

Abstract
The yeast DEAD-box protein Mss116p functions as a general RNA chaperone in splicing mitochondrial group I and group II introns. For most of its functions, Mss116p is thought to use ATP-dependent RNA unwinding to facilitate RNA structural transitions, but it has been suggested to assist folding of one group II intron (aI5γ) primarily by stabilizing a folding intermediate. Here we compare three aI5γ constructs: one with long exons, one with short exons, and a ribozyme construct lacking exons. The long exons result in slower splicing, suggesting that they misfold and/or stabilize non-native intronic structure. Nevertheless, Mss116p acceleration of all three constructs depends upon ATP and is inhibited by mutations that compromise RNA unwinding, suggesting similar mechanisms. Results of splicing assays and a new two-stage assay that separates ribozyme folding and catalysis indicate that maximal folding of all three constructs by Mss116p requires ATP-dependent RNA unwinding. ATP-independent activation is appreciable for only a subpopulation of the minimal ribozyme construct and not for constructs containing exons. As expected for a general RNA chaperone, Mss116p can also disrupt the native ribozyme, which can refold after Mss116p removal. Finally, using yeast strains with mtDNA containing only the single intron aI5γ, we show that Mss116p mutants promote splicing in vivo to degrees that correlate with their residual ATP-dependent RNA-unwinding activities. Together, our results indicate that, although DEAD-box proteins play multiple roles in RNA folding, the physiological function of Mss116p in aI5γ splicing includes a requirement for ATP-dependent local unfolding, allowing conversion of non-functional to functional RNA structure.
Keywords: Ribozyme, RNA chaperone, RNA folding, RNA helicase, RNA-protein interaction
INTRODUCTION
RNAs fold into a diverse array of functional structures and participate in an equally diverse array of cellular processes, from gene expression to protein trafficking and the replication of chromosome ends.1–4 To form these structures, RNAs traverse energy landscapes that include deep valleys, representing stable intermediate conformations, and large barriers between and within folding pathways.5–7 In vitro, nearly every large RNA that has been studied folds through intermediates with non-native structures, referred to as ‘kinetic traps’.5 To continue folding productively, the non-native structures within these trapped intermediates must be disrupted in processes that can also require disruption of native structural elements.8,9
Accumulating evidence indicates that RNA folding in vivo is assisted by chaperone proteins,10–13 which promote folding to functional RNA structures and are subsequently dispensable.14 A critical group of RNA chaperones is the DEAD-box proteins, the largest family of superfamily 2 ‘helicases’.15,16 These proteins participate broadly in RNA-mediated processes, and indeed, at least one DEAD-box or related protein is required for essentially all processes carried out by structured RNAs.15–18 At the most general level, DEAD-box proteins use cycles of ATP binding and hydrolysis to generate tight, regulated binding to a short segment of single-stranded RNA.19–23 This cycle leads to a catalytic repertoire that includes RNA unwinding, annealing, and protein displacement activities in vitro and is thought to underlie in vivo roles in folding, conformational rearrangements, and more diverse functions such as stable clamping.24–30
Autocatalytic group I and group II introns have proven to be valuable model systems for understanding RNA folding, structure, and function.5,31–34 The mitochondrial (mt) genome of S. cerevisiae encodes nine group I introns and four group II introns, all of which require the DEAD-box protein Mss116p for efficient splicing in vivo.35 These introns differ substantially in their structural features and global architectures, suggesting that the roles played by Mss116p in their splicing reflect non-specific interactions with RNA. Further, other DEAD-box proteins are able to mitigate the defects from loss of functional Mss116p.36–39 These proteins include the cytoplasmic S. cerevisiae protein Ded1p, which functions in cellular compartments that lack group I and II introns and therefore does not function naturally in folding of these introns.
The mechanism by which DEAD-box proteins promote group I intron splicing has been analyzed biochemically. Mss116p and its Neurospora crassa homolog CYT-19 interact with cognate and non-cognate group I introns and accelerate conformational transitions, including those from kinetically-trapped, misfolded intermediates to the native states.10,38,40–42 These observations and the established propensity of group I introns to misfold5,7,43 have led to models in which the principal function of DEAD-box proteins in group I intron folding is to disrupt structure non-specifically, allowing misfolded intermediates additional opportunities to fold to the native state.7,10,11,39,44
Analogous models have been proposed for Mss116p and CYT-19 in group II intron folding.27,34–36,39 Group II introns consist of six domains with a complex set of local and long-range tertiary contacts that generate a functional structure with an active site for splicing, which is in many cases stabilized by the binding of specific proteins.34,45–47 To understand how Mss116p functions, attention has focused on the yeast mtDNA group II introns that are its natural substrates. Two of these introns, aI1 and aI2, are closely related group IIA introns and encode maturase proteins, which are required for structural stabilization during RNA splicing. In the absence of Mss116p in vivo, unspliced precursor RNA accumulates in a complex with the maturase, suggesting that Mss116p can function at a step after stable maturase binding, hypothesized to be the resolution of kinetic traps.35 The other two yeast mt group II introns, aI5γ and bI1, are small subgroup IIB introns that do not encode maturases, and their splicing is accelerated by Mss116p and CYT-19 in vivo and under near-physiological conditions in vitro.35–38 Although the specific folding steps accelerated by Mss116p remain to be established, biochemical studies showed that Mss116p functions on bI1 as an RNA chaperone, as it promotes ATP-dependent formation of an active intron structure and is then dispensable for activity.36
The mechanisms of acceleration for the remaining yeast group II intron, aI5γ, have been the subject of debate. This intron was the first for which self-splicing via lariat formation was demonstrated48 and thereafter has been used as a model for RNA folding and catalysis.33 Early studies used a shortened version of the intron termed D135, which lacks exons and some intron domains and functions as a ribozyme by cleaving an RNA oligonucleotide substrate, and were done at elevated temperature and high ion concentrations (42 °C, 100 mM Mg2+, 500 mM KCl). Under these conditions, hydroxyl radical footprinting and catalytic activity measurements suggested concerted folding in 1–2 min (0.6 min−1).49,50 The denaturant urea did not accelerate folding, consistent with the absence of a rate-limiting kinetic trap.49
In contrast, aI5γ splicing is accelerated by Mss116p and other DEAD-box proteins in vitro under near-physiological conditions (30 °C, 8 mM Mg2+, 100 mM KCl).36–38,51 Under these conditions, a native gel shift assay showed that folding of a D1356 ribozyme derivative to compact species is slower and more complex, giving as many as four kinetic phases with rate constants spanning at least three orders of magnitude (>1 min−1 to <10−3 min−1).52,53 The multiple phases suggest multiple pathways and rate-limiting steps, and the time scale of hours for major populations would be surprising for intramolecular diffusive processes. Nevertheless, the compaction rates of the slow, dominant phases are unaffected by urea, suggesting that if there are rate-limiting kinetic traps, they do not require substantial regions of buried RNA to become exposed to solvent in the transition state ensemble.52
Starting with the conclusion that the folding of aI5γ is not rate-limited by the resolution of kinetic traps, Solem et al suggested that Mss116p and other DEAD-box proteins promote splicing of aI5γ without unwinding RNA by binding and stabilizing an on-pathway intermediate required for intron compaction.37 A central piece of evidence was the behavior of an Mss116p mutant in which the SAT sequence of motif III is mutated to AAA (abbreviated SAT/AAA).54 This mutant was reported to promote in vitro splicing of aI5γ with near-wild-type efficiency but to be inactive in unwinding of moderately-long RNA duplexes (12–18 bp).37 A subsequent analysis, however, revealed that this mutant retains residual ATP-dependent unwinding activity for shorter RNA duplexes (6–10 bp), of a length more representative of the helices in group II introns, and that the decrease in unwinding efficiency can be as large as the decrease in splicing activity under the same solution conditions (8–28-fold).51 Thus, this mutant did not provide experimental support for the suggestion that Mss116p promotes aI5γ splicing without unwinding RNA.51
The experiments above used a standard construct of aI5γ, which includes the 887-nt intron and relatively long exons (~300 nt, termed LE construct). Recently, the same authors compared splicing of the LE construct and a construct containing short exons of <30 nt (SE). They concluded that Mss116p functions on the LE construct as an ATP-dependent RNA chaperone to resolve kinetic traps involving exons but promotes splicing of the SE construct by binding and stabilizing an on-pathway intermediate.55 A key basis for this conclusion was the finding that although the SAT/AAA mutant was compromised in promoting splicing of the LE construct, it appeared to be as efficient as wild-type Mss116p for the SE construct. Other work showed that Mss116p accelerates compaction of an RNA that consists only of aI5γ domain I (DI),53 suggesting that this step is accelerated during folding of the SE construct.37,53 However, acceleration of DI compaction is ATP-independent, whereas acceleration of splicing for the SE construct, like the LE construct, requires ATP. This difference suggests either that one or more other folding steps for the SE construct require ATP-dependent RNA unwinding or that ATP hydrolysis is required to accelerate dissociation of Mss116p following an ATP-independent role.53,55 Finally, recent single molecule studies with the D135 ribozyme revealed that Mss116p-promoted folding involves an initial ATP-independent step, presumably compaction of DI, and at least one later ATP-dependent step.56 The authors favored the interpretation that this later step involves dissociation of bound Mss116p to permit further RNA folding but left open the possibility of ATP-dependent RNA unwinding to resolve a kinetic trap.
To delineate the roles of Mss116p in aI5γ splicing, here we compare Mss116p-promoted folding of three constructs, all under the same near-physiological conditions. In addition to the standard LE construct, with 5′ and 3′ exons of 293 and 321 nt, respectively,57 we use a version with greatly shortened exons of 28 and 15 nts (SE construct),53,55,58 and the D135 ribozyme (Fig. 1).59 We show that the ability of Mss116p to promote folding is ATP-dependent for all of the constructs, and SAT/AAA and other mutants that are deficient in RNA-unwinding activity are deficient in promoting splicing of both the SE and LE constructs. Catalytic activity measurements with the D135 ribozyme and additional splicing assays with the SE and LE constructs indicate that a major role of ATP is to promote the disruption of RNA structure by Mss116p. Last, the relative abilities of Mss116p mutants to support ATP-dependent splicing and RNA unwinding in vitro correlate well with their abilities to support aI5γ splicing in vivo in a strain that lacks all other mt group I and group II introns. Together our results indicate that the physiological function of Mss116p in aI5γ splicing includes a critical role for ATP-dependent RNA unwinding to resolve inactive structures.
Fig. 1.

RNA constructs. (a) Long exon (LE) and short exon (SE) splicing constructs. Each intron domain is shown in a different color and indicated with a label, and exon lengths are indicated. (b) D135 ribozyme construct. Truncated and intact domains are shown in the same colors as in panel a, and the intact domains are labeled. As indicated, the ribozyme includes 37 nts at the 3′ end, which are derived from a multiple cloning site.59 The secondary structures were generated by modifying a diagram from the Comparative RNA Website.75
RESULTS
Splicing of LE and SE constructs and Mss116p acceleration
To probe how exons influence the mechanisms of Mss116p-promoted folding of the aI5γ intron, we first compared self-splicing of the LE and SE constructs. Under near-physiological conditions in vitro, splicing was considerably faster for the SE construct, consistent with a recent report using essentially the same constructs and conditions (Fig. 2a and Fig. S1).55 At 30 °C, the rate constant for the SE construct determined from the disappearance of precursor and appearance of lariat and linear intron was 1.4 (± 0.5) × 10−3 min−1, whereas splicing of the LE precursor was 80-fold slower (1.8 (± 0.5) × 10−5 min−1). Both constructs spliced faster at higher temperatures, with the LE construct displaying a greater temperature dependence (Fig. 2b). Thus, one or both long exons slow splicing and add to the enthalpic barrier, consistent with previous findings that exon sequences can strongly affect aI5γ splicing.58 Fig. 2b also shows that the splicing rates of both constructs level and begin to decrease with increasing temperature, suggesting that the active structures are unstable at higher temperatures. It is notable that this transition occurs at higher temperature for the LE construct, suggesting that the longer exons stabilize the native structure in addition to slowing its formation.
Fig. 2.
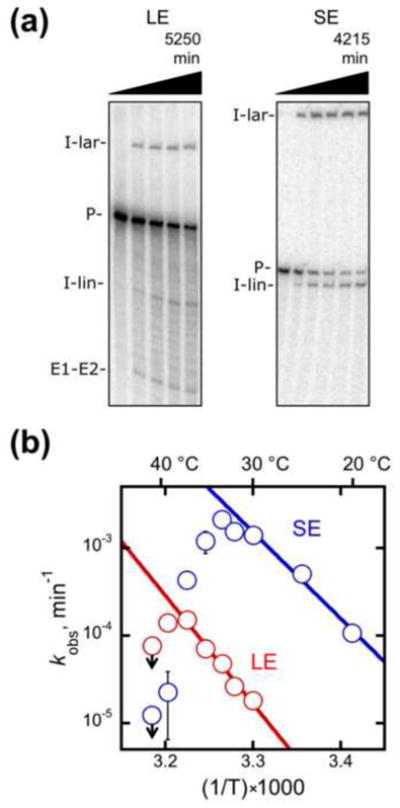
Self-splicing of the LE and SE constructs. (a) Gel images from splicing reactions performed at 33 °C. Splicing was monitored as the fraction of radiolabeled material present as precursor RNA (see Materials and Methods). Bands in this and subsequent gel images are precursor (P), excised intron lariat (I-lar), excised linear intron (I-lin), and ligated exons (E1–E2). (b) Temperature dependences for self-splicing of the LE (red) and SE (blue) constructs. Rate constants, as determined from single exponential fits or from initial rates of slower reactions, are plotted on a log scale against the inverse of temperature (in Kelvin) multiplied by 1000. Reaction conditions were 50 mM Na-MOPS, pH 7.0 (determined at 25 °C), 8 mM MgCl2, 100 mM KCl (including a contribution from Mss116p storage buffer), 1 mM ATP-Mg2+, and 10% Mss116p storage buffer (see Materials and Methods). All values shown reflect the average and standard error from 2–4 independent determinations except those at 41 °C, which reflect a single determination. Downward arrows indicate that non-specific products were observed under those conditions, allowing determination of only an upper limit on the rate constant for splicing. The regions in which splicing rate increased with temperature gave ΔH values of 57 kcal/mol and 44 kcal/mol for the LE and SE constructs, respectively.
We then measured Mss116p-accelerated splicing of the SE construct. Consistent with a recent report,55 the concentrations of Mss116p used previously for the LE construct39,51 gave strong inhibition with the SE construct, and we therefore used lower concentrations of Mss116p and RNA (1 nM RNA). For comparison, we performed experiments with the LE construct under these conditions. Splicing of both constructs was stimulated by low concentrations of Mss116p, just slightly in excess of RNA, in the presence of ATP (Fig. 3a and Fig. S2). In general, the time courses were well described by single rate constants, with endpoints of >90% for reactions that reached completion within the observation time (Fig. 3b). Both constructs required ATP for maximal stimulation, although SE construct splicing was weakly stimulated in the absence of ATP (Fig. 3b). Analogous ATP-independent stimulation was observed previously for group I intron splicing by Mss116p38 and at a very low level for the aI5γ LE construct by the E. coli DEAD-box protein SrmB.39
Fig. 3.

Mss116p-stimulated splicing of the LE and SE constructs. (a) Gel images of LE and SE splicing in the presence and absence of Mss116p (10 nM and 2 nM Mss116p for the LE and SE constructs, respectively). In this and all other experiments with Mss116p, the total intensity of radiolabeled RNA in the quantified region of the gel decreased less than 2-fold during the experiment, indicating minimal RNA degradation. (b) Progress curves of LE and SE splicing in the presence of various Mss116p concentrations, as indicated by color. Circles show results from reactions in the presence of 1 mM ATP and triangles indicate reactions without ATP. Points represent the fraction of labeled material present as precursor as a function of time, and the curves reflect the best fit by a first-order rate equation. (c) Mss116p concentration dependences for splicing of the LE and SE constructs in the presence of ATP. For the SE construct, Mss116p was saturating or nearly saturating even at low concentrations equivalent to that of the radiolabeled RNA (~1 nM), preventing determination of a second-order rate constant for activation.
As a quantitative benchmark for comparison, we measured the Mss116p concentration dependences (Fig. 3c). In earlier work with higher RNA and protein concentrations, acceleration of the LE construct by Mss116p and other DEAD-box proteins gave sigmoidal dependences, suggesting roles for multiple protomers,39 although the upward curvature was more obvious for the other proteins and had previously gone undetected for Mss116p.51 Here, across the limited range of stimulatory protein concentrations, the dependence was approximately linear, giving a second-order rate constant of 4.7 × 106 M−1 min−1. Although the possibility of a higher-order dependence cannot be excluded, for simplicity we use the linear dependence for comparison with the SE construct and Mss116p mutants.*
We found that Mss116p stimulates splicing of the SE construct at least as efficiently as it stimulates splicing of the LE construct (>4.2 × 106 M−1 min−1, Fig. 3c).51 Only a lower limit could be determined for the SE construct because even at the lowest attainable protein concentration, the splicing rate was not readily distinguished from the maximal value. This plateau for the SE construct results from strong inhibition of splicing by Mss116p at concentrations as low as 4 nM, considerably lower than for the LE construct (Fig. 3b,c and S2).51 Together, these results indicate that the long exons cause or exacerbate slow folding, Mss116p stimulates splicing of both the LE and SE constructs, and that maximal Mss116p stimulation of both constructs requires ATP.
Mutants that are deficient in RNA-unwinding activity
To explore further the properties of Mss116p required for acceleration of aI5γ splicing, we tested three mutants compromised in ATP-dependent RNA unwinding for stimulation of splicing of the LE and SE constructs. The SAT/AAA mutant gave an efficiency of 4.2 × 105 M−1 min−1 for the LE construct (Fig. 4), 10-fold lower than wild-type Mss116p (WT) and within the range of relative rate constants found previously.51 For the SE construct, the efficiency for the SAT/AAA mutant was 8.9 × 105 M−1 min−1, at least 4- to 5-fold lower than WT (a minimum estimate because, as noted above, we could only determine a lower limit for WT). Thus, the decrease in efficiency for the SAT/AAA mutant with the SE construct is comparable and could be even larger than for the LE construct. Acceleration of the SE construct by Mss116p SAT/AAA is ATP-dependent, with little or no ATP-independent acceleration up to at least 10 nM protein (Fig. 4b). Additionally, two mutants in motif I, K158A and K158R, which are defective in ATP binding and hydrolysis and in RNA unwinding,37 failed to stimulate splicing of either the LE or SE construct at any concentration tested (≤15 nM; Fig. S3).
Fig. 4.

Stimulation of splicing by the SAT/AAA mutant of Mss116p. (a) Progress curves of splicing of LE (left) and SE (right) constructs in the presence and absence of the Mss116p SAT/AAA. Circles indicate reactions in the presence of 1 mM ATP and triangles indicate reactions without ATP. (b) Concentration dependence of Mss116p SAT/AAA for stimulation of splicing of the LE and SE constructs in the presence of ATP.
These results show that mutations that decrease RNA-unwinding activity commensurately reduce the ability of Mss116p to stimulate splicing of both the SE and LE constructs. Our conclusion that the SAT/AAA mutant is similarly deficient in splicing of both constructs disagrees with the conclusion but not the data from the earlier work55 (see Discussion). The similar requirements for the SE and LE constructs could reflect that Mss116p accelerates a common folding step of these two constructs or that different folding steps coincidentally give similar dependences on Mss116p and ATP.
Two-stage, discontinuous catalytic activity assay for folding of D135 RNA
Previously, the folding of the D135 ribozyme was measured by catalytic activity using a continuous assay, in which folding and substrate cleavage occur simultaneously,50 but this assay can give information on the folding rate only under conditions in which the rate of substrate cleavage exceeds that of folding (e.g. 100 mM Mg2+ at 42 °C).49 To follow folding at lower Mg2+ concentrations, where catalytic activity is weaker, we designed a two-stage, discontinuous activity assay (Fig. 5a). Folding takes place in stage 1, and then portions of the folding reaction are transferred at various times into a second set of conditions (stage 2). This second set of conditions must allow substrate cleavage by ribozyme that has already folded to the native state at the time of transfer but inhibit the remainder of the population from reaching the native state on the time scale of the cleavage reaction.60
Fig. 5.
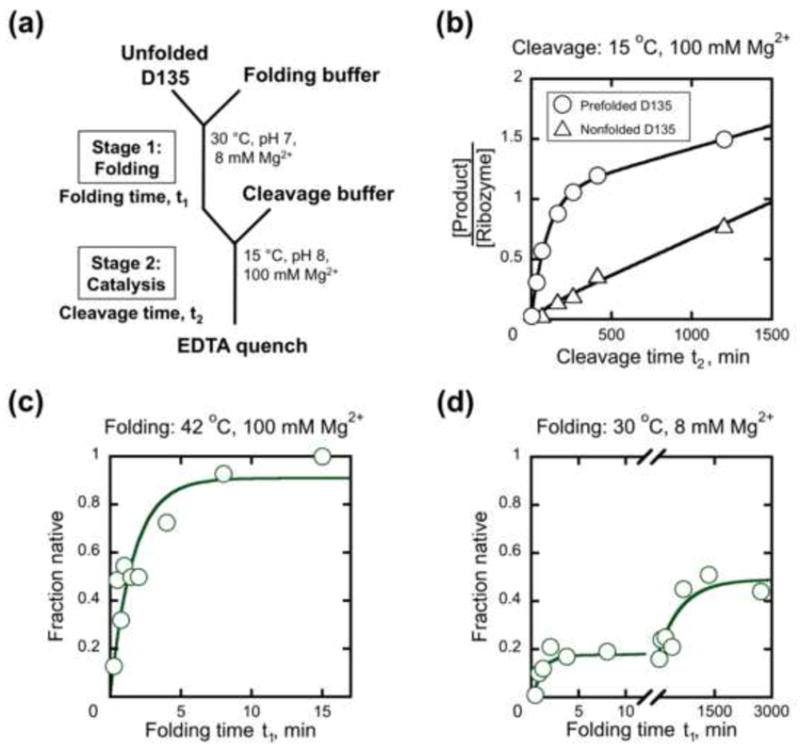
Two-stage catalytic activity assay to monitor folding of D135 ribozyme. (a) Reaction schematic depicting standard, near-physiological conditions for stage 1. In subsequent panels, results are shown plotted against folding time in stage 1 (t1) or cleavage time in stage 2 (t2). (b) Prefolded and nonfolded controls. Ribozyme was either prefolded (circles, 42 °C, 100 mM MgCl2, 500 mM KCl, 50 mM Na-MOPS, pH 7.0, 20 min) or transferred to stage 2 directly from buffer solution (triangles, 30 °C, 50 mM Na-MOPS, pH 7.0). As expected for a single turnover followed by slow product release, the reaction with prefolded ribozyme gave a stoichiometric burst of product (see Materials and Methods) and a second phase reflecting subsequent turnovers of the ribozyme. The nonfolded ribozyme reaction gave less substrate cleavage, with five determinations giving an average burst corresponding to 15% of the ribozyme population. (c) Progress of D135 RNA folding in stage 1 under non-physiological conditions similar to those used previously (42 °C, 100 mM Mg2+, pH 8.1),49 with the fraction of native ribozyme determined from the burst amplitude of substrate cleavage in stage 2. (d) Progress of D135 folding under near-physiological conditions. Burst amplitudes are normalized by the maximal burst amplitude from prefolded ribozyme, as shown in panel b. Fifteen determinations gave a fast phase with an amplitude of 0.22 ± 0.02 and a rate constant of 1.0 ± 0.2 min−1. A slower phase was also present, and six determinations for this phase gave a rate constant of 1.4 (± 0.6) × 10−3 min−1 and a final amplitude of ~0.6.
Based on previous work on group I intron ribozymes,9,61,62 we tested whether low temperature and high Mg2+ concentration would provide suitable conditions for stage 2. When D135 was prefolded to the native state under established conditions (42 °C, 100 mM Mg2+, 20 min), then transferred to 15 °C with 100 mM Mg2+, and a small excess of substrate was added, an initial phase of substrate cleavage gave a rate constant of 8.7 (± 0.4) × 10−3 min−1 and an amplitude approximately equal to one turnover of the ribozyme (Fig. 5b). Cleavage of the remainder of the substrate was slower, presumably because at least one of the cleavage products is released slowly from the ribozyme and limits subsequent turnovers. The 5′ portion of the substrate base pairs with exon-binding site (EBS) 1 and 2 of the ribozyme and is therefore a strong candidate for slow release.
Although a cleavage rate constant of ~9 × 10−3 min−1 indicates a rather slow reaction at 15 °C, it is within the expected range considering that the reaction proceeds at 1 min−1 under the same solution conditions at 42 °C. We reasoned that 15 °C and 100 mM Mg2+ could be suitable for stage 2 if folding to the native state under these conditions is even slower than substrate cleavage by the native ribozyme. Indeed, we found that ribozyme that was transferred directly from buffer solution in the absence of Mg2+ into stage 2 gave substantially less product formation (Fig. 5b, termed ‘nonfolded’ control), indicating that most of the ribozyme did not fold to the native state on the time scale of the cleavage reaction. The low temperature of stage 2 was critical, as a parallel experiment in which stage 2 was 30 °C gave minimal difference between the prefolded and nonfolded reactions (Fig. S4). Nevertheless, even at 15 °C a small amount of product (15%) appeared in the nonfolded control with roughly the same rate constant as product in the prefolded reaction (Fig. 5b). This product formation most likely reflects a subpopulation of the ribozyme that was able to fold rapidly to the native state in stage 2. In subsequent experiments, we normalized the data to account for this subpopulation (see Materials and Methods).
We next measured the kinetics of native folding in stage 1 by incubating the D135 ribozyme for various times and then transferring portions into stage 2 and measuring the burst amplitude from time courses of substrate cleavage (see Fig. 5a). As an initial control, we monitored folding under conditions similar to those used previously in the continuous activity assay (42 °C, 100 mM Mg2+). As expected, this experiment gave essentially the same result as the continuous assay, with complete folding to the native state occurring with a rate constant of 0.5 ± 0.1 min−1 (Fig. 5c).49 As indicated above, data were normalized by the fraction of ribozyme that reached the native state rapidly upon transfer from buffer solution in the absence of Mg2+, which was larger when the ribozyme was transferred from 42 °C (Fig. S5).
We next measured folding of D135 at the near-physiological conditions used for the LE and SE constructs. Under these conditions, folding was slower and more complex, with at least two phases (Fig. 5d). A minor fast phase gave a rate constant of 1.0 (± 0.2) min−1, and a second phase gave a rate constant of 1.4 (± 0.6) × 10−3 min−1 with an apparent endpoint of ~50–60% native ribozyme. These results are crudely consistent with those from a published gel-shift assay used to measure compaction of the related D1356 ribozyme under similar conditions, which gave two observed phases (0.16 min−1 and 0.006 min−1), an unresolved fast phase (>1 min−1), and a slower phase (~10−3 min−1, complete in 24 h).53 It is not clear whether the apparent endpoint in our experiment reflects equilibration of native and non-native forms or whether an unobserved third phase would give still more native RNA at longer folding times. Regardless, the results show that under the near-physiological conditions, D135 folds in a complex, multiphasic process that differs from the single-phase folding at higher temperature and Mg2+ concentrations.49,50,63
Acceleration of D135 folding by Mss116p
We next used the discontinuous activity assay to probe the effects of Mss116p on D135 ribozyme folding under near-physiological conditions. Addition of Mss116p and ATP with Mg2+ gave rapid accumulation of the native ribozyme (Fig. 6a), reducing the folding process to a single phase with a rate constant of 0.61 ± 0.07 min−1 and an apparent endpoint of 0.76 ± 0.04 (Fig. 6b). While this rate was slightly slower than that for the minor fast phase without Mss116p, the major effect of Mss116p was to increase the amplitude of the rapid native state formation. The rate constant did not change systematically across the accessible range of Mss116p concentrations (Fig. S6a), preventing determination of a second-order rate constant for acceleration. However, as observed with the LE and SE constructs, higher Mss116p concentrations were inhibitory. In contrast to the inhibition of splicing, which gave a decreased rate constant, here the inhibition gave a decreased endpoint. This result suggested either that higher concentrations of Mss116p can bind and trap folding intermediates, limiting the extent of native ribozyme formation, or that Mss116p can unfold the native ribozyme, generating a steady-state mixture of native and non-native ribozyme.
Fig. 6.
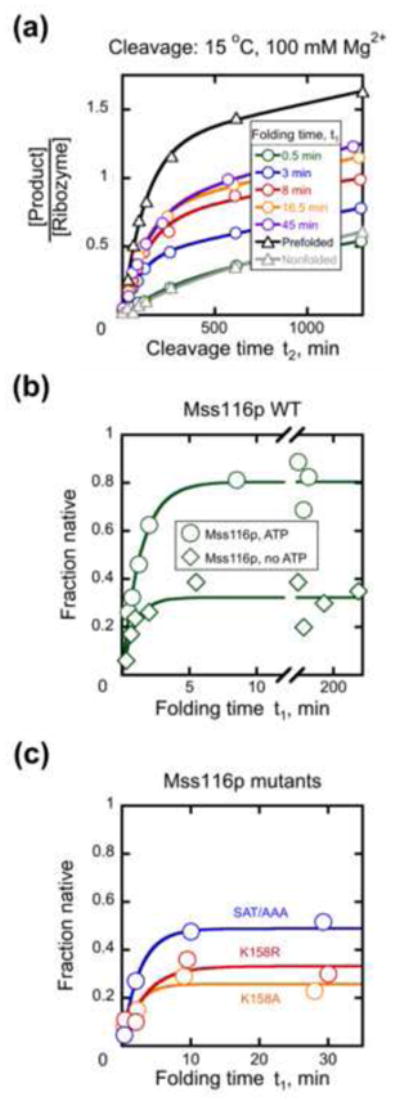
Mss116p accelerates native folding of the D135 ribozyme. (a) Time courses of substrate cleavage in stage 2 after incubation with Mss116p under near-physiological conditions in stage 1. D135 ribozyme (50 nM) was incubated with 100 nM Mss116p and 1 mM ATP for the indicated times before substrate cleavage was measured under stage 2 conditions (see Materials and Methods). Also shown are a ‘nonfolded’ control reaction, in which D135 was added directly to stage 2 conditions, and a prefolded control, in which D135 was incubated at 42 °C for 20 min in the presence of 100 mM Mg2+ to form native ribozyme. (b) Progress of D135 folding in stage 1 in the presence of 100 nM Mss116p with 1 mM ATP (circles, kobs = 0.61 ± 0.07 min−1, amplitude = 0.76 ± 0.04) or without ATP (diamonds, kobs = 0.64 ± 0.07 min−1, amplitude = 0.28 ± 0.01). The fraction of native ribozyme was determined by the burst amplitude in stage 2, as described in Materials and Methods. (c) Progress of D135 folding in stage 1 with Mss116p mutants in the presence of 1 mM ATP and 100 nM protein. Proteins are Mss116p SAT/AAA (blue, kobs = 0.5 ± 0.2 min−1, amplitude = 0.50 ± 0.09), Mss116p K158A (orange, kobs = 0.7 ± 0.1 min 1, amplitude = 0.34 ± 0.06), or Mss116p K158R (red, kobs = 0.7 ± 0.2 min−1, amplitude = 0.33 ± 0.01).
We then used the endpoint for the single fast phase as a diagnostic tool to examine the effects of ATP and Mss116p mutations. The enhanced folding by Mss116p was strongly dependent upon ATP, as 100 nM Mss116p by itself gave only a small increase in endpoint relative to the fast phase of a reaction without protein (Fig. 6b, cf., with Fig. 5d). As expected from the ATP requirement, the motif I mutants K158A and K158R also gave only small increases in endpoint (Fig. 6c). The SAT/AAA mutant was partially active, giving higher endpoints than the other mutants but lower than WT Mss116p, especially at lower protein concentrations (Fig. 6c and S6b, cf., with Fig. 6b). Because the rate constants did not depend systematically on the concentration of Mss116p SAT/AAA, we could not determine quantitatively whether the mutant is compromised for D135 folding relative to the WT protein (Fig. S6). Nevertheless, the lower endpoints suggest either that the mutant protein is unable to promote folding of a fraction of the ribozyme population or that it gives a lower steady-state level of native ribozyme.
The role of ATP in acceleration of D135 folding by Mss116p
Next we used the discontinuous assay to probe the role of ATP in Mss116p-mediated folding. It has been suggested that for D135 and the splicing constructs, acceleration of the critical folding step by Mss116p is ATP-independent, but ATP is required to promote dissociation of Mss116p, allowing rapid folding to the native state.53,55,56 The discontinuous activity assay afforded an incisive test of this model, which predicts that Mss116p would be active in the absence of ATP if an alternative means were provided to remove Mss116p prior to the determination of catalytic activity.
Thus, we incubated Mss116p with D135 and Mg2+ for various times in the absence of ATP and then added proteinase K (1 mg/ml) to degrade Mss116p (Fig. 7 and Fig. S7). After additional incubation of up to 60 min to allow further folding of any ribozyme that was ‘poised’ to form the native state following Mss116p dissociation, we transferred aliquots to stage 2 and measured the fraction of native ribozyme by activity. The prediction from the model above was that this reaction would give accumulation of ribozyme that could quickly reach the native state upon removal of Mss116p. However, with 100 nM Mss116p (2-fold excess over D135 ribozyme), removal of Mss116p by proteolysis did not promote rapid native ribozyme formation (Fig. 7a, closed symbols) beyond the level in an equivalent reaction without the proteolysis step (open symbols). Further, inclusion of ADP or the non-hydrolyzable analog AMP-PNP with Mss116p did not lead to significant native ribozyme formation after proteolysis (Fig. S8a). These results most simply suggested that the role of the ATPase cycle is not solely to accelerate Mss116p dissociation.
Fig. 7.
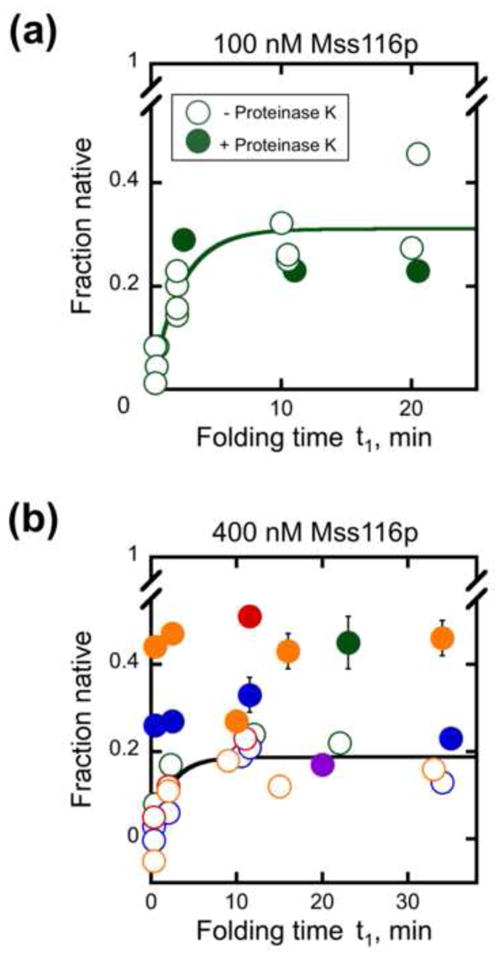
Proteolysis of Mss116p after incubation with D135 RNA in the absence of ATP. Mss116p was added in stage 1 at 100 nM (panel a) or 400 nM (panel b) and incubated for the indicated times under near-physiological conditions. Proteinase K (1 mg/ml) was then added at the indicated times (closed symbols) and incubated for an additional 8 – 60 min to permit further folding before aliquots were transferred to stage 2 and the fraction of native ribozyme was determined by measuring the substrate cleavage burst amplitude. The fraction of native ribozyme did not depend on the incubation time after proteinase K addition (8–60 min), and the symbols show the average values. Open symbols show equivalent reactions with Mss116p and without nucleotide, to which proteinase K was not added. Including or omitting 0.5% SDS with proteinase K to ensure removal of peptide fragments had no significant effect on the results (data not shown). For the experiments shown in panel b, 0.5% SDS was added immediately after proteinase K. Results from independent determinations are shown in different colors. It can be seen that the increase in native ribozyme upon proteinase K treatment is variable and that the variation is larger between experiments than within experiments.
We next considered an extension of the model in which the ATP requirement stems from Mss116p being sequestered at non-productive sites within the RNA, requiring multiple cycles of binding and release to bind productively. Thus, we increased the Mss116p concentration so that sufficient protein would be available even after non-productive sites were filled. With 400 nM Mss116p, 8-fold excess over D135, we observed a variable increase in native ribozyme upon proteolysis (10–25% of the population; Fig. 7b, closed symbols, different colors indicate replicate experiments), which reached an intermediate level between reactions with Mss116p in the presence and absence of ATP (compare with Mss116p+ATP level of ~0.8 shown in Fig. 6b). As above, inclusion of ADP or AMP-PNP with Mss116p gave results that were the same within error as those in the absence of added nucleotide (Fig. S8b). These results indicate that for a fraction of the ribozyme, ATP-independent activity of Mss116p and removal by proteolysis is indeed sufficient to promote folding to the native state. However, much of the ribozyme population remains non-native, most likely because it forms additional intermediates that require further activity of Mss116p.56 Still higher concentrations of Mss116p (up to 1200 nM) did not give more native ribozyme upon proteolysis than observed with 400 nM Mss116p (data not shown).
The variable but significant increase in native D135 ribozyme after Mss116p binding and proteolysis in the absence of ATP prompted us to investigate whether this might also occur with the splicing constructs. Incubation of 1 nM RNA with 2 nM or 10 nM Mss116p in the absence of nucleotide, or in the presence of ADP or AMP-PNP, followed by proteolysis gave no detectable splicing for the LE construct and at most a small increase in splicing rate for the SE construct (0–10%; data not shown). Together the results suggest that for a subpopulation of the ribozyme, ATP hydrolysis and product release are necessary only to accelerate Mss116p dissociation and allow continued folding as proposed.53 Nevertheless, ATP is required for additional steps in Mss116p-dependent folding of much of the D135 population and most or all of the populations of the SE and LE constructs.
Disruption of native D135 ribozyme by Mss116p
A strong prediction of models in which Mss116p functions as a general chaperone is that it would not specifically recognize non-native structure and would therefore be able to use ATP to unfold the native intron structure. This inherently non-specific mechanism can nevertheless lead to native ribozyme accumulation if the native state is more stable than populated misfolded states and therefore unfolded less efficiently or is biased to form intermediates after protein-induced unfolding that preferentially refold to the native state.41
Thus, we used the discontinuous assay to probe for an Mss116p-dependent decrease in the fraction of native D135 ribozyme.60 We prefolded D135 to the native state and then added Mss116p at a concentration that inhibited productive D135 folding (Fig. 8). At various times thereafter, we transferred portions of the reaction to stage 2 and determined the fraction of native ribozyme by activity. In the presence of Mss116p, the fraction of native ribozyme decreased rapidly, suggesting that Mss116p can unfold the native D135 ribozyme to give intermediates that do not readily refold to the native state in stage 2. Although substantial loss of native ribozyme was also observed for Mss116p in the absence of ATP, the decrease was faster and larger with ATP. Mss116p was then proteolyzed and the ribozyme was again incubated under conditions known to give native folding (42 °C, 100 mM Mg2+, 20 min). The fraction of native ribozyme returned to near its original value (Fig. 8, diamonds), confirming that the Mss116p-dependent decrease was due primarily to disruption of the native structure rather than a process giving irreversible inactivation.
Fig. 8.
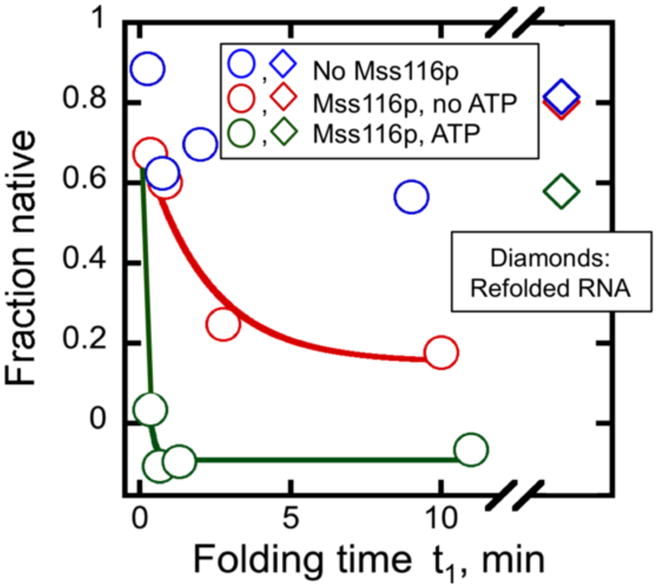
Unfolding of native D135 RNA by Mss116p. The ribozyme (1.8 μM) was prefolded at 42 °C, 100 mM Mg2+ and then diluted to 30 °C and 8 mM Mg2+ prior to addition of Mss116p (1.2 μM Mss116p; 74 nM D135 after dilution). Reactions included 1 mM ATP (green) no ATP (red) or the same volume of storage buffer without Mss116p (blue). Reactions were incubated for the indicated times and the fraction of native ribozyme was determined by transferring aliquots to stage 2 and measuring substrate cleavage. After 12 min, proteinase K (1 mg/ml) and additional Mg2+ (100 mM) were added and the RNA was again folded to the native state by incubation at 42 °C for 20 min prior to determining the fraction of native ribozyme as above (diamonds). This refolding step was included to ensure that the decrease in native ribozyme upon incubation with Mss116p arose from unfolding rather than an irreversible process such as RNA degradation.
Mss116p-promoted splicing of aI5γ in vivo
Finally, to determine which activities of Mss116p are physiologically relevant for aI5γ splicing, we tested whether the Mss116p mutants can promote splicing of aI5γ in vivo (Fig. 9). We used a previously developed in vivo splicing assay in which WT or mutant Mss116p is expressed from a centromere-containing (CEN) plasmid in an MSS116 deletion strain (mss116Δ).35 Because Mss116p is required for the synthesis of mt respiratory components, null mutants cannot grow on non-fermentable carbon sources, such as glycerol, but grow well on raffinose, a non-repressing fermentable sugar, enabling their splicing phenotype to be assessed by Northern hybridization. To eliminate the possibility of indirect effects from defects in splicing of other introns in the COX1 gene, we used isogenic strains containing mtDNAs with the single intron aI5γ.35 The COX1 pre-mRNAs in these strains contain aI5γ with long 5′ and 3′ exons (~1.5 and 0.5 kb, respectively, to the 5′ and 3′ ends of COX1 mRNA), and thus most closely resemble the LE construct used in vitro.
Fig. 9.

Northern hybridization and correlated immunoblot comparing the ability of wild-type and mutant Mss116p to promote splicing of aI5γ in vivo. (a) Northern hybridization. The blot shows whole-cell RNAs (1.0 μg) from the indicated strains separated in a 1.5% agarose gel and hybridized with a 32P-labeled oligonucleotide complementary to COX1 exon 6. Lanes: (1) WT 161-aI5γ (WT); (2) mss116Δ-aI5γ transformed with CEN plasmid pRS416 (empty vector); (3–6) mss116Δ-aI5γ transformed with CEN plasmids expressing Mss116p mutants K158A, K158R, and SAT/AAA. (b) Immunoblot. The blot shows TCA-precipitated proteins (~60 μg) from the same strains as in panel (a) separated in a 4–20% polyacrylamide gradient gel and probed with an anti-Mss116p antibody. (c) Immunoblot stained with AuroDye Forte to confirm equal loading. The numbers to the left of the gel in (b) and (c) indicate the positions of size markers (Precision Plus Protein Dual Color Standards; Bio Rad).
Fig. 9a shows a Northern blot comparing the abilities of WT and mutant versions of Mss116p to promote splicing of aI5γ in vivo. The blot was hybridized with a COX1 exon probe to detect spliced mRNA and unspliced precursor RNA. Because the COX1 gene contains only aI5γ, the blot shows two major COX1 transcripts, corresponding to spliced mRNA and an unspliced precursor RNA, with the ratio of the bands providing a measure of the splicing efficiency. As expected, the wild-type strain, which has a functional chromosomal copy of MSS116, spliced aI5γ efficiently, yielding a predominant band corresponding to COX1 mRNA (lane 1), while the mss116Δ strain accumulates unspliced precursor RNA (lane 2). Expression of WT Mss116p from the CEN-plasmid efficiently complemented the splicing defect in the mss116Δ strain, restoring aI5γ splicing to nearly the level of the wild-type strain (lane 3). By contrast, the K158A and K158R mutants were unable to promote splicing of aI5γ substantially above the low residual level in the mss116Δ strain (null phenotype; lanes 4 and 5), although K158A gave a very small increase that paralleled a small amount of residual ATPase activity.37 The SAT/AAA mutant gave an intermediate phenotype, with approximately equal amounts of unspliced precursor and spliced mRNA (lane 6). We verified by immunoblotting that the mutant proteins were expressed at or near the level of WT Mss116p (Fig. 9b,c). Previous experiments using strains with multiple COB and COX1 introns similarly showed that K158A and other motif I mutants gave a null phenotype, and that the SAT/AAA mutant gave an intermediate phenotype for splicing of aI5γ and all other mt group I and II introns examined.35,54,64 Collectively, the findings for the motif I mutants show that ATP binding and hydrolysis are essential for Mss116p-promoted in vivo splicing of all S. cerevisiae mt group I and II introns. Further, the splicing efficiencies of the motif I and SAT/AAA mutants agree with their activities in splicing and folding aI5γ RNAs in vitro and, as for all other Mss116p mutants examined, correlate with their residual RNA-unwinding activities for appropriately sized duplexes.51
DISCUSSION
Here we used in vitro and in vivo approaches to probe the mechanisms by which the yeast DEAD-box protein Mss116p promotes splicing of the group II intron aI5γ. Our results indicate that a major physiological role of Mss116p in splicing this intron, as with other group I and group II introns, is to use ATP to promote conformational transitions that require the transient disruption of RNA structure.
Requirement for ATP binding and hydrolysis and effects of exon length on Mss116p-mediated splicing in vitro
To probe the functional requirements of Mss116p in aI5γ splicing, we tested mutants that are compromised in ATP binding, hydrolysis, and RNA unwinding, and we used the SE and LE constructs to test whether the ATP requirement depends on the lengths of the flanking exons. The experimental design was essentially the same as in a recent report,55 and where overlapping experiments were performed, the data are largely consistent. Thus, the LE construct splices much slower than the SE construct; both constructs require Mss116p and ATP for accelerated splicing; and with higher concentrations of Mss116p, the splicing rates level and then decrease. The plateau between activation and inhibition occurs at a higher Mss116p concentration and a higher splicing rate for the LE construct than the SE construct. Also consistent with prior work, the mutants K158A and K158R, which are deficient in ATP binding and hydrolysis, are unable to stimulate splicing of either construct.37 Similar results were obtained for other Mss116 and CYT-19 motif I mutants with the LE construct.36,38
However, key conclusions differ from those in previous work.55 Zingler et al compared the rate constants for the LE and SE constructs at Mss116p concentrations near the plateau regions and concluded that the LE construct is stimulated to a greater extent by Mss116p, perhaps reflecting Mss116p recruitment by the exons or facilitation of protein oligomerization. In our view, it is dangerous to interpret these maximum values as a measure of activation because they reflect the intersection of activation and inhibition activities and can be influenced by changes in either or both activities. Instead, we interpreted the slope of the rising portion of the Mss116p concentration dependence, which reflects the overall reaction efficiency — i.e. the free energy change in going from free protein and RNA in solution to the rate-limiting transition states — and is the part of the concentration dependence that is least affected by inhibition. Making this comparison, we observe that Mss116p stimulates splicing of the SE construct at least as efficiently as the LE construct. Thus, rather than stimulating the SE construct less strongly, we conclude that Mss116p inhibits it more strongly.
The slower, more temperature-dependent splicing of the LE construct suggests that the long exons cause or contribute to kinetic barriers in RNA folding. This conclusion is consistent with previous work indicating that sequences in the 5′ exon upstream of the intron-binding sequences (IBS1 and IBS2) can have large effects on aI5γ splicing.58 The inhibition of splicing from exon sequences could reflect the formation of misfolded exon structures that must be resolved prior to splicing, as demonstrated for group I introns.65,66 Alternatively or in addition, the long exons may stabilize structure within the aI5γ intron, presumably by interacting with the intron, thereby increasing barrier heights for disruption of both non-native and native structure. Evidence in support of this possibility comes from the lower, more temperature-dependent splicing rate of the LE construct in the absence of Mss116p, indicating a higher enthalpic barrier, and from the greater stability of the LE construct. The latter is suggested by the higher temperature optimum for LE construct splicing and its greater resistance to inhibition by Mss116p, which presumably reflects disruption of the native intron structure. Our finding that stimulation of both the SE and LE constructs by Mss116p depends upon ATP and is affected similarly by Mss116p mutations raises the possibility of common rate-limiting steps and barriers for these two constructs, which may be hindered by exon stabilization in the LE construct. Further work will be necessary to define the structures of folding intermediates for these constructs and the roles played by exons.
The SAT/AAA mutant is compromised for splicing SE and LE constructs
The motif III mutant of Mss116p (SAT/AAA) follows a design used with eIF4A and other DEAD-box proteins67–69 and was originally described in a Ph.D. thesis from H.R. Huang in Philip Perlman’s lab, where it was classified as a ‘weak allele’ based on in vivo analysis of translation and splicing.54 This mutant has since been the subject of substantial study and contributed to physical models of Mss116p function.37,51 Recently, the Mss116p SAT/AAA mutant was reported to be impaired in splicing of the aI5γ LE construct but as efficient as WT Mss116p for splicing of the SE construct.55 Because it is deficient in RNA-unwinding activity, this result was taken as evidence that RNA unfolding is required for the exons but not within the intron. In contrast, our data show that Mss116p SAT/AAA is reduced in activity by at least a comparable amount for the SE construct as for the LE construct. The difference in conclusions can be understood from inspection of both sets of data. The previous conclusion that Mss116p SAT/AAA promotes splicing of the SE construct with WT efficiency came from a comparison of splicing rates with 15 nM protein, which is well into the inhibitory regime for WT but much closer to the plateau for the SAT/AAA mutant (Fig. 2 of ref. 55). The reduced efficiency of Mss116p SAT/AAA with the SE construct is visible at lower protein concentrations, both in our data (Fig. 3 and 4) and in Fig. S2B of ref. 55. Thus, the SAT/AAA mutant again fails to provide evidence that Mss116p can promote splicing of aI5γ without unwinding RNA.
The roles of ATP in Mss116p-promoted intron folding
Using a new two-stage, discontinuous catalytic activity assay, we found that Mss116p accelerates native folding of the D135 ribozyme under near-physiological conditions, supporting and extending previous studies of Mss116p on folding of aI5γ derivatives.38,53,56 We then used the discontinuous assay to gain new insights into the roles of ATP in Mss116p-mediated folding of aI5γ. It was suggested previously that Mss116p promotes folding by stabilizing an on-pathway folding intermediate in a reaction that is inherently ATP-independent, but that ATP is needed to promote release of Mss116p after this step.55 A key prediction of this model is that removal of Mss116p by proteolysis after binding in the absence of ATP would allow the ribozyme to fold to the native state, which could be detected by activity in stage 2 of our assay. In contrast to this expectation, we found that with two-fold excess Mss116p over D135, proteolysis of Mss116p does not give additional native ribozyme. However, with 8-fold excess Mss116p, proteolysis does allow 10–25% of the ribozyme to reach the native state. The partial recovery indicates that for a subpopulation of the ribozyme, Mss116p accelerates native folding by promoting a step, presumably compaction of domain I,53 without requiring ATP, and it provides experimental support for models in which Mss116p dissociation is required for productive folding.55 Acceleration of domain I compaction may arise from transient binding and stabilization of a folding intermediate, as suggested by the ATP independence and the ability of several basic proteins to promote this step with low efficiency.37,55,56 Alternatively or in addition, Mss116p and other DEAD-box proteins may accelerate this step by disrupting local structure in a process that does not strictly require ATP, perhaps giving transient unstacking of coaxially stacked helices to allow bending of an internal loop within D1.20,51,70
Nevertheless, even under the most favorable conditions, the yield of native D135 ribozyme upon incubation with Mss116p followed by proteolysis is substantially smaller than in the presence of Mss116p and ATP, indicating that most of the population requires Mss116p and ATP for at least one additional step. The fraction that is able to avoid this additional requirement is smaller for the SE construct and undetectable for the LE construct. The increased dependence on ATP with increasing exon length supports the hypothesis that the additional ATP-dependent step(s) include localized RNA unwinding, because the larger size and complexity of these RNAs are expected to lead to folding via more complex pathways with more trapped intermediates, and likely with intermediates of greater stability due to contributions from the exons. It is also reasonable to expect that binding of Mss116p to at least some sites within the intron would be inhibitory throughout the folding process, and thus it is likely that acceleration of Mss116p dissociation is an additional role of the ATPase cycle independent of any structural stabilization.
Disruption of the native D135 ribozyme
Models for DEAD-box proteins as general RNA chaperones postulate that they disrupt RNA structure non-specifically, generating a kinetic redistribution of folding intermediates and additional chances for productive folding.41 A corollary is that there is no absolute mechanism for distinguishing native from non-native structure. In support of this hypothesis, we used the discontinuous activity assay to show that Mss116p can disrupt the native D135 ribozyme in an ATP-dependent manner. This result parallels previous findings for a native group I intron ribozyme41,51 and indicates that Mss116p is capable of promoting native folding of aI5γ by disrupting misfolded intermediates, which are less stable than the native state. Although it was reported that Mss116p cannot unfold the native structure of a domain I construct of aI5γ,53 unfolding may have gone undetected by non-denaturing gel assays if the isolated domain I refolds rapidly to the native structure or to alternative compact forms that migrate similarly to the native structure.
Requirement for ATP binding and hydrolysis by Mss116p in vivo
To investigate which activities of Mss116p are required for its in vivo function, we used yeast strains that lack all mt group I and group II introns except aI5γ.35 The major COX1 pre-mRNA in these strains contains aI5γ with long 5′ and 3′ exons (~1.5 and 0.5 kb, respectively) and thus most closely resembles the LE construct used to analyze the effect of Mss116p in vitro. In other yeast strains that contain multiple COX1 introns, the COX1 pre-mRNAs are expected to be even longer and more heterogenous, depending upon the order in which the introns upstream of aI5γ are spliced.
Our Northern blot analysis indicates that the motif I mutants are essentially inactive for aI5γ splicing, with a very low level of residual activity for K158A, and that Mss116p SAT/AAA is more active than the motif I mutants but nevertheless significantly compromised relative to WT Mss116p, as expected from its decreased RNA-unwinding activity.51 These effects mirror those of the same mutations in vitro, most simply suggesting similar barriers to group II intron folding in vivo and in vitro.
Our results are in agreement with previous in vivo studies of Mss116p mutations in strains containing multiple introns, which showed that K158A and other motif I mutations give a null phenotype for splicing of aI5γ and all other group I and II introns examined, and that SAT/AAA and other motif III mutations give an intermediate splicing-defective phenotype for aI5γ and all other group I and II introns examined.35,54,64 We note in particular that the initial characterization of the SAT/AAA mutation in a strain with three COX1 introns showed that —this mss116 allele does not function as well as the wild-type MSS116 on splicing 54 (in contrast to statements in the Discussion of ref. 55). Although another mutant, Q412A, has been suggested to promote efficient RNA splicing in vivo despite low RNA-unwinding activity,55,64 its unwinding activity was assayed only with a relatively long duplex (17 bp) and was greater than that of a motif III mutant (T307A) in the same study. Thus, in our view, the simplest interpretation is that Q412A supports splicing by virtue of its residual RNA-unwinding activity, which is expected to be higher for shorter duplexes of the type found in group I and II intron RNAs.
Conclusions and implications
Together, our results indicate that a critical activity in Mss116-mediated folding of the aI5γ intron is ATP-dependent RNA-unwinding activity. ATP is required for maximal stimulation of folding and splicing of all constructs tested, and Mss116p is capable of disrupting even the most stable global structure of the intron, the native state. Further, for all Mss116p mutants analyzed to date, the ability to promote splicing of aI5γ in vitro and in vivo correlates with RNA-unwinding activity with appropriately sized duplexes, suggesting that this unwinding activity is necessary for stimulation of aI5γ splicing. Nevertheless, it remains likely that different RNAs and different folding intermediates require different activities and that DEAD-box proteins use multiple mechanisms to promote RNA folding. The recent advances in understanding the folding of aI5γ make this RNA an attractive system for further dissection of the specific folding transitions accelerated by DEAD-box proteins and the detailed mechanisms through which the accelerations are achieved.
MATERIALS AND METHODS
Recombinant Plasmids
pMAL-Mss116p, used to express Mss116p from E. coli for biochemical studies, contains the Mss116p coding sequence (codons 37–664) with an in-frame N-terminal MalE fusion cloned downstream of a tac promoter in the expression vector pMAL-c2t.38,39 pHRH197, used to express Mss116p in S. cerevisiae for genetic analysis, contains the MSS116 gene with its endogenous promoter cloned in the centromere-containing (CEN) plasmid vector pRS416.71 Sequences encoding Mss116p mutations (K158A, K158R, S305A/T307A) were introduced into these plasmids by Quikchange mutagenesis (Stratagene). pJD20 encodes the long-exon (LE) construct downstream of a phage T7 promoter in Bluescribe.57 The LE construct is a 1501-nt precursor RNA that includes a 293-nt 5′ exon, the 887-nt aI5γ intron, and a 321-nt 3′ exon. The 5′ exon consists of 20 nt derived from the vector (plus three guanosines from the T7 promoter) and 270 nt of COX1 5′-exon sequence, and the 3′ exon consists of 291 nt of COX1 exon sequence followed by 30 nt of vector sequence. Plasmid pUC19::aI5γ-SE, which encodes the SE construct, was created by PCR amplification of pJD20 using the primers 5′-TAATACGACTCACTATAGGGACTTACTACGTGGTGGGAC-3′ and 5′-TTGATAATACATAGTATCCCGATAGGTAGACC-3′, which added a T7 promoter (underlined). The resulting PCR product was re-amplified with the primers 5′-GCCCcatatgTAATACGACTCACTATAGGG and 5′-GGGCaagcttAATACATAGTATCCCGATAGG to add NdeI and HindIII sites (lowercase), and cloned between the corresponding sites of pUC19. The SE construct is a 930-nt precursor RNA that includes a 28-nt 5′ exon, the 887-nt aI5γ intron, and a 15-nt 3′ exon. The 5′ exon consists of three guanosines from the T7 promoter and 25 nt of COX1 5′-exon sequence, and the 3′ exon consists of 11 nt of COX1 exon sequence followed by 4 nt of vector sequence. pQL71 encodes the D135 ribozyme (see Fig. 1b).49
RNA preparation
The LE and SE RNA constructs were transcribed in vitro from HindIII-digested plasmids by T7 RNA polymerase in the presence of [α-32P]-UTP (Perkin Elmer) using a Megascript kit (Ambion). RNA was isolated by phenol-chloroform extraction and size exclusion chromatography using two consecutive G-50 columns. D135 RNA was transcribed in vitro from HindIII-digested pQL71 using T7 RNAP and purified via an RNeasy column (Qiagen). The RNA oligonucleotide substrate for D135 (CGUGGUGGGACAUUUUCGAGCGGU) was 5′-end-labeled with [γ-32P]-ATP (Perkin Elmer) by using T4 polynucleotide kinase (New England Biolabs). RNA concentrations were determined by specific activity of the [α-32P]-UTP precursor or spectrophotometrically using extinction coefficients at 260 nm of 1.76 × 107 M−1 cm−1 for the LE construct, 1.17 × 107 M−1 cm−1 for the SE construct, 5.86 × 106 M−1 cm−1 for the D135 construct, and 2.36 × 105 M−1 cm−1 for the D135 substrate oligonucleotide.
Preparation of Mss116p
Wild-type and mutant versions of Mss116p were expressed and purified as described.38,51 After purification, protein was dialyzed overnight against storage buffer solution (20 mM Tris-Cl, pH 7.5, 500 mM KCl, 1 mM EDTA, 1 mM DTT, 50% glycerol), flash frozen, and stored at −80 °C.
Splicing reactions
Splicing reactions were performed in a thermal cycler in 20 or 50 μl (50 mM Na-MOPS, pH 7.0, 100 mM KCl, 8 mM MgCl2, and 5% glycerol). The RNA concentration was 20 nM for reactions shown in Fig. 2 and S1, and it was 1 nM for reactions in the presence of Mss116p (Fig. 3, 4, and S3). Control reactions at the lower concentration in the absence of Mss116p gave rate constants within 2-fold of those at the higher concentration (Fig. 3a and data not shown). When indicated, reactions also included 1 mM ATP, added as a stoichiometric complex with Mg2+. RNA was heated briefly in the absence of Mg2+ (92 °C, 1–2 min), cooled rapidly, and then splicing reactions were initiated by adding RNA to a pre-incubated tube containing splicing buffer solution or by adding RNA and then MgCl2. Portions of reactions were quenched at various times by adding 4 μl of 100 mM EDTA to 2 μl of reactions in Fig. 2 and S1 or by adding 5 μl of 50 mM EDTA, 0.1% SDS, 1 mg/ml proteinase K to 3 μl of reactions in Fig. 3, 4, S2, and S3. Splicing products were separated on a denaturing 4% polyacrylamide gel and quantified using a phosphorimager and ImageQuant TL (GE Healthcare). Rate constants were obtained by fitting the time-dependent decrease in precursor RNA, relative to reaction products, either by an exponential decay or by a line for slow reactions of the LE construct in the absence of Mss116p (Kaleidagraph, Synergy Software). In the latter case the rate constants were inferred from the initial splicing rate. Unless otherwise indicated, rate constants are reported as the average and standard error from 2–4 independent determinations. In the presence of higher concentrations of Mss116p, some splicing reactions included a lag phase. The lag was included in the analysis by allowing the y-intercept of a single exponential equation to vary. Inclusion or exclusion of the lag phase did not significantly affect the rate constant for the slower, major phase of splicing. The origin of the lag is unclear, but its appearance at higher, inhibitory Mss116p concentrations suggests that it originates from a molecular process associated with inhibition by Mss116p.
Discontinuous catalytic activity assay for D135 RNA folding
The discontinuous activity assay for monitoring D135 ribozyme folding consisted of two stages. In stage 1, D135 ribozyme (50 nM) was allowed to fold for various times. The ribozyme was first denatured at 90 °C for 1 min in buffer solution containing 50 mM Na-MOPS, pH 7.0, and then folding was initiated by addition of D135 to a stage 1 reaction (30 °C, 50 mM Na-MOPS, pH 7.0, 100 mM KCl, and 8 mM MgCl2 unless otherwise indicated). ATP was also present (1 mM) when indicated, added as a stoichiometric complex with Mg2+. In stage 2, substrate cleavage was monitored by diluting portions of the stage 1 folding reaction 5-fold into a solution with higher Mg2+ concentration and pH and lower temperature (15 °C, 500 mM KCl, 100 mM MgCl2, 80 mM HEPES, pH 8.1, 1 mg/ml proteinase K). This solution also included the radiolabeled oligonucleotide substrate (30 nM, 3-fold in excess of D135 ribozyme after dilution). Time points (2 μl) were quenched by adding 4 μl of 100 mM EDTA, and the substrate and product were separated in a denaturing 20% polyacrylamide gel.
Cleavage time courses were fit by an exponential phase followed by a linear phase, with the amplitude of the exponential phase reflecting the fraction of native D135 ribozyme.72 After early experiments established the rate constants of the fast and slow phases, the burst amplitude was determined from a single time point at 300 min.61 This time was chosen because it allows completion of the fast phase while minimizing the contribution from the slow phase. Burst amplitudes were normalized and scaled to reflect the fraction of the population that was native at the time of transfer to stage 2. First, the raw burst amplitude was corrected by subtracting an amount representing the fraction of unfolded molecules that fold rapidly in stage 2 (~0.15 under standard conditions). This value was then divided by the value reflecting full native folding, determined in reactions with pre-folded ribozyme, after subtracting an equivalent amount (e.g. 1 − 0.15 = 0.85). Folding rate constants are reported as the average and standard error from at least two independent determinations.
S. cerevisiae Northern hybridizations and immunoblotting
The S. cerevisiae wild-type strain d 161/U7-aI5γ is a derivative of 161-U7 MATa ade1 lys1 ura3, in which the mtDNA contains a single intron aI5γ.35 In the isogenic strain mss116Δ-aI5γ, the MSS116 gene was replaced by a kanr cassette and the resident mtDNA was replaced by cytoduction with mtDNA containing only aI5γ.35 Wild-type and mutant versions of Mss116p with a C-terminal myc tag were expressed from the CEN plasmid pHRH197, which carries a ura3 marker.71 Transformants of the mss116Δ-aI5γ strain containing the CEN-plasmids were selected by plating on 2% agar plates containing 2% dextrose and Hartwell’s complete (HC) media (1X YNB solution, 1X HC dropout 6 amino acid solution, plus 1% each of lysine, tryptophan, and histidine; 0.1% adenine; and 2% leucine) lacking uracil.73
For Northern hybridizations, cells were grown at 30 °C to O.D.600 of 1.0 – 1.6 in Hartwell’s Complete liquid medium containing 2% raffinose and lacking uracil. RNA was isolated as previously described.71 Samples containing 1.0 μg of RNA were denatured by incubating with 5.6% glyoxal in 50% DMSO, 0.1 M NaPO4 at 65°C for 15 min and electrophoresed in a 1.5% agarose gel with RNA-grade 1X TAE (40 mM Tris-acetate, pH 8, 1 mM EDTA) at 25°C. The gels were blotted onto a nylon membrane (Hybond-XL, GE Healthcare) overnight, hybridized with a 5′-end-labeled DNA oligonucleotide probe complementary to COX1 exon 6 (GAATAATGATAATAGTGCAAATGAATGAACC), and scanned with a phosphorimager.
For immunoblotting, cells were grown as above, and proteins were precipitated with trichloroacetic acid as described,74 except that the resulting protein pellets were washed once with 0.5 ml of 1 M Tris base before being resuspended in 150 μl of SDS-PAGE sample buffer. Samples (~60 μg of protein measured by O.D.280) were run in a pre-cast 4–20% polyacrylamide gradient gel (BioRad) with 0.1% SDS in the running buffer and transferred to a Sequi-Blot™ PVDF membrane (BioRad) using a BioRad Criterion blotter apparatus. The blot was probed with anti-Mss116p guinea pig primary antibody (1:5,000 dilution),54 developed using an ECL Plus Western Blotting-kit (GE Healthcare), and imaged using Kodak Biomax XAR film. To confirm equal loading of protein samples, the PVDF membrane was stripped of antibodies using Restore™ Plus Western Blot Stripping Buffer (ThermoScientific) and stained with AuroDye Forte (GE Healthcare) following the manufacturer’s directions.
Supplementary Material
Acknowledgments
We thank members of the Russell and Lambowitz labs and Philip Perlman (Howard Hughes Medical Institute) for comments on the manuscript. This work was supported by NIH grants GM70456 to R.R. and GM37951 to A.M.L.
ABBREVIATIONS
- bp
base pair(s)
- DTT
dithiothreitol
- EDTA
ethylenedinitrilotetraacetic acid
- LE construct
self-splicing construct of the aI5γ intron with long 5′ and 3′ exons of 293 and 321 nucleotides, respectively
- MOPS
3-(N-morpholino)-propanesulfonic acid
- Mss116p SAT/AAA
mutant of Mss116p with S305A and T307A substitutions in motif III
- mt
mitochondrial
- nt(s)
nucleotide(s)
- PVDF
polyvinylidene fluoride
- SDS
sodium dodecyl sulfate
- SE construct
self-splicing construct of the aI5γ intron with short 5′ and 3′ exons of 28 and 15 nucleotides, respectively
- Tris
tris(hydroxymethyl)aminomethane
- WT
wild-type
Footnotes
The efficiency reported in the current work is approximately 5-fold larger than previously reported.39,51 In principle, this difference could be caused by the decreased RNA and protein concentrations, changes in buffer from Tris to MOPS, small accompanying changes in counterion identity and concentration, and/or recent preparations of protein being somewhat more active than earlier preparations. Further experiments indicate that the difference is not caused by the change in buffer conditions, which result in a small decrease in activity (2-fold), whereas there may be contributions from the changes in RNA and protein concentration (≤ 2-fold) and differences in activity between different protein preparations (data not shown).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roth A, Breaker RR. The structural and functional diversity of metabolite-binding riboswitches. Annu Rev Biochem. 2009;78:305–334. doi: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noller HF. RNA structure: reading the ribosome. Science. 2005;309:1508–1514. doi: 10.1126/science.1111771. [DOI] [PubMed] [Google Scholar]
- 3.Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol. 2006;7:484–494. doi: 10.1038/nrm1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egea PF, Stroud RM, Walter P. Targeting proteins to membranes: structure of the signal recognition particle. Curr Opin Struct Biol. 2005;15:213–220. doi: 10.1016/j.sbi.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Treiber DK, Williamson JR. Exposing the kinetic traps in RNA folding. Curr Opin Struct Biol. 1999;9:339–345. doi: 10.1016/S0959-440X(99)80045-1. [DOI] [PubMed] [Google Scholar]
- 6.Tinoco I, Jr, Bustamante C. How RNA folds. J Mol Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 7.Russell R. RNA misfolding and the action of chaperones. Front Biosci. 2008;13:1–20. doi: 10.2741/2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Treiber DK, Rook MS, Zarrinkar PP, Williamson JR. Kinetic intermediates trapped by native interactions in RNA folding. Science. 1998;279:1943–1946. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- 9.Russell R, Das R, Suh H, Travers KJ, Laederach A, Engelhardt MA, Herschlag D. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J Mol Biol. 2006;363:531–544. doi: 10.1016/j.jmb.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 10.Mohr S, Stryker JM, Lambowitz AM. A DEAD-box protein functions as an ATP-dependent RNA chaperone in group I intron splicing. Cell. 2002;109:769–779. doi: 10.1016/s0092-8674(02)00771-7. [DOI] [PubMed] [Google Scholar]
- 11.Lorsch JR. RNA chaperones exist and DEAD box proteins get a life. Cell. 2002;109:797–800. doi: 10.1016/s0092-8674(02)00804-8. [DOI] [PubMed] [Google Scholar]
- 12.Schroeder R, Grossberger R, Pichler A, Waldsich C. RNA folding in vivo. Curr Opin Struct Biol. 2002;12:296–300. doi: 10.1016/s0959-440x(02)00325-1. [DOI] [PubMed] [Google Scholar]
- 13.Zemora G, Waldsich C. RNA folding in living cells. RNA Biol. 2010;7:8–15. doi: 10.4161/rna.7.6.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem. 1995;270:20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 15.Linder P. Dead-box proteins: a family affair--active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linder P. mRNA export: RNP remodeling by DEAD-box proteins. Curr Biol. 2008;18:R297–299. doi: 10.1016/j.cub.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 18.Jankowsky E, Bowers H. Remodeling of ribonucleoprotein complexes with DExH/D RNA helicases. Nucleic Acids Res. 2006;34:4181–4188. doi: 10.1093/nar/gkl410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorsch JR, Herschlag D. The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry. 1998;37:2180–2193. doi: 10.1021/bi972430g. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Potratz JP, Tijerina P, Del Campo M, Lambowitz AM, Russell R. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc Natl Acad Sci USA. 2008;105:20203–20208. doi: 10.1073/pnas.0811075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F, Putnam A, Jankowsky E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc Natl Acad Sci USA. 2008;105:20209–20214. doi: 10.1073/pnas.0811115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henn A, Cao W, Hackney DD, De La Cruz EM. The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J Mol Biol. 2008;377:193–205. doi: 10.1016/j.jmb.2007.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao W, Coman MM, Ding S, Henn A, Middleton ER, Bradley MJ, Rhoades E, Hackney DD, Pyle AM, De La Cruz EM. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-box proteins. J Mol Biol. 2011;409:399–414. doi: 10.1016/j.jmb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 25.Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Jankowsky E, Fairman ME. RNA helicases--one fold for many functions. Curr Opin Struct Biol. 2007;17:316–324. doi: 10.1016/j.sbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Potratz JP, Tijerina P, Russell R. Mechanisms of DEAD-box proteins in ATP-dependent processes. In: Jankowsky E, editor. RNA helicases. 19. Royal Society of Chemistry; 2010. pp. 61–98. Vol. RSC Biomolecular Sciences. [Google Scholar]
- 28.Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol Chem. 2009;390:1237–1250. doi: 10.1515/BC.2009.135. [DOI] [PubMed] [Google Scholar]
- 29.Jarmoskaite I, Russell R. DEAD-box proteins as RNA helicases and chaperones. WIREs: RNA. 2011;2:135–152. doi: 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cech TR. The generality of self-splicing RNA: relationship to nuclear mRNA splicing. Cell. 1986;44:207–210. doi: 10.1016/0092-8674(86)90751-8. [DOI] [PubMed] [Google Scholar]
- 32.Narlikar GJ, Herschlag D. Mechanistic aspects of enzymatic catalysis: lessons from comparison of RNA and protein enzymes. Annu Rev Biochem. 1997;66:19–59. doi: 10.1146/annurev.biochem.66.1.19. [DOI] [PubMed] [Google Scholar]
- 33.Pyle AM, Fedorova O, Waldsich C. Folding of group II introns: a model system for large, multidomain RNAs? Trends Biochem Sci. 2007;32:138–145. doi: 10.1016/j.tibs.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Lambowitz AM, Zimmerly S. Group II introns: mobile ribozymes that invade DNA. In: Gesteland RF, Cech TR, Atkins JF, editors. RNA Worlds: From Life’s Origins to Diversity in Gene Regulation. Cold Spring Harb Perspect Biol; 2010. doi: 1.a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang HR, Rowe CE, Mohr S, Jiang Y, Lambowitz AM, Perlman PS. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc Natl Acad Sci USA. 2005;102:163–168. doi: 10.1073/pnas.0407896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohr S, Matsuura M, Perlman PS, Lambowitz AM. A DEAD-box protein alone promotes group II intron splicing and reverse splicing by acting as an RNA chaperone. Proc Natl Acad Sci USA. 2006;103:3569–3574. doi: 10.1073/pnas.0600332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solem A, Zingler N, Pyle AM. A DEAD protein that activates intron self-splicing without unwinding RNA. Mol Cell. 2006;24:611–617. doi: 10.1016/j.molcel.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 38.Halls C, Mohr S, Del Campo M, Yang Q, Jankowsky E, Lambowitz AM. Involvement of DEAD-box proteins in group I and group II intron splicing. Biochemical characterization of Mss116p, ATP hydrolysis-dependent and -independent mechanisms, and general RNA chaperone activity. J Mol Biol. 2007;365:835–855. doi: 10.1016/j.jmb.2006.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Del Campo M, Mohr S, Jiang Y, Jia H, Jankowsky E, Lambowitz AM. Unwinding by local strand separation is critical for the function of DEAD-box proteins as RNA chaperones. J Mol Biol. 2009;389:674–693. doi: 10.1016/j.jmb.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tijerina P, Bhaskaran H, Russell R. Nonspecific binding to structured RNA and preferential unwinding of an exposed helix by the CYT-19 protein, a DEAD-box RNA chaperone. Proc Natl Acad Sci USA. 2006;103:16698–16703. doi: 10.1073/pnas.0603127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature. 2007;449:1014–1018. doi: 10.1038/nature06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bifano AL, Caprara MG. A DExH/D-box protein coordinates the two steps of splicing in a group I intron. J Mol Biol. 2008;383:667–682. doi: 10.1016/j.jmb.2008.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duncan CD, Weeks KM. SHAPE analysis of long-range interactions reveals extensive and thermodynamically preferred misfolding in a fragile group I intron RNA. Biochemistry. 2008;47:8504–8513. doi: 10.1021/bi800207b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan C, Russell R. Roles of DEAD-box proteins in RNA and RNP folding. RNA Biol. 2010;7:28–37. doi: 10.4161/rna.7.6.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai L, Chai D, Gu SQ, Gabel J, Noskov SY, Blocker FJ, Lambowitz AM, Zimmerly S. A three-dimensional model of a group II intron RNA and its interaction with the intron-encoded reverse transcriptase. Mol Cell. 2008;30:472–485. doi: 10.1016/j.molcel.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michel F, Costa M, Westhof E. The ribozyme core of group II introns: a structure in want of partners. Trends Biochem Sci. 2009;34:189–199. doi: 10.1016/j.tibs.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Peebles CL, Perlman PS, Mecklenburg KL, Petrillo ML, Tabor JH, Jarrell KA, Cheng HL. A self-splicing RNA excises an intron lariat. Cell. 1986;44:213–223. doi: 10.1016/0092-8674(86)90755-5. [DOI] [PubMed] [Google Scholar]
- 49.Swisher JF, Su LJ, Brenowitz M, Anderson VE, Pyle AM. Productive folding to the native state by a group II intron ribozyme. J Mol Biol. 2002;315:297–310. doi: 10.1006/jmbi.2001.5233. [DOI] [PubMed] [Google Scholar]
- 50.Su LJ, Brenowitz M, Pyle AM. An alternative route for the folding of large RNAs: apparent two-state folding by a group II intron ribozyme. J Mol Biol. 2003;334:639–652. doi: 10.1016/j.jmb.2003.09.071. [DOI] [PubMed] [Google Scholar]
- 51.Del Campo M, Tijerina P, Bhaskaran H, Mohr S, Yang Q, Jankowsky E, Russell R, Lambowitz AM. Do DEAD-box proteins promote group II intron splicing without unwinding RNA? Mol Cell. 2007;28:159–166. doi: 10.1016/j.molcel.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fedorova O, Waldsich C, Pyle AM. Group II intron folding under near-physiological conditions: collapsing to the near-native state. J Mol Biol. 2007;366:1099–1114. doi: 10.1016/j.jmb.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fedorova O, Solem A, Pyle AM. Protein-facilitated folding of group II intron ribozymes. J Mol Biol. 2010;397:799–813. doi: 10.1016/j.jmb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang HR. PhD thesis. The University of Texas Southwestern Medical Center; Dallas: 2004. Functional studies of intron- and nuclear-encoded splicing factors in the mitochondria of Saccharomyces cerevisiae. [Google Scholar]
- 55.Zingler N, Solem A, Pyle AM. Dual roles for the Mss116 cofactor during splicing of the ai5γ group II intron. Nucleic Acids Res. 2010;38:6602–6609. doi: 10.1093/nar/gkq530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karunatilaka KS, Solem A, Pyle AM, Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature. 2010;467:935–939. doi: 10.1038/nature09422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jarrell KA, Dietrich RC, Perlman PS. Group II intron domain 5 facilitates a trans-splicing reaction. Mol Cell Biol. 1988;8:2361–2366. doi: 10.1128/mcb.8.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nolte A, Chanfreau G, Jacquier A. Influence of substrate structure on in vitro ribozyme activity of a group II intron. RNA. 1998;4:694–708. doi: 10.1017/s1355838298980165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swisher J, Duarte CM, Su LJ, Pyle AM. Visualizing the solvent-inaccessible core of a group II intron ribozyme. EMBO J. 2001;20:2051–2061. doi: 10.1093/emboj/20.8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wan Y, Mitchell D, Russell R. Catalytic activity as a probe of native RNA folding. Methods in Enzymology. 2009;468:195–218. doi: 10.1016/S0076-6879(09)68010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Russell R, Herschlag D. Probing the folding landscape of the Tetrahymena ribozyme: commitment to form the native conformation is late in the folding pathway. J Mol Biol. 2001;308:839–851. doi: 10.1006/jmbi.2001.4751. [DOI] [PubMed] [Google Scholar]
- 62.Wan Y, Suh H, Russell R, Herschlag D. Multiple unfolding events during native folding of the Tetrahymena group I ribozyme. J Mol Biol. 2010;400:1067–1077. doi: 10.1016/j.jmb.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su LJ, Waldsich C, Pyle AM. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res. 2005;33:6674–6687. doi: 10.1093/nar/gki973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bifano AL, Turk EM, Caprara MG. Structure-guided mutational analysis of a yeast DEAD-box protein involved in mitochondrial RNA splicing. J Mol Biol. 2010;398:429–443. doi: 10.1016/j.jmb.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woodson SA, Cech TR. Alternative secondary structures in the 5′ exon affect both forward and reverse self-splicing of the Tetrahymena intervening sequence RNA. Biochemistry. 1991;30:2042–2050. doi: 10.1021/bi00222a006. [DOI] [PubMed] [Google Scholar]
- 66.Woodson SA, Emerick VL. An alternative helix in the 26S rRNA promotes excision and integration of the Tetrahymena intervening sequence. Mol Cell Biol. 1993;13:1137–1145. doi: 10.1128/mcb.13.2.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pause A, Sonenberg N. Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 1992;11:2643–2654. doi: 10.1002/j.1460-2075.1992.tb05330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karow AR, Klostermeier D. A conformational change in the helicase core is necessary but not sufficient for RNA unwinding by the DEAD box helicase YxiN. Nucleic Acids Res. 2009;37:4464–4471. doi: 10.1093/nar/gkp397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Banroques J, Doere M, Dreyfus M, Linder P, Tanner NK. Motif III in superfamily 2 “helicases” helps convert the binding energy of ATP into a high-affinity RNA binding site in the yeast DEAD-box protein Ded1. J Mol Biol. 2010;396:949–966. doi: 10.1016/j.jmb.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 70.Waldsich C, Pyle AM. A folding control element for tertiary collapse of a group II intron ribozyme. Nat Struct Mol Biol. 2007;14:37–44. doi: 10.1038/nsmb1181. [DOI] [PubMed] [Google Scholar]
- 71.Mohr G, Del Campo M, Mohr S, Yang Q, Jia H, Jankowsky E, Lambowitz AM. Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro. J Mol Biol. 2008;375:1344–1364. doi: 10.1016/j.jmb.2007.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russell R, Herschlag D. New pathways in folding of the Tetrahymena group I RNA enzyme. J Mol Biol. 1999;291:1155–1167. doi: 10.1006/jmbi.1999.3026. [DOI] [PubMed] [Google Scholar]
- 73.Amberg DC, Burke D, Strathern JN. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. CSHL Press; 2005. [Google Scholar]
- 74.Yaffe MP, Schatz G. Two nuclear mutations that block mitochondrial protein import in yeast. Proc Natl Acad Sci USA. 1984;81:4819–4823. doi: 10.1073/pnas.81.15.4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, Pande N, Shang Z, Yu N, Gutell RR. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.