

Abstract

Using an amino acid-based imidazole catalyst, a regiodivergent silylation of chiral diols in cases where there is not a significant steric and electronic difference between the regioisotopic hydroxyl groups has been developed. This transformation allows for the conversion of racemic diols into regioisomeric, enantiomerically enriched, monosilylated products. The utility of this process is highlighted in the efficient enantioselective preparation of a useful synthetic intermediate and the natural product, sapinofuranone A.
Amino acid-modified imidazole 1 was shown to be an effective catalyst for the enantioselective silylation of meso-diols (eq 1, Scheme 1).1 Additional investigations extended the utility of this transformation to the kinetic resolution of racemic 1,2-diols (eq 2).2 In this case, the steric differences between R and R’ of the racemic diols allowed the catalyst 1 to discriminate between the two hydroxyl groups, preferentially silylating the hydroxyl group with the appropriate configuration adjacent to the less hindered R group. Herein, we describe a method for catalytic enantioselective silylation of racemic syn-diols where the two heteroatomic sites are sterically and electronically similar (i.e., R ≈ R’; Scheme 1, eq 3), but not identical. With these substrates, a regiodivergent reaction of the racemic mixture (RRRM) is performed,3 where both enantiomers of the racemic diol are mono-silylated leading to two unique silyl ether regioisomers that can be isolated with appreciable levels of enantiomeric purity (up to 97% ee).
Scheme 1.

Enantioselective silylations of diols
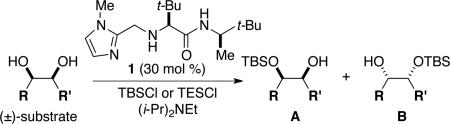
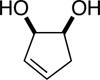
Initial experiments focused on the regioselective silylation of 3-cyclohexene-syn-1,2-diol (2; eq 4). When racemic diol 2 was subjected to imidazole-catalyzed silylation, mono-silylation proceeded without any regio-selectivity. This finding stands in contrast to our earlier study regarding the resolution of racemic diols (ie. eqs 2 & 5). For example, the subtle steric difference between a methyl and an ethyl group within substrate 3 provided sufficient dissimilarity to give rise to preferential silylation of the hydroxyl group that is proximal to the smaller methyl unit (eq 5).
 |
(4) |
 |
(5) |
Since direct access to enantiomerically enriched diol 2 by catalytic enantioselective dihydroxylation of 1,3-cyclohexadiene furnishes products of low enantiomeric purity (e.g., 37% ee),4 and to the best of our knowledge, there are no other direct enantioselective methods available for synthesis of such organic molecules, we decided to investigate enantioselective catalytic silylations of this substrate class. We expected that reactions of diols, such as 2, would provide access to enantiomerically enriched, differentially functionalized, mono-silylated derivatives that could be used in a variety of synthetic applications.
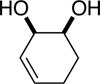
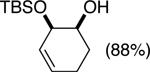
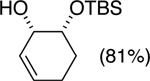
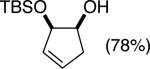
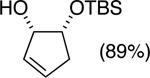
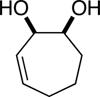
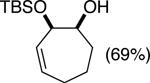
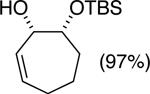
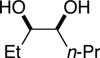
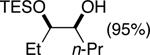
The data shown in Table 1 summarizes our findings in connection with the RRRM of racemic diols through a catalytic enantioselective silylation. In these examples, the catalyst promotes efficient site-selective silylation of each enantiomer of the starting diol.5 Accordingly, when the racemic starting material is subjected to the silylation conditions, the diol substrate is consumed entirely (>98%). It is important to note that none of the bissilylated products are formed under the reaction conditions (<2% by 400 MHz 1H NMR analysis). As illustrated in Table 1, five-, six- or seven-membered ring unsaturated diols serve as suitable substrates (entries 1–3 and 5). Acyclic syn-diols undergo efficient and selective silylation as well (entries 4 and 6).6
Table 1.
RRRM through catalytic enantioselective silylation
 | |||||
|---|---|---|---|---|---|
| entry | substrate | A (ee) | B (ee) | yield (comb.) | ratio A/B |
| (1) |
|
|
|
92%a | 50/50 |
| (2) |
|
|
|
89%b | 58/42 |
| (3) |
|
|
|
89%c | 64/36 |
| (4) |
|
|
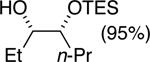
|
90%d | 50/50 |
| (5) |
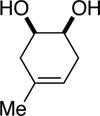
|
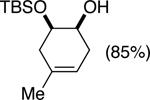
|
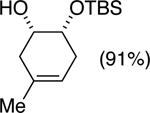
|
75%e | 52/48 |
| (6) |
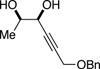
|
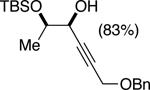
|
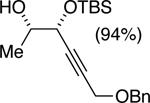
|
50%f | 52/48 |
Conditions:
TBSCl (2 equiv), DIPEA (1 equiv), toluene [1 M], -30 °C, 120 h.
TBSCl (1.5 equiv), DIPEA (1 equiv), toluene [1 M], -60 °C, 120 h.
TBSCl (1.5 equiv), DIPEA (1 equiv), toluene [1 M], -30 °C, 120 h.
TESCl (1.5 equiv), DIPEA (1 equiv), THF [1 M], -60 °C, 72 h.
TBSCl (1 equiv), DIPEA (1 equiv), toluene [1 M], -60 °C, 120 h.
TBSCl (1 equiv), DIPEA (1.25 equiv), THF [1 M], -30 °C, 110 h.
In an ideal RRRM process, both enantiomers of the starting diols are consumed at the same rate and undergo highly site-selective transformation, leading to the formation of two differentially functionalized enantiomerically enriched products. Any deviation from the above scenario can lead to unequal amounts of products, which does affect the enantiomeric purities of the products. This limitation is observed in the reactions illustrated in entries 2 and 3 of Table 1. In these cases, a low level of substrate control during the silylation leads to one regioisomer being formed in greater than 50% yield, but with diminished enantioselectivity. The minor regioisomer, on the other hand, is obtained with higher levels of enantiomeric purity.7
The present regiodivergent silylation strategy can be applied to instances where enantiomerically enriched mixture of substrates, such as those illustrated in Scheme 2, are used as substrates; this process is referred to as a regiodivergent reaction on an enantioenriched mixture (RREM).8 The non-racemic diols (+)-2 and (–)-2 were prepared through the use of the complimentary Sharpless enantioselective dihydroxylation involving AD-mix-α and AD-mix-β with 1,3-cyclohexadiene. The Os-catalyzed transformations thus provide the desired ene-diol enantiomers in high yield, albeit in low enantiomeric purity.4 This limitation can be addressed by employing a second catalytic, enantioselective reaction performed sequentially such that the final product is obtained in higher levels of enantiopurity. For example, when the asymmetric silylation of diol (+)-2 is carried out with catalyst 1, mono-silyl ether (+)-4 can be isolated in 64% yield and 97% ee. If the same transformation is performed with enantiomerically enriched (–)-2, silylated allylic alcohol (+)-5 is generated in 64% yield and 90% ee. It is important to note that, although the idealized case for RRRM can provide a maximum of 50% yield of each product, performing the two aforementioned processes serves to increase the theoretical yield of the products 4 or 5 (since the starting diol 2 is enantiomerically enriched).
Scheme 2.

Combination of RRRM with enantioselective dihydroxylation
The value of the approach outlined above is highlighted through the utilization of (+)-4 in the first total synthesis of sapinofuranone A. This butanolide natural product was first isolated from the liquid cultures of the phytotoxic fungus Sphaeropsis sapinea in 1999 by Evidente and co-workers.11 Our approach to sapinofuranone A begins with ozonolytic cleavage/oxidation12 of enantiomerically enriched monosilyl diol (+)-4 to furnish lactone 6 (Scheme 3); the resulting sensitive α-siloxy aldehyde is used directly in Wittig olefination. After optimization, we were able to obtain the desired diene in 78% yield and 2.4:1 Z:E selectivity for the newly formed olefin. Subsequent removal of the silyl group with TBAF delivers sapinofuranone A in 94% yield.
Scheme 3.

Synthesis of sapinofuranone A
In another demonstration of utility of the method, silyl ether (-)-5 can be converted readily into γ-siloxy-β-methylcyclohexenone 8 (Scheme 4), which has previously found applications in enantioselective syntheses of natural products, such as the kinamycins15 and karahana lactone16. Formation of enantiomerically enriched 8 is accomplished through allylic oxidation (PCC) of mono-silylether (-)-5, generating α-siloxy ketone 7. Alkylation of 7 (MeMgBr) affords a 2:1 mixture of diastereomers that is subjected directly to another oxidation to furnish 8 in 70% yield (ee = 90%).
Scheme 4.

Representative application of RREM products
In summary, the present study demonstrates the enantioselective catalytic silylation of racemic diols that offers access to enantiomerically enriched mono-silylated regioisomers. We also show, through combination with catalytic enantioselective dihydroxylation in a RREM process, that the theoretical yield and enantiopurity of the products can be significantly enhanced. Finally, the utility of the method is demonstrated through the synthesis of the enantioenriched γ-siloxy-β-methylcyclohexenone, as well as the first total synthesis of sapinofuranone A.
Supplementary Material
Acknowledgment
Financial support was provided by the NIH (GM-57212). Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available Experimental details and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org..
References
- 1.Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67–70. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Y, Mitra A, Hoveyda AH, Snapper ML. Angew. Chem., Int. Ed. 2007;44:8471–8474. doi: 10.1002/anie.200703650. For related examples, see: Weickgenannt A, Mewald M, Muesmann TWT, Oestreich M. Angew. Chem., Int. Ed. 2010;49:2223–2226. doi: 10.1002/anie.200905561.Patel SG, Wiskur SL. Tetrahedron Lett. 2009;50:1164–1166.
- 3.Kagan HB. Tetrahedron. 2001;57:2449–2468.Kumar RR, Kagan HB. Adv. Synth. Catal. 2010;352:231–242. The nomenclature associated with this type of reaction manifold, which has also been referred to as parallel kinetic resolution or divergent kinetic resolution, has been the subject of recent discussion. See references 5 and 6.
- 4.Wang Z, Kakiuchi K, Sharpless KB. J. Org. Chem. 1994;59:689–6897. [Google Scholar]
- 5.Jure M, Vedejs E. Angew. Chem., Int. Ed. 2005;26:3974–4001. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 6.For recent representative examples of RRRM, see: Webster R, Böing C, Lautens M. J. Am. Chem. Soc. 2009;131:444–445. doi: 10.1021/ja807942m.; Miller LC, Ndungu MJ, Sarpong R. Angew. Chem., Int. Ed. 2009;48:2398–2402. doi: 10.1002/anie.200806154.Wu B, Parquette JR, RajanBabu TV. Science. 2009;326:1662. doi: 10.1126/science.1180739.
- 7.a Vigneron JP, Dhaenes M, Horeau A. Tetrahedron. 1973;29:1055–1059. [Google Scholar]; b Heller G. Angew. Chem. Int. Ed. 2000;39:495–499. doi: 10.1002/(sici)1521-3773(20000204)39:3<495::aid-anie495>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 8.For related examples, see: Gansäuer A, Fan C-A, Keller F, Keil J. J. Am. Chem. Soc. 2007;129:3484–3485. doi: 10.1021/ja0686211.Gansäuer A, Lei S, Otte M. J. Am. Chem. Soc. 2010;132:11858–11859. doi: 10.1021/ja105023y.
- 11.a Evidente A, Sparapano L, Fierro O, Bruno G, Motta A. J. Nat. Prod. 1999;62:253–256. doi: 10.1021/np980318t. [DOI] [PubMed] [Google Scholar]; b Cimino P, Bifulco G, Evidente A, Abouzeid M, Riccio R, Gomez-Paloma L. Org. Lett. 2002;4:2779–2782. doi: 10.1021/ol026310z. [DOI] [PubMed] [Google Scholar]
- 12.Claus RE, Schreiber SL. Org. Synth. 1986;64:150–156. [Google Scholar]
- 15.Nicolaou KC, Li H, Nold AL, Pappo D, Lenzen A. J. Am. Chem. Soc. 2007;129:10356–10357. doi: 10.1021/ja074297d. [DOI] [PubMed] [Google Scholar]
- 16.Galano J-M, Audran G, Monti H. Tetrahedron. 2000;56:7477–7481. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.