

Abstract
Background/Aims
The response rates of HCV infection to interferon therapy vary depending on viral and host factors. We hypothesized that key regulators of the IFN signaling pathway are predictive of treatment outcome.
Methods
We measured the expression of signal transducer and activator of transcription 1 (STAT1) and suppressor of cytokine signaling 3 (SOCS3) in pretreatment liver biopsies. Staining quantitation was compared to treatment outcomes.
Results
Forty-nine patients with HCV and 25 patients with HCV/HIV infection treated with peginterferon/ribavirin were analyzed. Pretreatment hepatic SOCS3 expression was higher in non-responders than responders. Genotype 1 responders had similar levels of SOCS3 as genotype 2/3 responders. African Americans (AA) had higher hepatic SOCS3 than non-AA. Pretreatment hepatic SOCS3 was the most powerful independent predictor of sustained virologic response (SVR), even more so than genotype by logistic regression analysis. Failure to achieve SVR and AA race were independently associated with high hepatic SOCS3 levels. The hepatic expression of STAT-1 did not differ between responders and non-responders.
Conclusions
Our data indicate that hepatic SOCS3 is a stronger baseline predictor of antiviral response than viral genotype. Poor response to antiviral therapy in AA may be associated with higher hepatic SOCS3 expression.
Keywords: Suppressor of cytokine signaling 3, Hepatitis C virus, HCV/HIV coinfection, Race, Immunohistochemistry, Signal transducer and activator of transcription 1
1. Introduction
Hepatitis C virus (HCV) infection is a major cause of chronic liver disease, affecting 170 million persons world-wide, and can lead to serious outcomes such as end-stage liver disease and hepatocellular carcinoma [1]. The most effective current treatment for chronic hepatitis C is the combination of peginterferon (PEG-IFN) and ribavirin (RBV) [2], but this regimen is expensive, has substantial side effects, and variable efficacy depending on viral and host factors. Genotype 1, high viral load, older age, obesity, and advanced hepatic fibrosis are associated with diminished treatment response [3,4], but there is no single reliable pretreatment predictor of virologic response. Serum HCV RNA measurements at 4 and 12 weeks of antiviral therapy appear to be the current best predictor of treatment response but cannot be determined prior to treatment initiation [3,5]. Therefore, it is essential to identify accurate pretreatment predictors for treatment outcome in patients with chronic hepatitis C.
The molecular mechanisms of poor or variable response to IFN-based therapy have been explored. It is likely that both viral and host factors influence the response to HCV treatment. There is now abundant evidence that several HCV proteins interfere with host IFN signaling. For example, HCV polyprotein expression in replicon cell culture models is associated with a decrease in 2′–5′oligoadenylate synthetase, MxA and signal transducer and activator of transcription 1 (STAT1) [6–9]. Suppressor of cytokine signaling 3 (SOCS3), which is thought to antagonize IFN signaling by interfering with Jak-STAT signaling at the level of STAT1 phosphorylation, is increased by HCV core protein overexpression in cell culture [10] and is associated with treatment outcome in in vivo studies [11,12].
In this context, we hypothesized that key regulators of the IFN signaling pathway would be predictive of treatment outcome. We measured the hepatic protein expression of STAT1 and SOCS3, each of which plays a key role in IFN signaling and is known to be altered in HCV infection, and correlated their expression with treatment outcome.
2. Materials and methods
2.1. Patients
Two groups of patients were analyzed. The first group was composed of patients with chronic hepatitis C infection who had been treated with PEG-IFN-based regimens at a single center between 2002 and 2005. Subjects were included in the current study if they fulfilled the following criteria: (a) chronic HCV with detectable circulating HCV RNA, (b) a pretreatment liver biopsy consistent with chronic viral hepatitis, (c) compensated liver disease, and (d) treatment with the combination of PEG-IFN-α and RBV for at least 12 weeks. Patients with other forms of chronic liver disease or antibodies to human immunodeficiency virus (HIV) were excluded from this group. Clinical and laboratory data were collected from the medical records at the time of treatment and/or liver biopsy. The study protocol was approved by the MGH Institutional Review Board.
The second group consisted of chronic hepatitis C patients coinfected with HIV who were enrolled in the Adult AIDS Clinical Trials Group A5071. A5071 was a prospective, randomized, phase II/III trial evaluating the efficacy of PEG-IFN plus RBV in HCV/HIV-coinfected individuals with stable HIV disease who had not previously been treated. The study has previously been described in detail [13].
2.2. Laboratory data
HCV genotyping was performed with the use of a line-probe assay (LiPA, Innogenetics) before treatment. HCV RNA was quantified by using the Amplicor Monitor HCV assay (Roche Molecular Diagnostics, Branchburg, New Jersey), which has a detection limit of 600 IU/mL. Sustained virologic response (SVR) was defined as undetectable HCV RNA by the Roche Amplicor 2.0 assay (lower limit of detection 60 IU/mL) 24 weeks after the completion of treatment. Patients who did not achieve SVR were classified as non-responders.
2.3. Histological examination
Liver biopsy sections were reviewed by a single hepatopathologist (AKB) who was blinded to the laboratory parameters and clinical data. The grade of inflammation and the stage of fibrosis were scored according to the method of Ishak et al. [14].
Steatosis was assessed semiquantitatively by scoring the proportion of hepatocytes affected in the whole sample, as follows: 0 = none; 1 = minimal (up to 5% hepatocytes containing fat); 2 = mild, (>5%, and ≤33%); 3 = moderate (>33%, and ≤66%); and 4 = severe (>66%).
2.4. Immunohistochemistry
Liver sections (4 μm) were cut from paraffin blocks. Sections were deparaffinized with three dips of xylene and rehydrated in decreasing concentrations of ethanol. Slides were heated in a microwave for 20 min in 10 mM sodium citrate buffer (pH 6) for antigen retrieval and allowed to cool for 40 min. Endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide for 20 min. Nonspecific binding was blocked by applying 10% goat serum (Invitrogen, Carlsbad, CA) for 1 h. Sections were incubated with monoclonal mouse anti-STAT1 antibody (Cell Signaling Technology, Beverly, MA; dilution 1:100) and polyclonal rabbit anti-SOCS3 antibody (Santa Cruz Biotechnology, Santa Cruz, CA; dilution 1:100) in humid chambers overnight at 4 °C. After three washes in PBS, peroxidase-conjugated secondary antibody and DAB substrate (Dako, Carpinteria, CA) were applied for 30 and 3 min, respectively at room temperature. Sections were counterstained with methyl green (Dako), dehydrated and coverslipped. Negative control sections were incubated either with nonimmune serum or without any primary antibody. To demonstrate IFN inducibility of STAT1, Huh7 cells treated with 100 IU interferon α-2a for 24 h were used as a positive control.
Sections were examined with a Nikon Eclipse 50i microscope (Nikon, Melville, NY) and photographed using a Nikon DS-Fi1 camera (Nikon) and software (NIS-Elements F version 2.30; Nikon). Five to twenty non-overlapping random high power fields were photographed per section at a magnification of 400×. Identical imaging conditions, including illumination intensity and camera exposure time, were applied to all photographs. All images were analyzed with Image J software (available at http://rsb.info.nih.gov/ij). DAB-stained areas were selected using the Image J thresholding tool followed by conversion to a binary image. Thresholding conditions were set identically for all images. The percent positive area was defined as the ratio of the selected DAB-positive pixels to the total number of pixels within a defined area of interest.
Each step of immunohistochemistry and image processing was performed blinded to the clinical data.
2.5. Statistical analysis
Statistical analyses were performed using non-parametric methods (Mann-Whitney U, Kruskal–Wallis, and χ2 tests). Correlation was assessed by Pearson’s correlation coefficients. All p values were two-tailed and were considered significant if the associated value was less than 0.05. For multivariate analysis, two-way ANOVA was performed between response status, genotype, race, and infection/coinfection status to determine which variables independently associated with hepatic SOCS3 staining and with SVR.
3. Results
3.1. Patient characteristics
A total of 49 patients with HCV monoinfection and 22 with HCV/HIV coinfection were divided into responders and non-responders depending on the outcome of PEG-IFN and RBV therapy (Tables 1 and 2). Responders were defined as those achieving sustained virologic response (undetectable HCV RNA at 24 weeks after completion of therapy). Non-responders were defined as failure to clear HCV RNA despite a minimum of 12 weeks of PEG-IFN and RBV therapy. Age, gender, race, ALT, histologic grade, and steatosis score did not differ significantly between responders and non-responders both in patients with HCV monoinfection and HIV/HCV coinfection. Genotype 1 was more common in non-responders compared with responders. Non-responders had higher serum HCV RNA levels, and more advanced fibrosis in patients with HCV monoinfection, but not in patients with HCV/HIV coinfection.
Table 1.
Baseline characteristics of patients with hepatitis C monoinfection.
| Characteristic | Responders (n = 30) | Non-responders (n = 19) | p Value |
|---|---|---|---|
| Age, mean (SEM) | 45.6 (1.8) | 48.4 (1.5) | 0.349 |
| Gender (male/female) | 20/10 | 13/6 | 1.0 |
| Race | 0.323 | ||
| African American | 1 | 3 | |
| White | 27 | 15 | |
| Others | 2 | 1 | |
| Genotype | |||
| 1/2, 3 | 18/12 | 18/1 | 0.025 |
| Viral load | |||
| ≥800,000/< 800,000 IU/mL | 14/14 | 14/3 | 0.03 |
| ALT, mean (SEM) | 104.4 (12.6) | 95.2 (13.4) | 0.711 |
| Histology | |||
| Inflammatory grade, median (interquartile) | 6 (5,6) | 6 (5,6.5) | 0.707 |
| Fibrosis stage, median (interquartile) | 2.5 (1.75,3) | 3 (2,5) | 0.039 |
| Presence/absence of cirrhosis | 4/26 | 6/13 | 0.569 |
| Steatosis | |||
| None | 17 | 11 | 0.609 |
| Mild | 8 | 7 | |
| Moderate/severe | 5 | 1 | |
Table 2.
Baseline characteristics of patients with HCV/HIV coinfection.
| Characteristic | Responders (n = 10) | Non-responders (n = 15) | p value |
|---|---|---|---|
| Age, mean (SEM) | 41.4 (2.2) | 44.7 (1.5) | 0.311 |
| Gender (male/female) | 9/1 | 10/5 | 0.345 |
| Race | 0.167 | ||
| African American | 2 | 7 | |
| White | 8 | 6 | |
| Hispanic | 0 | 2 | |
| Genotype | |||
| 1,4/2,3 | 4/6 | 14/1 | 0.007 |
| Viral load | |||
| ≥800,000/< 800,000 IU/mL | 7/3 | 13/2 | 0.175 |
| Histology | |||
| Inflammatory grade, median (interquartile) | 6 (5,6) | 6 (3.5,6) | 0.557 |
| Fibrosis stage, median (interquartile) | 3 (2,3) | 2 (1,3.5) | 0.516 |
| Presence/absence of cirrhosis | 1/8 | 1/14 | 1.0 |
| Steatosis | 0.558 | ||
| None | 2 | 5 | |
| Mild | 4 | 5 | |
| Moderate/severe | 3 | 5 | |
3.2. Hepatic SOCS3 expression was higher in nonresponders than responders
For all patients, SOCS3 staining within hepatocytes was diffusely cytoplasmic (Fig. 1A–C). In HCV monoinfection, non-responders exhibited significantly higher pretreatment hepatic SOCS3 expression compared with responders in patients with HCV monoinfection (21.6% ± 2.7 vs. 10% ± 1.3, p < 0.001) (Fig. 2A and B). Among patients with HCV genotype 1 infection, non-responders again had increased SOCS3 expression compared with responders (23.0% ± 2.9 vs. 12.6% ± 1.8, p < 0.05) (Fig. 2C). Patients with genotype 1 infection showed greater SOCS3 expression than those with genotype 2 or 3 (17.2% ± 1.9 vs. 7.7% ± 1.9, p < 0.005) (Fig. 2D). However, among responders, there was no significant difference in SOCS3 expression between genotypes 1 and 2/3 (11.7 ± 1.8 vs. 7.4 ± 1.4, p = 0.124) (Fig. 2C).
Fig. 1.
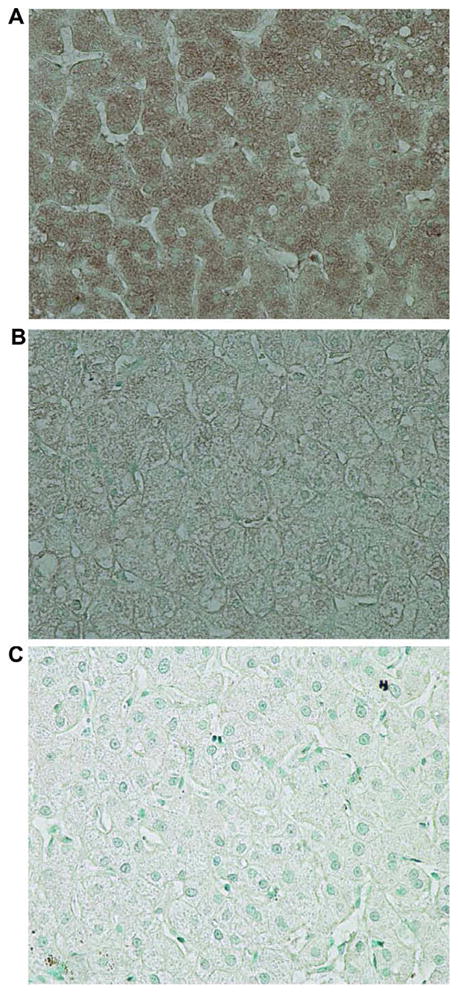
Immunohistochemistry of SOCS3 in patients with chronic hepatitis C. (A) Typical staining in a non-responder. Strong SOCS3 staining is seen in hepatocyte cytoplasm. (B) Typical staining in a responder. SOCS3 is faintly stained in hepatocyte cytoplasm. (C) Negative control using normal rabbit serum in a non-responder. Original magnification 400×. [This figure appears in colour on the web.]
Fig. 2.

Comparison of hepatic SOCS3 expression in patients with chronic hepatitis C monoinfection. (A and B) Pretreatment hepatic SOCS3 expression is higher in non-responders (NR; n = 19) than responders (R; n = 30) across all genotypes. (C) Pretreatment hepatic SOCS3 expression is higher in nonresponders (n = 17) than responders (n = 18) among patients with genotype 1 infection. SOCS3 expression is not significantly different between genotype 1 (G1, n = 18) and genotype 2/3 responders (G2/3, n = 12). (D) Patients with genotype 1 infection showed greater SOCS3 expression than those with genotype 2 or 3. Data are presented as means ± SEM. *p < 0.001, **p < 0.05, ***p < 0.005. [This figure appears in colour on the web.]
None of the clinical, virological or pathological factors listed in Table 1 other than viral genotype were significantly associated with hepatic SOCS3 expression in patients with HCV monoinfection.
For patients with HCV/HIV coinfection, SOCS3 expression was also increased in non-responders compared to responders (21.6% ± 3.2 vs. 10.5% ± 3.7, p < 0.05) (Fig. 3A). Patients with genotype 1/4 infection tended to have higher SOCS3 expression than those harboring genotype 2 or 3 infection (19.7% ± 3.0 vs. 10.1% ± 2.3, p = 0.097); however, among responders, there was again no significant difference in SOCS3 expression between genotypes 1 and 2/3 (11.5 ± 4.6 vs. 9.2 ± 2.5, p = 0.762) (data not shown). Logistic regression analysis of the overall group revealed that pretreatment hepatic SOCS3 expression was the most powerful independent predictor of SVR (p = 0.002), even more so than genotype (p = 0.056).
Fig. 3.

Comparison of SOCS3 expression in patients with HCV–HIV coinfection. (A) Non-responders (n = 15) exhibit higher pretreatment hepatic SOCS3 expression than responders (n = 10) in patients with HCV/HIV coinfection. (B) AA (n = 9) have higher pretreatment SOCS3 expression than non-AA (= 16). Data are presented as means ± SEM. *p < 0.05. [This figure appears in colour on the web.]
In patients with HCV/HIV coinfection, SOCS3 expression was higher in African Americans (AA) compared to non-AA (25.4 ± 4.4 vs. 12.2 ± 2.1, p = 0.017) (Fig. 3B). Similarly, for all patients, SOCS3 expression was higher in AA compared with non-AA (23.2 ± 3.5 vs. 13.5 ± 1.3, p = 0.003). There were too few AA patients in the HCV monoinfected group (n = 4) to assess for meaningful differences in SOCS3 expression.
Using two-way ANOVA analysis, responder status was independently associated with increased SOCS3 staining (p < 0.0001). AA race was also significantly associated with elevated SOCS3 staining, even after adjustment for responder status (p = 0.05).
In separate control studies on independently collected fresh liver tissue, we confirmed that staining of SOCS3 correlated with tissue levels of protein by Western blot (data not shown), supporting the ability of staining to quantitate SOCS3 levels.
3.3. Hepatic STAT1 expression is similar between responders and non-responders
As with SOCS3, STAT1 staining was diffusely cytoplasmic (Fig. 4A). There was no difference in pretreatment hepatic STAT1 expression between responders and non-responders (Fig. 4A–C and E), in patients with HCV monoinfection (Fig. 4E) and HCV–HIV coinfection (data not shown). Hepatic STAT1 levels did not correlate with serum HCV RNA levels, viral genotype, histologic activity or fibrosis stage. Phospho-STAT1 immunoreactivity was not observed in any of the samples (data not shown), likely because they were obtained prior to the administration of IFN, a potent stimulus for STAT1 phosphorylation.
Fig. 4.
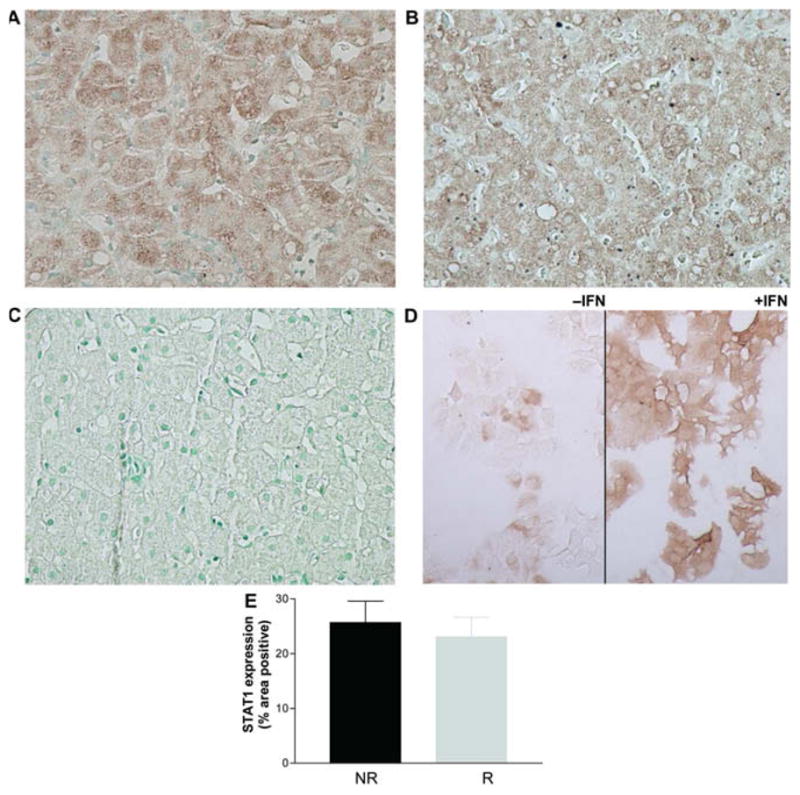
Immunohistochemistry of STAT1. (A) STAT1 antibodies diffusely stained the hepatocyte cytoplasm in a responder. (B) STAT1 staining in a non-responder. (C) Negative control using normal mouse serum in a responder. (D) STAT1 staining in Huh7 cells treated with IFN as a positive control. (E) There was no difference in pretreatment hepatic STAT1 expression between non-responders (n = 19) and responders (n = 30) in patients with HCV monoinfection. Data are presented as means ± SEM. [This figure appears in colour on the web.]
4. Discussion
IFN-based therapy is the only effective treatment for chronic hepatitis C at present, but produces variable outcomes depending on host and viral factors. Considering its cost and side effects, it would be extremely useful to be able to predict treatment outcome prior to the initiation of treatment. In addition, identifying factors that influence treatment outcome would be helpful in elucidating the mechanism of action and resistance to IFN in HCV infection. We therefore investigated the hepatic expression of STAT1 and SOCS3, proteins known to play key roles in IFN signaling and/or action and known to be altered in HCV infection, and correlated their expression with treatment outcome.
SOCS3 is a member of the suppressors of cytokine signaling family and is thought to interfere with intracellular IFN signaling by directly inhibiting JAK tyrosine kinase activity [15]. Studies in cell culture models have suggested a role for SOCS3 in HCV IFN resistance. First, HCV core protein overexpression has been reported to induce SOCS3 in cell culture models, resulting in impaired IFN signaling [10]. Second, HCV replicon cells resistant to IFN therapy have been shown to produce higher levels of SOCS3; silencing of SOCS3 in these resistant cells partially restored IFN sensitivity [16]. Furthermore, several in vivo studies have shown that hepatic SOCS3 expression is associated with response to IFN treatment. One study showed that high pretreatment hepatic SOCS3 mRNA and protein expression was associated with non-response to IFN therapy and obesity [11]. A second study also showed that SOCS3 mRNA expression in peripheral lymphocytes was higher in non-responders than responders [16]. In contrast, a third study reported that pretreatment hepatic SOCS3 mRNA expression was higher in responders than non-responders, but fell significantly more in response to IFN therapy in responders compared to non-responders [12]. Finally, a genetic polymorphism in the SOCS3 gene was reported to influence the level of SOCS3 expression and interferon treatment outcome in chronic hepatitis C [17].
Our study has the advantage of measuring SOCS3 protein, which is a more direct readout of SOCS3 activity than mRNA levels. In this study, we show that nonresponse to PEG-IFN-based therapy is associated with a significantly higher pretreatment hepatic SOCS3 expression in patients with chronic hepatitis C, which is consistent with the majority of previous studies. Moreover, we investigated SOCS3 expression in patients with HCV/HIV coinfection, and found results similar to those seen in HCV monoinfection. SOCS3 expression was significantly higher in genotype 1 than genotype 2/3 infections, but when compared only among responders, there was no difference in SOCS3 expression between genotypes. These data suggest that elevated pretreatment hepatic SOCS3 expression can explain the higher rate of IFN non-response in patients with genotype 1 infection, and can potentially distinguish future responders from non-responders, even within unfavorable treatment groups. Ours is the first study to identify a possible basis for increased rates of non-response in genotype 1 infection. We found that SOCS3 expression between genotype 2,3 responders and non-responders was not significantly different. However, it should be cautioned that there were only two genotype 2,3 non-responders. Further study is therefore needed in larger numbers of genotype 2,3 patients to determine whether SOCS3 levels are predictive of outcome.
Another remarkable finding of our study is that African Americans had higher pretreatment hepatic SOCS3 expression than Caucasians or Hispanics in patients with HCV/HIV coinfection, independent of response status. This finding may help to explain why African Americans exhibit consistently poor responses to IFN-based therapy for chronic hepatitis C. It suggests that an intrinsic impairment of IFN responsiveness underlies the poor kinetic responses to IFN-based therapies observed in this group. Whether increased SOCS3 levels themselves are the driving force for limited responsiveness or rather reflect a compensatory response to a primary block elsewhere in the IFN pathway is not known. As SOCS3 can be induced by IFN-α [18], higher SOCS3 expression may reflect preactivation of the endogenous IFN system. Indeed, several studies have shown that non-responders exhibit higher pretreatment levels of interferon-stimulated genes than responders [19,20]. Further confirmation of this observation in larger populations and additional mechanistic exploration are warranted.
STAT1 is a key molecule mediating type I IFN signal transduction against HCV. HCV core protein decreases STAT1 levels in cell culture models, which is thought to be one mechanism of IFN resistance mediated by hepatitis C [9]. However, pretreatment hepatic STAT 1 was reported to be higher in non-responders than responders in chronic hepatitis C in vivo [20]. In this study, we showed that treatment outcome was not associated with pretreatment hepatic STAT1 protein expression. In addition, pretreatment hepatic STAT1 expression was not correlated with serum HCV RNA levels (data not shown), which has been suggested by others to correlate with the hepatic HCV RNA level [21].
Our study is limited by its retrospective design and the relatively small number of samples studied. However, in examining two distinct patient populations, HCV monoinfection and HCV/HIV coinfection, we observed strikingly concordant results, supporting the reliability of the finding.
This study is the first report to show that hepatic SOCS3 levels differ between responders and nonresponders with HCV/HIV coinfection and has more discriminatory value than genotype itself. While there is overlap between responder and non-responder groups (Fig. 2B), it will be of interest to determine in future studies whether there is a quantifiable threshold that clearly predicts non-response. Irrespective of its utility as a clinical tool, these findings provide biological insight into the driving forces for non-response.
In summary, pretreatment hepatic SOCS3 protein levels are correlated with outcomes of interferon-based treatment in patients with chronic HCV monoinfection and HCV/HIV coinfection. The finding that SOCS3 levels were comparable among responders irrespective of genotype suggests that SOCS3 can be a more powerful pretreatment predictor of response than genotype. Higher hepatic SOCS3 expression in AA may be an explanation for poor response to antiviral therapy in this group. These data strongly implicate SOCS3 as an important antagonist of the in vivo response to IFN. Prospective studies to evaluate the utility of this measurement should be performed to confirm its predictive power.
Acknowledgments
We gratefully acknowledge the NIAID-AIDS Clinical Trials Group for biopsy material for these studies (NIH AI38858 and AI38855).
Abbreviations
- IFN
interferon
- RBV
ribavirin
- STAT1
signal transducer and activator of transcription 1
- SOCS3
suppressor of cytokine signaling 3
- HCV
hepatitis C virus
- AA
African Americans
- PEG-IFN
peginterferon
Footnotes
Financial support: NIH AI069939, DK078772 (to R.T.C.) and Massachusetts Biomedical Research Corporation Tosteson Postdoctoral Fellowship (to A.W.T.). The authors declare that they do not have anything to disclose regarding funding from industries or conflict of interest with respect to this manuscript.
References
- 1.Dienstag JL, McHutchison J. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology. 2006;130:231–264. doi: 10.1053/j.gastro.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Di Bisceglie AM, Hooofnagle JH. Optimal therapy of hepatitis C. Hepatology. 2002;36:S121–S127. doi: 10.1053/jhep.2002.36228. [DOI] [PubMed] [Google Scholar]
- 3.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 4.Zeuzem S, Feinman SV, Rasenack J, Heathcote EJ, Lai MY, Gane E, et al. Peginterferon alfa-2a in patients with chronic hepatitis C. N Engl J Med. 2000;343:1666–1672. doi: 10.1056/NEJM200012073432301. [DOI] [PubMed] [Google Scholar]
- 5.Davis GL, Wong JB, McHutchison JG, Manns MP, Harvey J, Albrecht J. Early virologic response to treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C. Hepatology. 2003;38:645–652. doi: 10.1053/jhep.2003.50364. [DOI] [PubMed] [Google Scholar]
- 6.Murashima S, Kumashiro R, Ide T, Miyajima I, Hino T, Koga Y, et al. Effect of interferon treatment on serum 2′,5′-oligoadenylate synthetase levels in hepatitis C-infected patients. J Med Virol. 2000;62:185–190. doi: 10.1002/1096-9071(200010)62:2<185::aid-jmv9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 7.Knapp S, Yee LJ, Frodsham AJ, Hennig BJ, Hellier S, Zhang L, et al. Polymorphisms in interferon-induced genes and the outcome of hepatitis C virus infection: roles of MxA, OAS-1 and PKR. Genes Immun. 2003;4:411–419. doi: 10.1038/sj.gene.6363984. [DOI] [PubMed] [Google Scholar]
- 8.MacQuillan GC, de Boer WB, Platten MA, McCaul KA, Reed WD, Jeffrey GP, et al. Intrahepatic MxA and PKR protein expression in chronic hepatitis C virus infection. J Med Virol. 2002;68:197–205. doi: 10.1002/jmv.10182. [DOI] [PubMed] [Google Scholar]
- 9.Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128:1034–1041. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, et al. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17:488–490. doi: 10.1096/fj.02-0664fje. [DOI] [PubMed] [Google Scholar]
- 11.Walsh MJ, Jonsson JR, Richardson MM, Lipka GM, Purdie DM, Clouston AD, et al. Non-response to antiviral therapy is associated with obesity and increased hepatic expression of suppressor of cytokine signalling 3 (SOCS-3) in patients with chronic hepatitis C, viral genotype 1. Gut. 2006;55:529–535. doi: 10.1136/gut.2005.069674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y, Feld JJ, Sapp RK, Nanda S, Lin JH, Blatt LM, et al. Defective hepatic response to interferon and activation of suppressor of cytokine signaling 3 in chronic hepatitis C. Gastroenterology. 2007;132:733–744. doi: 10.1053/j.gastro.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung RT, Andersen J, Volberding P, Robbins GK, Liu T, Sherman KE, et al. Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med. 2004;351:451–459. doi: 10.1056/NEJMoa032653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 15.Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273:35056–35062. doi: 10.1074/jbc.273.52.35056. [DOI] [PubMed] [Google Scholar]
- 16.Persico M, Capasso M, Persico E, Svelto M, Russo R, Spano D, et al. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: Insulin resistance and response to antiviral therapy. Hepatology. 2007;46:1009–1015. doi: 10.1002/hep.21782. [DOI] [PubMed] [Google Scholar]
- 17.Persico M, Capasso M, Russo R, Persico E, Croce L, Tiribelli C, et al. Elevated expression and polymorphisms of SOCS3 influence patient response to antiviral therapy in chronic hepatitis C. Gut. 2008;57:507–515. doi: 10.1136/gut.2007.129478. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerer JM, Lesinski GB, Kondadasula SV, Karpa VI, Lehman A, Raychaudhury A, et al. IFN-alpha-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol. 2007;178:4832–4845. doi: 10.4049/jimmunol.178.8.4832. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128:1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 20.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci USA. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White PA, Pan Y, Freeman AJ, Marinos G, French RA, Lloyd AR, et al. Quantification of hepatitis C virus in human liver and serum samples by using LightCycler reverse transcriptase PCR. J Clin Microbiol. 2002;40:4346–4348. doi: 10.1128/JCM.40.11.4346-4348.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]