhibit minimum uptake by macrophages and maximum uptake by HUVEC cells or KB cells.
2. Materials and Methods
2.1 Materials
Poly(D,L-lactide–co–glycolide)-co-Poly(ethylene glycol) with terminal methoxy groups (PLGA27K-mPEG5K) was obtained from Boehringer Ingelheim (Ingelheim am Rhein, Germany). Poly(D,L-lactide–co–glycolide) (50/50) with terminal carboxylate groups (PLGA, inherent viscosity 0.67 dL/g, MW ~ 45 kDa) was obtained from Lactel (Pelham, AL, USA). tBOC-NH-PEG-NH2 (MW 5000) and tBOC-NH-PEG-NHS, (MW 5000) were purchased from Laysan Bio, Inc (Arab, AL, USA). cRGD-PEG4 peptide was obtained from Peptides International (Louisville, KY, USA) and Folic Acid from Sigma-Aldrich (St. Louis, MO, USA). Molecular biology buffers and tissue culture reagents were purchased from Invitrogen (Carlsbad, CA, USA). KB cell line was obtained from American Type Culture Collection (Manassas, VA, USA), and HUVEC cell line together with its growth medium was purchased from Lonza, Inc (Allendale, NJ, USA)
2.2 Methods
2.2.1 Synthesis of PLGA-PEG-RGD
The synthesis of PLGA45K-PEG5K-RGD was accomplished first by the conjugation of a modified cyclic RGD to PEG followed by the conjugation of the resultant RGD-PEG to PLGA. First, cRGD-PEG4 (Arg-Gly-Asp-D-Phe-Lys-PEG4) (8 mg, 8.94 μmol) was dissolved in 400 μL of dimethylformamide (DMF) and reacted with tBOC-NH-PEG-NHS (22 mg, 4.47 μmol) and N,N-Diisopropylethylamine (DIEA, 10 μL) for 12 h. The reaction product was dialyzed 24 h in water to remove unreacted RGD. The resultant tBOC-PEG-RGD was lyophilized and reconstituted in 400 μL of trifluoroacetic acid (TFA) to remove the protecting group. After 4 h of reaction, product was dried under vacuum and dissolved in 200 μL of DMSO. Concurrently, PLGA-COOH (100 mg, 2.22 μmoL) was reacted with N-hydroxysuccinimide (NHS) in the presence of 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC) in dichloromethane (DCM) for 2 h. PLGA-NHS was dried under vacuum and dissolved in 300 μL of DMSO. Finally, PEG-RGD and PLGA-NHS in DMSO were mixed with DIEA, allowed to react for 24 h, precipitated in ice cold methanol, and dried under vacuum. PLGA-PEG-RGD was characterized with GPC to confirm the presence of the triblock PLGA-PEG-RGD. To verify conjugation of PEG to RGD Maldi-TOF was used, and it showed that only PEG-RGD was present while free RGD was absent (see supporting information).
2.2.2 Synthesis of PLGA-PEG-Folate
The synthesis of PLGA45K-PEG5K-Folate was accomplished first by the conjugation of an activated Folate-NHS to PEG followed by the conjugation of the resultant Folate-PEG to PLGA. First, Folic acid (60 mg, 0.14 mmol) was dissolved in 2mL of DMF and reacted with NHS in the presence of EDC for 12 h. Folate-NHS was precipitated in a mixture of 30/70 acetone/diethyl ether and dried under vacuum. Dry Folate-NHS was dissolved in 500 μL of DMSO and reacted with tBOC-NH-PEG-NH2 (92.4 mg, 28 μmol) for 12 h in the presence of DIEA. The reaction product was then precipitated under cool ether, reconstituted in an aqueous solution of NaOH and dialyzed for 48 h. The resultant tBOC-PEG-Folate was lyophilized and reconstituted in 700 μL of TFA to remove the protecting group. After 4 h of reaction, the solution was dried under vacuum and then dissolved in 500 μL of DMSO. Concurrently, PLGA-COOH (420 mg, 9.33 μmoL) was reacted with NHS under the presence of EDC in DCM for 2 h. PLGA-NHS was dried under vacuum and dissolved in 500 μL of DMSO. Finally, PEG-folate and PLGA-NHS in DMSO were mixed and allowed to react for 24 h. The final product was precipitated in ice cold methanol and dried under vacuum. PLGA-PEG-Folate was characterized with NMR to verify the conjugation of PLGA to PEG, and GPC to confirm the presence of PLGA-PEG-Folate. To verify conjugation of PEG to folate Maldi-TOF was used, and it showed that only PEG-folate was present while free folate was absent. Finally to quantify the amount of PLGA-PEG-Folate compared to PEG-folate, UV-VIS was used and it was found that 98% of the final reaction product was PLGA-PEG-Folate and 2% PEG-Folate (see supporting information).
2.2.3 Synthesis of PLGA-Alexa-488
The synthesis of PLGA45K-Alexa488 was accomplished by the conjugation of an activated PLGA-COOH with Alexa Fluor-488 Cadaverine through EDC/NHS conjugation. First, 1mg of Alexa-488 was dissolved in 1 mL of DMF. Concurrently, PLGA-COOH (50 mg, 1.1 μmol) was mixed with EDC and NHS in 500 μL of DMF. Finally, PLGA solution was reacted with Alexa-488 solution for 16 h at room temperature. The reaction product was precipitated in ice cold methanol and dried under vacuum.
2.2.4 Synthesis and Characterization of NPs
NPs were prepared by the nanoprecipitation method [20, 21]. In brief, PLGA-mPEG was mixed with a predefined amount of PLGA-PEG-Folate or PLGA-PEG-RGD and PLGA-Alexa488 in acetonitrile to a final polymer concentration of 10 mg/mL. For all experiments the weight percent of PLGA-Alexa488 in the polymer mixture was fixed to 2.5%. At this concentration, PLGA-Alexa488 did not affect the size and zeta potential of the NPs and was readily detected by FACS. For each NP formulation, 200 μL of the polymer mixture was added drop-wise to water with constant stirring, giving a final NP concentration of 1 mg/ml. The NPs were stirred for 2 h, and the remaining organic solvent was removed by washing the NPs three times using an Amicon centrifugation filtration membrane. Particle sizing was performed using dynamic light scattering (DLS) with a Zetasizer Nano ZS instrument (Malvern Instruments Ltd., U.K.). For each measurement, 100 μL or more of the sample was loaded in a disposable low-volume cuvette. The NP surface zeta potential was measured using the same instrument used for the size measurements. For each measurement, particles were washed with water and reconstituted in 1 mL of 1× PBS (0.5 mg/mL). TEM experiments were carried out on a JEOL 2011 instrument at an acceleration voltage of 200 kV. The TEM sample was prepared by depositing 10 μL of the NP suspension (1.0 mg/mL) onto a 200-mesh carbon-coated copper grid. Samples were blotted away after 30 min incubation and grids were negatively stained for 20 min at room temperature with sterile-filtered 2% (w/v) uranyl acetate aqueous solution.
2.2.5 Quantification of RGD and Folate on Nanoparticle Surface
Detection and quantification of RGD on the surface of the NP was accomplished using the Lava Prep Fluorescence-based Peptide Quantification Kit (Gel Company Inc, San Francisco, CA, USA). Briefly, NP-RGD were prepared and washed three times with DI water to a final concentration of 1mg/mL. Then, 100 μL of NP solution was mixed with 100 μL of the assay working solution and incubated for 60 minutes under dark. Fluorescence of the resulting solution was measured using a PerkinElmer plate reader VICTOR3 (Waltham, MA, USA) according to manufacturer’s instructions. Using a calibration curve of free cRGD-PEG4, the amount of RGD present in solution was determined. Each assay was repeated 3 times (each with n = 8 samples) and the average value was taken as the representative.
Detection and quantification of folate on the NP surface was accomplished first by incubating NP-Folate with folate antibodies and then determining the amount of antibody using the Low Protein BCA Assay (Lamda Biotech Inc, St. Louis, MO, USA). Briefly, NP-Folate was prepared and washed three times with DI water and re-suspended in PBS to a final concentration of 1 mg/mL. Then, 5 μL of NP solution was mixed with 95 μL of excess folate antibody in PBS, and the resultant solution incubated for 2 h with slow shaking. After 2 h, excess antibody was removed by washing the NP solution 3 times using a 300 kDa cutoff centrifugal filter. The final product was re-suspended in 100 μL of PBS followed by the addition of 100 μL of BCA working solution. The resultant solution was heated at 60 °C for 30 minutes and analyzed using a UV spectrophotometer according to the manufacturer’s instructions. Using a calibration curve of free folate antibody, the amount of folate present in the NP solution was determined. Each assay was repeated 5 times (each with n = 4 samples) and the average value was taken as the representative. Low BCA assays were done on free antibody solutions before and after centrifugal filtration to verify that no antibody remained left on the filter and that the initial amount was recovered after filtration.
2.2.6 NP Binding and Uptake by RAW264.7, KB and HUVEC cells
RAW 264.7 cells were cultured in DMEM medium with 10% fetal bovine serum, 50 units/mL penicillin and 50 mg/mL streptomycin. KB cells were cultured in Folate-free RPMI medium with 10% fetal bovine serum, 50 units/mL penicillin and 50 mg/mL streptomycin. HUVEC cells were cultured in EGM-2 BulleKit medium. All cells were cultured at 37 °C and 5% CO2. Cells were seeded at a density of 50,000 cells per well on a 24-well plate. After 24 h, the cell medium was removed and replaced with NPs dissolved in the same medium at a concentration of 1 mg/mL. Cells and NPs were incubated for 4 h for the case of RAW 264.7 and HUVEC cells and 6 h for case of KB cells followed by three washes with 1% BSA solution in PBS to remove excess NPs. RAW264.7 cells were removed from the plate using a cell scraper and centrifuged, while KB and HUVEC cells were treated with trypsin, removed from the plate, and centrifuged. The resultant cell pellets were reconstituted in 250uL of PBS and placed on a 96-well plate for FACS analysis. Flow cytometry analysis was performed on a BD Biosciences LSR II with High-Throughput sampler (HTS) option, with 10,000 cells were collected for each measurement.
2.2.7 Microscopy of Cells
For fluorescence microscopy studies, KB or HUVEC cells were seeded on a four-well chamber slide with 20,000 cells/well and incubated at 37°C and 5% CO2 for 24 h. Next, the cells were incubated in fresh complete medium with NPs. After incubation, cells were washed three times in PBS and fixed in 4% paraformaldehyde at room temperature. For imaging, slides were mounted with a coverslip using H-1000 Vectorshield mounting medium with DAPI. Fluorescence imaging was carried out using a Zeiss Fluorescence Microscope.
3. Results and Discussions
3.1 Synthesis of PLGA-PEG-RGD and PLGA-PEG-Folate
Figure 1A outlines the synthesis procedure of PLGA-PEG-RGD and PLGA-PEG-Folate block co-polymers. The underlying principles of the synthesis of these two compounds are the same: conjugation of an amine group (NH2) with a succinimide group in the presence of a base such a DIEA. The succinimide end group can be obtained from a carboxyl group (COOH) in the presence of EDC, such as the conversion of PLGA-COOH to PLGA-NHS. For the case of PLGA-PEG-RGD, the initial materials were the commercially available modified cyclic RGD (cRGD-PEG4-NH2) and tBOC-NH-PEG-NHS. For the case of PLGA-PEG-Folate, it is known that the γ-COOH group of folic acid is more reactive when derivatized and it allows folic acid to maintain its binding properties with the folate receptor [22]. Therefore, modification of the γ-COOH group of folic acid to NHS was the preferred strategy. Once folate-NHS was obtained, it was reacted with commercially available tBOC-NH-PEG-NH2. The chemistry presented here for the synthesis of PLGA-PEG-RGD and PLGA-PEG-Folate may allow for the conjugation of many targeting ligands (e.g. peptides, proteins, aptamers, small molecules, etc..), provided there is an available NH2 or COOH group to be conjugated to PEG, with the PEG conjugate subsequently conjugated to PLGA.
Figure 1.
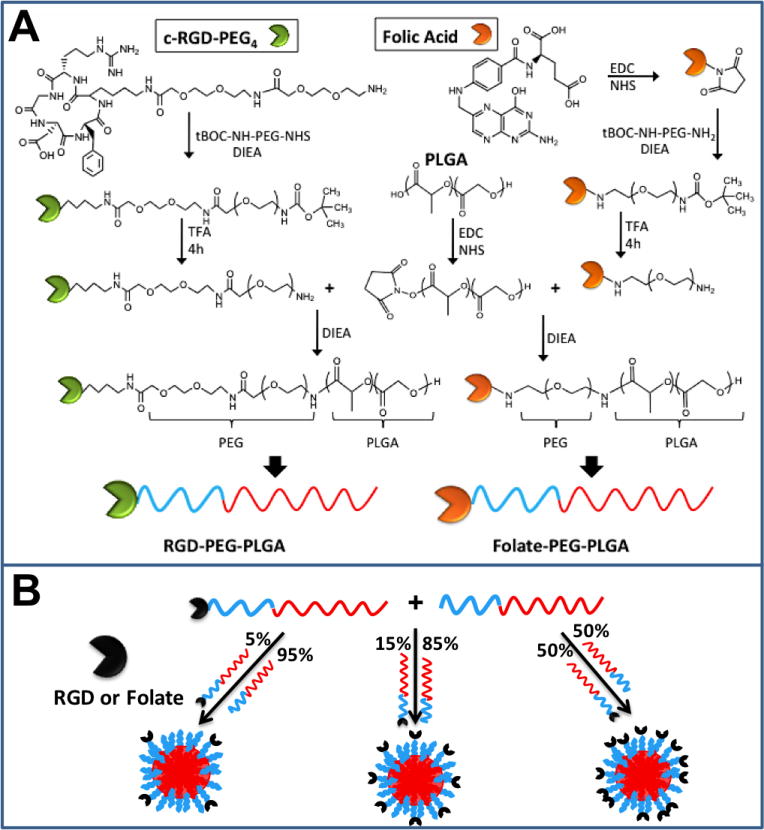
(A) Synthesis of PLGA-PEG-RGD and PLGA-PEG-Folate. (B) Preparation of targeted NPs with different surface ligand densities by mixing PLGA-mPEG with functionalized PLGA-PEG-TL.
3.2 Synthesis of RGD- and folate-targeted NPs
RGD and folate-targeted NPs were formulated by dissolving PLGA-PEG-RGD or PLGA-PEG-Folate copolymers with PLGA-mPEG in acetonitrile followed by precipitation in water. Ligand density was varied by using different percentages of TL-conjugated polymers in the total polymer formulation (Figure 1B). In principle, the resulting NPs are composed of a PLGA hydrophobic core, in which hydrophobic drugs can be encapsulated, and a PEG hydrophilic corona-like shell decorated with targeting moieties. The single-step synthesis eliminates conjugation steps after particle preparation and enhances batch-to-batch reproducibility. Prepared targeted NPs were characterized using DLS to measure size and surface zeta potential, and TEM to observe NP morphology and confirm size.
For NP-RGD it was observed that as the amount of PLGA-PEG-RGD was increased in the formulation, the surface zeta potential remained close to neutral, which was expected since PLGA-mPEG has a charge-neutral methoxy end group and RGD in PLGA-PEG-RGD is also neutral at a pH of 7.4 [23] (Figure 2A). A similar effect was observed for NP-Folate (Figure 2B). From DLS measurements, NP-RGD had a constant size of ~50 nm for formulations with 0%, 7%, and 22% PLGA-PEG-RGD. For formulations with 50%, and 74% of PLGA-PEG-RGD a gradual size increase of the NP was observed to 66 nm and 73 nm, respectively (Figure 2C). For NP-Folate, as the amount of PLGA-PEG-Folate was increased, size increase of NPs was observed for each formulation: 50 nm at 0%, 60 nm at 7%, 76 nm at 22%, 96 nm at 50% and 122 nm at 74% of PLGA-PEG-Folate (Figure 2D). Considering that both functionalized polymers PLGA-PEG-RGD and PLGA-PEG-Folate had the similar molecular weight according to GPC ( Mn = 50,005 Da and 49,551 Da, respectively) and the PLGA-mPEG of same MW was used for both NP formulations, this disparity in size for NP-Folate suggests that the effect is due to the differences between folate and RGD (i.e. presence of folate affects self-assembly). TEM images of both targeted NPs at a composition of 7% TL-conjugated polymer showed homogenous populations of NPs with minimal particle-particle aggregation (Figure 2E for NP-RGD and Figure 2F for NP-Folate).
Figure 2.

Characterization of targeted NP-RGD and NP-Folate: Zeta Potential (A,B) Size (C, D) and TEM micrographs (E,F). Error bars represent standard deviation of n = 3 samples.
3.3 NP-RGD and NP-Folate ligand densities
To investigate the effect of targeting ligand hydrophobicity on the surface composition of NPs, we determined the percentage of the initial RGD or Folate in the form of PLGA-PEG-RGD and PLGA-PEG-folate present on the surface of the NP in solution. For the case of NP-RGD we used the Lava Prep Peptide assay, a fluorescence-based method capable of detecting lysine, arginine, and histidine on peptides. For NP-Folate we first incubated the NPs with folate antibodies, followed by a washing step to remove excess antibody. Finally, we used Low BCA protein assay to determine the amount of antibody (and hence the corresponding amount of folate) on the NP surface. For the case of NP-RGD, all of the RGD present in the form of PLGA-PEG-RGD was detected on the NP surface (Figure 3A). For the cases of 75% and 100% RGD, the average fraction of starting RGD presented on the NP surface appeared to be 0.93 and 0.89, respectively. We believe that at large percentages of PLGA-PEG-RGD there might be an overlap of RGD molecules that result on the NP surface which may limit the detection accuracy of the assay, though for percentages of PLGA-PEG-RGD below 75% this ligand overlap may not occur. In contrast, for NP-Folate a maximum of 21% of the folate in the form of PLGA-PEG-Folate was detected on the NP surface for all formulations (Figure 3B). Although the accuracy of this assay may be compromised at high concentrations of PLGA-PEG-Folate due to steric hindrance of antibodies on the NP surface, results were found to be reliable for PLGA-PEG-Folate percentages below 74% at the given NP sizes, since others have detected ~ 4 times the number of antibodies we found on NPs of similar size [24].
Figure 3.

Quantification of targeted NP surface composition. (A) Fraction of initial RGD that resulted on the NP surface. (B) Fraction of initial folate that resulted on the NP surface. (C) Estimated ligand Densities and (E) estimated number of ligands per NP of NP-RGD as a function of the percentage of PLGA-PEG-RGD in formulation. (D) Estimated ligand Densities and (F) estimated number of ligands per NP of NP-Folate as a function of the percentage of PLGA-PEG-Folate in formulation. Theoretical (□) and experimental (Δ). Error bars represent standard deviation in n = 8 samples for NP-RGD and n = 4 samples for NP-Folate.
From these results, we hypothesize that the poor water solubility of folate ligands causes interactions of folate and PLGA when PLGA-PEG-Folate self-assembles into a NP, resulting in a significant fraction of folate buried in the core of the NP. Conversely, because RGD has high water solubility, it interacts minimally with PLGA and has minumum thermodynamic drive to enter the hydrophobic core of the NP. The hypothesis that a significant fraction of folate molecules from PLGA-PEG-Folate result in the NP core, may explain the larger NP size observed for higher fractions of PLGA-PEG-Folate. The low water solubility of folate may present a lower barrier to polymer and NP aggregation during self-assembly of the NPs, resulting in larger NPs with cores that are more loosely packed due to inclusion of PEG-Folate blocks of PLGA-PEG-Folate into the NP core. A similar increase in size of NPs formed by nanoprecipitation was observed previously when PLGA was added to PLGA-PEG prior to nanoprecipitation [25].
Knowing the size and size distribution of the targeted NPs and assuming an estimated NP density of 1.27 mg/mL [26], we calculated the ligand density (number of ligands per μm2 of NP surface) and the number of ligands per NP. Figures 3C and 3D show the theoretical, or expected, ligand densities calculated assuming 100% of ligands used in NP formulation are presented on the particle surface, as well as the experimentally determined amount of targeting ligands detected on the particle surface. For the case of NP-RGD, as the amount of PLGA-PEG-RGD was increased, the NP ligand density increased, and the observed ligand densities correlated well with the theoretical calculations. For NP formulations with 5%, 10%, 16%, 22%, 50%, 76% and 100% of PLGA-PEG-RGD the average number of RGD molecules on the surface of the NP were calculated to be 45, 97, 144, 230, 1030, 2270, and 3731, respectively (Figure 3E). Similarly, for the case of NP-Folate, as the amount of PLGA-PEG-Folate was increased, the NP ligand density also increased but the theoretical ligand densities did not correlate well the experimentally determined values. With 7%, 22%, 50%, and 74% of PLGA-PEG-Folate in the initial formulations, NPs were expected to have 107, 632, 2707, and 12127 ligands on their surface; however, only 23, 89, 368, and 1874 ligands, respectively, were detected on the surface (Figure 3F). The reduced presence of folate on the surface of the particle may have potential repercussions on the targeting capabilities of these NPs, which needs to be accounted for future experimental design. These results highlight the importance of considering the chemical properties of targeting ligands, such as solubility, as factors that may affect the single-step self-assembly of targeted NPs.
3.5 Cellular binding and uptake of targeted NPs
To examine the effect of ligand solubility on NP binding and uptake we investigated in vitro the detection of NPs by macrophages and their targeting capabilities to cell models with over-expressed ligand receptors was investigated in vitro. Phagocytosis of NPs by macrophages depends both on the size and the surface characteristics of the particle [27]. For instance, hydrophobic surfaces induce opsonization of NPs, which promotes NP phagocytosis [27, 28]. Conversely, PEG-functionalized and hydrophilic surfaces reduce NP uptake by macrophages [29]. We investigated the binding and uptake of RGD- and Folate-targeted NPs by macrophages as the fraction of RGD or folate biofunctional copolymer was varied. Flow cytometry was used to measure the association of NPs with macrophages, with 2.5% PLGA-Alexa488 added to the NP formulations as a fluorescent reporter. In the case of NP-RGD, it was observed that as the amount of PLGA-PEG-RGD was increased, there was no increase in uptake by macrophages when compared to non-targeted (i.e. fully PEG-functionalized) NPs, with the exception of NPs composed of 74% PLGA-PEG-RGD, which showed an almost 3-fold increase in uptake (Figure 4A). Although RGD is not known to be immunogenic [30], the disruption of the outer layer of the PEG shell at high RGD surface densities might induce an increase in phagocytosis. In contrast, for the case of NP-Folate, as the PLGA-PEG-Folate was increased, an augment in binding and uptake by macrophages was observed. Specifically, for NPs composed of 7%, 22%, 50% and 74% PLGA-PEG-Folate, a 2.0, 3.8, 4.7, and 8.5-fold increase in phagocytosis was observed, respectively (Figure 4B). It has been shown that NPs of larger size are more readily taken up by macrophages [31], and hence the observed increase in macrophage uptake could be due an increase in NP size. Alternatively, this increase in uptake may be due to the low hydrophilicity of folate which may induce opsonization of the NP surface. Finally, some reports have shown that NP-Folate target activated macrophages since they have over-expressed folate receptors [22, 32].
Figure 4.

Cellular binding and uptake of targeted NPs as a function of % of functionalized PLGA-PEG added in the NP formulation. (A) RGD targeted PLGA-PEG NPs and (B) Folate-targeted PLGA-PEG NPs. (C) Association of NP-RGD with HUVEC cells. (D) Association of NP-Folate with KB cells. (E) Representative images of non-targeted and NP-RGD (at 50% of PLGA-PEG-RGD) uptake by HUVEC cells. (F) Representative images of non-targeted and NP-Folate (at 50% PLGA-PEG-Folate) uptake by KB cells. Error bars represent standard deviation in n = 3 samples.
We determined the targeting capabilities of the nanoparticles with RGD and folate at different densities. We measured the binding and uptake of NP-RGD by HUVEC cells, which was taken as a model cell line with integrin-ανβ3 receptor over-expressed on the cell membrane. Similarly, binding and uptake of NP-Folate was investigated using KB cells, used as a model cell line with folate receptor over-expressed on the cell membrane. Flow cytometry was used to measure the association of NPs with HUVEC and KB cells, with 2.5% PLGA-Alexa488 added to the NP formulations as a fluorescent reporter. Increase in the amount of PLGA-PEG-RGD in the NP formulation (which correlates with a higher number of RGD molecules per NP) resulted in a corresponding increase in fluorescence associated with HUVEC cells up to ~200-fold when compared to non-targeted NPs (Figure 4C). The increase in RGD molecules on the NP surface may induce multivalent interactions with the integrin receptors in HUVEC cells leading to increased NP binding and uptake. On the other hand, for the case of NP-Folate, when the amount of PLGA-PEG-Folate was increased from 0% to 7%, to 22%, only marginal increase in uptake by KB cells was observed (Figure 4D). Only at 50% and 74% of PLGA-PEG-Folate was a substantial increase in uptake observed, with increase in fluorescence associated with the cells of approximately 4- and 9-fold, respectively, compared to non-targeted NPs. As mentioned before, we hypothesize that during self-assembly of PLGA-PEG-Folate and PLGA-mPEG into a NP, only ~20% of the folate ends up on the NP surface (Figure 3B) and the rest is embedded in the NP core due to interactions with PLGA. Thus, the amount of folate on the surface of the NPs may not be sufficient to significantly increase the uptake by KB cells for formulations with 7% and 22% of PLGA-PEG-Folate. The NP surface folate concentration was sufficient to elicit a significant increase in uptake by KB cells only when > 50% PLGA-PEG-Folate was used in NP formulation.
From these results one can determine optimal NP candidates for future experiments. For the case of NP-RGD, formulations composed of up to 50% PLGA-PEG-RGD could be used since they experienced marginal phagocytosis compared to non-targeted NPs and a ~70-fold increase in fluorescence when exposed to HUVEC cells. In contrast, for NP-Folate only those formulations with ~50% PLGA-PEG-Folate could be used since below this percentage only modest targeting effects were observed and above this percentage increased phagocytosis was observed. Microscopy images presented in figures 4E and 4F show that non-targeted NPs do not enter, or minimally enter, the cell while targeted NP-RGD and NP-Folate composed 50% TL-conjugated polymer readily enter HUVEC cells and KB cells, respectively. NP formulations with narrower ranges of the fraction of TL-conjugated biopolymer together with other in vitro experiments that investigate complement activation [33], cell cytotoxicity [34], among other factors [35], could be carried out to narrow the window of optimal NP candidates for in vivo experiments.
4. Conclusions
We synthesized PLGA-PEG copolymers functionalized with either folic acid (a ligand with poor water solubility) or RGD peptide (a ligand with high water solublity), for single-step preparation of targeted polymeric nanoparticles. By mixing PLGA-PEG-RGD or PLGA-PEG-Folate with PLGA-mPEG at different ratios we were able to prepare NPs with varying surface ligand densities. The surface of the targeted NPs was characterized, demonstrating that all of the RGD included in the form of PLGA-PEG-RGD was present on the surface of the NP-RGD while only ~20% of the folate in the form PLGA-PEG-Folate was found on the NP-Folate surface. Uptake of targeted NPs by macrophages was investigated showing that NP-Folate was increasingly taken up as the amount of PLGA-PEG-Folate in the formulation was increased, while NP-RGD experienced minimum phagocytosis even at 50% PLGA-PEG-RGD. Finally, we studied the association of NP-RGD with HUVEC cells and NP-Folate with KB cells showing that as the concentration of PLGA-PEG-RGD in the NP-RGD formulation was increased a corresponding increase in uptake was observed, while NP-Folate only showed significant targeting at ~50% PLGA-PEG-Folate. These studies provide insight into the behavior of targeted NPs prepared in a single-step with TLs of different solubilites and show an in vitro experimental design for finding NPs with favorable targeting ligand density.
Supplementary Material
Acknowledgments
We thank the Flow Cytometry Core facility of The David H. Koch Institute for Integrative Cancer Research at MIT for the use of FACS. We thank Eric Pridgen, Pamela Basto and Dr. Christopher Alabi for helpful discussions. Electron microscopy image acquisition was performed in the Center for Materials Science and Engineering (CMSE) imaging facility. This research was supported by the Koch-Prostate Cancer Foundation Award in Nanotherapeutics (R.L. and O.C.F.), the NCI Center of Cancer Nanotechnology Excellence (U54-CA151884; R.L. and O.C.F.) and the NHLBI Programs of Excellence in Nanotechnology (HHSN268201000045C; R.L. and O.C.F.). P.M.V. is supported by NSF graduate research fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Nanomedicine--challenge and perspectives. Angew Chem Int Ed Engl. 2009;48:872–97. doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mamo T, Moseman EA, Kolishetti N, Salvador-Morales C, Shi J, Kuritzkes DR, et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine (Lond) 2010;5:269–85. doi: 10.2217/nnm.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mulder WJ, Fayad ZA. Nanomedicine captures cardiovascular disease. Arterioscler Thromb Vasc Biol. 2008;28:801–2. doi: 10.1161/ATVBAHA.108.165332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farokhzad OC. Nanotechnology for drug delivery: the perfect partnership. Expert Opin Drug Deliv. 2008;5:927–9. doi: 10.1517/17425247.5.9.927. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–82. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 6.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–3. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 7.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexis F, Basto P, Levy-Nissenbaum E, Radovic-Moreno AF, Zhang L, Pridgen E, et al. HER-2-targeted nanoparticle-affibody bioconjugates for cancer therapy. Chem Med Chem. 2008;3:1839–43. doi: 10.1002/cmdc.200800122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133:11–7. doi: 10.1016/j.jconrel.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 10.Pan J, Liu Y, Feng SS. Multifunctional nanoparticles of biodegradable copolymer blend for cancer diagnosis and treatment. Nanomedicine (Lond) 2010;5:347–60. doi: 10.2217/nnm.10.13. [DOI] [PubMed] [Google Scholar]
- 11.Pirollo KF, Chang EH. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008;26:552–8. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107:17939–44. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, et al. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A. 2008;105:2586–91. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 15.Wu Z, Li XX, Hou CY, Qian Y. Solubility of Folic Acid in Water at pH Values between 0 and 7 at Temperatures (298.15, 303.15, and 313.15) K. Journal of Chemical and Engineering Data. 2010;55:3958–61. [Google Scholar]
- 16.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, et al. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci U S A. 2008;105:9343–8. doi: 10.1073/pnas.0803728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Low PS, Antony AC. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Deliv Rev. 2004;56:1055–8. doi: 10.1016/j.addr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Patil YB, Toti US, Khdair A, Ma L, Panyam J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials. 2009;30:859–66. doi: 10.1016/j.biomaterials.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles: preparation, properties and possible applications in drug delivery. Curr Drug Deliv. 2004;1:321–33. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 21.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–76. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41:147–62. doi: 10.1016/s0169-409x(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 23.Ghandehari H, Sharan R, Rubas W, Killing WM. Molecular modeling of arginine-glycine-aspartic acid (RGD) analogs: relevance to transepithelial transport. J Pharm Pharm Sci. 2001;4:32–41. [PubMed] [Google Scholar]
- 24.Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–50. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 25.Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, et al. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008;8:2906–12. doi: 10.1021/nl801736q. [DOI] [PubMed] [Google Scholar]
- 26.Vauthier C, Schmidt C, Couvreur P. Measurement of the density of polymeric nanoparticulate drug carriers by isopycnic centrifugation. J Nanoparticle Res. 1999;1:411–8. [Google Scholar]
- 27.Owens DE, 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Vlerken LE, Vyas TK, Amiji MM. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm Res. 2007;24:1405–14. doi: 10.1007/s11095-007-9284-6. [DOI] [PubMed] [Google Scholar]
- 30.Gaillard C, Duval M, Dumortier H, Bianco A. Carbon nanotube-coupled cell adhesion peptides are non-immunogenic: a promising step toward new biomedical devices. J Pept Sci. 2010;17:139–42. doi: 10.1002/psc.1290. [DOI] [PubMed] [Google Scholar]
- 31.Epstein-Barash H, Gutman D, Markovsky E, Mishan-Eisenberg G, Koroukhov N, Szebeni J, et al. Physicochemical parameters affecting liposomal bisphosphonates bioactivity for restenosis therapy: internalization, cell inhibition, activation of cytokines and complement, and mechanism of cell death. J Control Release. 2010;146:182–95. doi: 10.1016/j.jconrel.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Turk MJ, Waters DJ, Low PS. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 2004;213:165–72. doi: 10.1016/j.canlet.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 33.Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev. 2009;61:428–37. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2:469–78. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 35.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5:487–95. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.