

Abstract
Chronic lymphocytic leukemia (CLL) cells feature a pronounced apoptotic resistance. The vascular endothelial growth factor (VEGF) possesses a role in this apoptotic block, although underlying functional mechanisms and the involvement of the microenvironment are unclear. In this study, the VEGF status in CLL was assessed by enzyme-linked immunosorbent assay and immunofluorescence. VEGF receptor 2 (VEGFR2) phosphorylation was determined flow cytometrically and by immunofluorescence. For co-culture, CLL cells were cultivated on a monolayer of the bone marrow–derived stromal cell (BMSC) line HS5. Secreted VEGF was neutralized using the monoclonal antibody mAb293 (R&D Systems, Minneapolis, MN, USA). To block protein secretion, we used Brefeldin A. VEGF was downregulated in BMSCs by small interfering RNA (siRNA), and we assessed survival by annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining. CLL cells express and secrete VEGF and possess phosphorylated VEGFR2. This positive VEGF status is not sufficient to prevent spontaneous apoptosis in vitro. Coculture with BMSCs, which secrete vast amounts of VEGF, maintains in vitro CLL cell survival. Blockage of secreted VEGF using the monoclonal antibody mAb293 significantly reduced the survival support for cocultured CLL cells. Both general blockage of protein secretion by Brefeldin A in BMSCs, but not in CLL cells, and siRNA-mediated downregulation of VEGF in BMSCs, significantly reduced the coculture-mediated survival support for CLL cells. It can be concluded that BMSC-derived proteins and VEGF, in particular, but not CLL cell–derived VEGF, is essentially involved in the coculture-mediated survival support for CLL cells. Hence, therapeutic targeting of VEGF signaling might be a promising approach to overcome the apoptotic resistance CLL cells feature within their natural microenvironment.
INTRODUCTION
Chronic lymphocytic leukemia (CLL) mainly arises from accumulation of malignant monoclonal CD5+ B-lymphocytes exhibiting a mature phenotype (1), which is mainly due to decreased programmed cell death (apoptosis) rather than increased proliferation of B-cells (2). Various signaling pathways have been associated with the initiation and course of CLL, including a variety of humoral factors and cytokines implicated in deregulating these pathways (3). Among other proteins, vascular endothelial growth factor (VEGF) was described as being involved in the pathophysiology of CLL. VEGF is a potent proangiogenic factor and, via activation of the VEGF receptor (VEGFR) family, regulates blood vessel growth and formation (4). CLL cells produce and secrete VEGF and display VEGFRs (5). Moreover, several studies noted elevated VEGF levels in serum or plasma of CLL patients to positively correlate with disease progression (6), advanced disease stage (7) or expression levels of the VEGF receptor 2 (VEGFR2) and shortened survival times (8). In agreement with this, compared with healthy tissue, microvessel density was higher in CLL bone marrow biopsies, a suggested effect of VEGF-induced increased angiogenesis, and was again positively correlated with the clinical stage (9). However, on the basis of these descriptive data, no statement can be made regarding the involvement of CLL cell–derived VEGF, since serum or plasma VEGF can originate from any other blood component as well. Furthermore, the mentioned studies focused mainly on the angiogenic aspects of VEGF.
Besides its role in angiogenesis, VEGF is a known survival factor for different kinds of cell types including endothelial cells, hematopoietic stem cells and solid tumor cells (10,11). In primary CLL cells, exogenous VEGF appeared to support cell survival and prevent drug-induced apoptosis (12,13). In addition, we have recently shown that targeting VEGF receptors effectively induces apoptosis in primary CLL cells and reduces tumor growth in a VEGF-positive CLL-like xenograft mouse model (14). Also, other compounds directed against VEGFR1 and VEGFR2 could be demonstrated to induce apoptosis in CLL cells (12,15). Another study showed VEGF to be involved in CD154 (CD40L)-mediated CLL cell survival (16). Hence, VEGF can be considered a prosurvival factor in CLL, although its actual source and mechanism of action is as yet unclear.
Although CLL cells supposedly support their own survival by expressing prosurvival factors, they are not completely autarkic, since they die rapidly when removed from their natural environment and when cultured in vitro (17). This is why their microenvironment is proposed to be crucially involved in their malignant phenotype (18,19). Because early stages of CLL are characterized by bone marrow infiltration (20), the bone marrow microenvironment can be considered a critical side of nurturing in the disease process. Bidirectional interactions between the malignant CLL cells and the nontransformed bystander cells, via both secretion of soluble factors as well as direct physical cell–cell contacts, lead to the establishment of an abnormal microenvironment favoring the survival of CLL cells. The microenvironment might also represent a niche for the CLL cell to retreat therapeutic interventions (21–23). Among accessory cells present in the natural microenvironment of CLL cells in vivo, bone marrow–derived stromal cells (BMSCs) have received a high level of attention. BMSCs secrete a variety of prosurvival factors, such as interleukins and also VEGF, therewith exerting a complex regulatory function on CLL cells (24). Further, since CLL cells exhibit receptors for many prosurvival cytokines, a high response potential toward these factors secreted by accessory nontumorigenic cells can be assumed.
Despite the proposed role of VEGF in CLL cell apoptotic resistance, little is understood about whether VEGF acts mainly as a microenvironmental stimulus and/or whether CLL cells themselves contribute to the enhanced apoptotic resistance by maintaining an autocrine VEGF loop. Thus, our aim was to get further insight into the role of VEGF in the apoptotic block of CLL cells within their cellular microenvironment mimicked by coculture with bone marrow–derived stromal cells. Indeed, our results point to the importance of VEGF derived from the microenvironment, rather than an autocrine prosurvival VEGF mechanism.
Because it has been suggested that protective niches in the microenvironment may largely account for disease relapse by guarding malignant cells from therapeutic interventions, knowledge about the complex interactions between CLL cells and nonmalignant accessory cells might reveal a path for more successful CLL treatment strategies.
MATERIAL AND METHODS
Patients and Sample Preparation
All CLL patients included in this study had a confirmed diagnosis according to standard criteria (25). Patients were either untreated or had not been treated for at least 3 months before blood withdrawal. Patients represented all Binet stages. All patients provided informed written consent. The study was in accordance with the declaration of Helsinki and was approved by the internal review board of the University Hospital Cologne.
CLL cells were enriched from peripheral blood of patients using RosetteSep® B-cell enrichment cocktail (Stem Cell Technologies, Vancouver, BC, Canada) before Ficoll density centrifugation. CLL cell purity was usually >96% after this process, as assessed by flow cytometry. Peripheral blood mononuclear cells (PBMCs) from healthy volunteers were isolated using Ficoll density gradient centrifugation. Fresh primary cells were used for all experiments.
Cell Culture of Primary Cells and Coculture
Primary cells and the BMSC line HS5 (in the following called BMSC) were maintained in RPMI 1640 supplemented with 1% penicillin/streptomycin and 10% fetal calf serum (FCS) (Biochrom AG, Berlin, Germany). Cells were cultured in a humidified atmosphere at 37°C with 5% CO2. For coculture experiments, BMSCs were seeded at 3 × 105 cells/mL and incubated overnight. Adherent BMSCs were washed with FCS free medium, and primary CLL cells were seeded onto BMSC monolayers at 4 × 106 cells per mL in RPMI 1640, 1% penicillin/streptomycin and 10% FCS. CLL cells were harvested after gentle agitation by removal of the supernatant. BMSC contamination in coculture supernatant was usually <1.5%, as assessed by flow cytometry.
Detection of Apoptotic Cell Death
The annexin V–FITC/PI apoptosis detection kit I (BD Bioscience Pharmingen, San Diego, CA, USA) was used following the manufacturer’s instruction. Fluorescence was measured on FACSCanto (BD Bioscience) and analyzed with FACSDiva® software. In one case, cell survival was illustrated by dot blots, where annexin V–FITC is displayed on the x axis and PI on the y axis. Double-negative cells in the first quadrant are considered alive, whereas double-positive cells in the third quadrant can be called dead.
In the BMSC/CLL coculture, survival of CLL cells in monoculture (%) was subtracted from the percentage of surviving CLL cells in the BMSC coculture to obtain a relative survival advantage (stated as survival advantage of coculture over monoculture).
Enzyme-Linked Immunosorbent Assay
Cell culture supernatants were used for enzyme-linked immunosorbent assay (ELISA) experiments. Supernatants of primary cells after 24 h in culture were concentrated using microcon filters (Ultracel YM-10; Millipore, Billerica, MA, USA). BMSC supernatant was diluted 1:10 before being loaded into the wells. ELISAs were prepared using the VEGF Duo Kit (R&D Systems, Minneapolis, MN, USA). The ELISA procedure was carried out following the recommended instructions. All analyses and calibrations were performed in duplicate.
Immunofluorescence
Cells were fixed using 4% formaldehyde for 20 min followed by a washing step using 1 × phosphate-buffered saline (PBS) and subsequently permeabilized with 1 × PBS containing 0.1% Triton X for 5 min. Cells were blocked for 30 min in 1 × PBS, 0.4% gelatin, 10% normal goat serum (both Sigma-Aldrich, Hamburg, Germany) and 1% fragment crystallizable receptor (FcR) blocking reagent (Miltenyibiotec, Bergisch-Gladbach, Germany) at room temperature. The primary antibody against VEGF was from Santa Cruz Biotechnology (sc-152). VEGFR2 and phosphorylated VEGFR2 were detected using anti-VEGFR2 (number 2479) and anti-phospho VEGFR2 Tyr951 (number 2478) (Cell Signaling Technology, Danvers, MA, USA). Primary antibody incubation was carried out overnight in 1 × PBS containing 0.4% gelatin, 2% normal goat serum and 0.1% FcR blocking reagent, in a humidified atmosphere at 4°C. A matching rabbit IgG isotype control was used.
After three washing steps (1 × PBS, 0.4% gelatin), cells were incubated with secondary fluorescently labeled antibody (number 4412, Cell Signaling Technology) in 1 × PBS, 0.4% gelatin, for 1 h at room temperature. After three washing steps with 1 × PBS, 0.4% gelatin, one washing step with 1 × PBS and one washing step with H2O CLL cells were resuspended in Mowiol (Carl Roth, Karlsruhe, Germany), including 4′6-diamidino-2-phenylindole·2HCl (DAPI) and spotted on slides. BMSCs were directly grown in chamber slides and fixed, permeabilized and incubated (without FcR blocking reagent) as described for primary cells. Imaging was carried out on a Zeiss Axioscope.
Intracellular Phospho-Flow Cytometry Analysis
VEGFR2 and phosphorylated VEGFR2 were detected using anti-VEGFR2 (number 2479) and anti-phospho VEGFR2 Tyr951 (number 2478) (Cell Signaling Technology). Unlabeled rabbit IgGs were used as controls (Southern Biotech, Birmingham, AL, USA). Cells were fixed in 2% formaldehyde, permeabilized in 90% methanol and either stored at −20°C for later use or washed and immediately incubated with the primary antibody or matched isotype control. A secondary fluorescently labeled antibody (Cell Signaling Technology) was used. Fluorescence was measured on FACSCanto (BD Bioscience) and analyzed using FACSDiva® software. Overlays were produced using the free flow cytometry data analysis software Cyflogic® version 1.2.1. Gray-shaded peaks represent the isotype controls.
Brefeldin A Treatment
Brefeldin A (BFA) (Sigma-Aldrich) was diluted in dimethyl sulfoxide (DMSO) to a stock concentration of 10 mg/mL. Primary CLL cells or BMSCs were treated with 50 and 200 ng/mL BFA for 8 h, respectively. These concentrations were chosen, since they efficiently block secretion while not influencing survival (assessed by ELISA and annexin V–FITC/PI staining). DMSO treatment functioned as the control. In the following, cells were washed and used for coculture experiments. Coculture was carried out for 16 h, and survival was determined using annexin V–FITC/PI staining on FACSCanto. Survival results are presented as survival advantage (difference of survival in monoculture and survival in coculture).
VEGF-Neutralizing Antibody
A monoclonal anti-VEGF antibody from R&D systems (mAb293) was used to neutralize VEGF activity in CLL/BMSC coculture. The effect of this antibody on VEGFR2 phosphorylation status was tested by intracellular phospho-flow cytometry. A concentration of 10 μg/mL significantly decreased VEGFR2 phosphorylation and was therefore used in subsequent experiments. Coculture was set up as described with the addition of the VEGF-neutralizing antibody. An isotype-matched (mouse IgG) antibody functioned as the control. After 24 h of culture, survival was measured by annexin V–FITC/PI staining on FACS -Canto. The survival advantage of coculture versus monoculture was calculated as specified before.
siRNA-Mediated Downregulation of VEGF
All small interfering RNAs (siRNAs), controls and the transfection reagent HiPerFect were purchased from Qiagen (Hilden, Germany). HiPerFect-mediated transfection is based on the cationic/neutral lipid technique of siRNA. BMSCs were seeded at 105/mL in RPMI 1640, 1% penicillin/streptomycin and 10% FCS 1 d before transfection. Transfection procedures were carried out as suggested by the manufacturer. The final siRNA concentration was 50 nmol/L. A fluorescently labeled nontarget siRNA functioned as a transfection and negative control and was used for optimization. Mock transfection served as a transfection reagent control. Secreted VEGF protein was measured using ELISA as described above and normalized to simultaneously measured numbers of living BMSCs at the indicated time points. At 24 h after transfection, BMSCs were washed to remove transfection complexes. CLL cells (4 × 106/mL in RPMI 1640, 10% FCS) were added to un-transfected, control or VEGF siRNA transfected confluent BMSCs, with and without the addition of recombinant human (rh) VEGF (5 ng/mL). CLL cells were also cultured as a monoculture. Viability of CLL cells was assessed as described above after 24 and 48 h of coculture. To calculate a relative survival advantage of coculture compared with monoculture, measured percentages of annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) double-negative cells in coculture and monoculture were subtracted from each other. Three independent siRNA experiments were carried out to calculate an average.
Statistical Analysis
Error bars represent standard error of the mean (SEM). Statistical differences between mean values were calculated using the appropriate test stated in the figure legends. Calculation was carried out using GraphPad Prism 5 software. A P value of <0.05 was considered statistically significant.
RESULTS
CLL Cells Secrete VEGF and Exhibit Phosphorylated VEGFR2
Analysis of secreted VEGF in medium of 24 h in vitro cell culture revealed higher amounts of secreted VEGF in CLL cells compared with PBMCs from healthy volunteers (Figure 1A). Further, VEGF protein could be detected in the cytoplasm of CLL cells (Figure 1B, i) by immunofluorescence. Also, the presence of VEGFR2 (see Figure 1B, ii) and its phosphorylation (see Figure 1B, iii) was confirmed. VEGFR2 was mainly membrane localized, whereas upon phosphorylation, VEGFR2 was predominantly present intracellularly.
Figure 1.

VEGF/VEGFR2 status in CLL cells. (A) CLL cells secrete higher VEGF levels (235.9 ± 29.1 pg/mL) than PBMCs from healthy volunteers (94.1 ± 12.1 pg/mL) (P = 0.0002, Mann-Whitney test) as assessed by ELISA after 24 h of culture. (B) VEGF protein (i ) is present in cytoplasm of primary CLL cells. Also the presence of VEGFR2 (ii ), mainly localized to the membrane, and its phosphorylation (iii ) can be demonstrated by immunofluorescence. For all three antibodies, rabbit IgG antibody functioned as isotype (iso) control (iv). Magnification is 100×. One representative sample of six independent experiments is displayed. ***P < 0.001.
Because of the ability of CLL cells to produce and secrete VEGF and the concomitant presence of phosphorylated VEGFR2, the existence of an autocrine survival-supporting VEGF loop can be suggested. Nevertheless, CLL cells die within a few days under culture conditions and are not capable of maintaining their most prominent pathophysiological feature, their apoptotic resistance, in vitro. It can be concluded that autocrine VEGF alone is not sufficient for apoptotic protection, and further factors, derived from the microenvironment, are required.
Survival Supporting BMSCs Have a Positive VEGF Status
As commonly accepted, CLL cells can be maintained in culture when cultured on a feeder layer, such as for example BMSCs. Using the BMSC line HS5 as a feeder layer, CLL cells are efficiently protected from spontaneous apoptosis under culture conditions in our experiments (Figure 2A).
Figure 2.

Impact of the BMSC line HS5 on CLL cell survival and the VEGF status of BMSCs. (A) CLL cells are largely protected from spontaneous apoptosis when cultured on the BMSC line HS5 as a feeder layer. Survival after 120 h in monoculture was 12.0% ± 5.9% (Mono), whereas CLL cell survival in a coculture with HS5 cells was 81.7% ± 1.6% (Co). Survival was assessed by annexin V–FITC/PI staining on FACSCanto. (B) BMSCs secrete approximately 18-fold higher VEGF than CLL cells (4301.1 ± 122.3 pg/mL versus 235.9 ± 29.1 pg/mL), as assessed by ELISA after 24 h in culture (P = 0.007, Mann-Whitney test). (C) VEGF protein is present in cytoplasm of HS5 cells as demonstrated by immunofluorescence (i ). The right panel represents the isotype control (ii ). Magnification is 100×. One representative sample of three independent experiments is displayed. **P < 0.01.
Up to now, several factors have been associated with the protective effect of BMSCs on CLL cell survival in vitro. In this work, we analyzed the impact of VEGF on CLL cell survival in BMSC co-cultures. The amount of secreted VEGF in the supernatant of 24-h cell culture was around 18-fold higher than that of CLL cells (Figure 2B). Also, VEGF protein could be detected in the cytoplasm of BMSCs (Figure 2C).
Coculture with BMSCs Maintains VEGFR2 Phosphorylation
CLL cells possess phosphorylated VEGFR2 after being isolated from peripheral blood (Figure 1B, iii), indicating active VEGF signaling, which has been ascribed a prosurvival role in hematological malignancies. Because CLL cells are not capable of maintaining their own survival under culture conditions, it can be assumed that VEGF signaling is possibly turned down or off with time in culture. We analyzed the presence of VEGFR2 and its phosphorylation at 24 and 72 h under culture conditions in monoculture and in coculture with BMSCs. Whereas no overall reduction of VEGFR2 could be detected (Figure 3A), its phosphorylation went down with time in a monoculture setting (Figure 3B). Going along with prevention of spontaneous apoptosis, coculture with BMSCs also prevented loss of VEGFR2 phosphorylation (Figure 3D). Also in a coculture setting, the total amount of VEGFR2 was largely unchanged (Figure 3C). Figure 3A, B, C, and D depict representative histograms. Results from five independent experiments are summarized in Figure 3E and F.
Figure 3.

Influence of coculture with BMSCs on VEGFR2 phosphorylation in CLL cells. Whereas the overall VEGFR2 levels remained stable with time (A), VEGFR2 phosphorylation was significantly reduced in CLL cells after 72 h of monoculture (B). Also, coculture with BMSCs did not alter total VEGFR2 levels (C) but largely prevented VEGFR2 phosphorylation loss (D). VEGFR2 and phosphorylated (p) VEGFR2 were detected using intracellular phospho-flow cytometry on FACSCanto. Mean fluorescence intensities (MFIs) are displayed. Results of five independent experiments are summarized for total VEGFR2 (E) and pVEGFR2 (F).
Secreted Proteins from BMSCs Have a Higher Impact on CLL Cell Survival in Coculture than CLL Cell–Derived Secreted Proteins
It has been demonstrated that the survival support CLL cells gain from being cultured together with a feeder layer such as BMSCs, is mediated by both secreted factors as well as direct physical cell–cell contact. These conclusions were drawn from experiments where CLL cells were physically separated from feeder cells using transwells. From these experiments, it cannot be concluded whether the secreted factors involved in the prevention of spontaneous apoptosis of CLL cells in vitro are derived from the feeder layer or from the CLL cells themselves. To investigate, we blocked secretion of either BMSCs or CLL cells using the exocytosis inhibitor BFA. A BFA concentration of 200 ng/mL for BMSCs and 50 ng/mL for CLL cells was determined to effectively prevent protein secretion, as exemplarily demonstrated by VEGF (Figure 4A), while at the same time, survival was not significantly affected (Figure 4B). Either CLL cells or BMSCs were treated for 8 h with BFA. Cells were washed to remove BFA, and co-cultures were set up (BFA-treated CLL cells with untreated BMSCs and BFA-treated BMSCs with untreated CLL cells). Because of the reversibility of the BFA effect after its removal, the observed time frame for manipulated co-cultures was reduced to 16 h. The survival advantage CLL cells gain from coculture was significantly reduced when protein secretion by BMSCs was inhibited (Figure 4C). When CLL cells were pretreated with BFA, the impact on CLL cell survival in coculture was not significantly changed compared with untreated coculture (Figure 4D).
Figure 4.

Prevention of secretion of either cell component of coculture using BFA. (A) BFA treatment (8 h) reduced VEGF protein secretion from 3905.4 ± 251.4 pg/mL (untreated control [UTC]) to 68.4 ± 21.3 pg/mL and from 193.4 ± 12.6 pg/mL (UTC) to 34.8 ± 10.9 pg/mL in stromal cells (left) and CLL cells (right), respectively (P = 0.002 and P = 0.01, DMSO versus BFA treatment for stromal cells and CLL cells, respectively, paired two-tailed t test). Secreted VEGF was analyzed by ELISA. (B) Survival was 83.4% ± 2.4% (UTC) and 76.3% ± 1.6% (BFA) for stromal cells (left) and 73.0% ± 2.9% (UTC) and 69.3% ± 3.1% (BFA) for CLL cells (right). (C) Coculture with BFA-treated stromal cells provided a significantly reduced survival advantage (2.3% ± 3.0%) when compared with coculture with untreated stromal cells (11.3% ± 5.7%) (P = 0.038, paired two-tailed t test). (D) BFA-treated CLL cells on untreated stromal cells did not show significantly reduced survival when compared with an untreated CLL cell survival in a coculture setting (4.1% ± 1.4% versus 5.5% ± 1.1%). DMSO functioned as a vehicle control. Survival advantage was calculated by subtracting survival (%) in monoculture from survival (%) in coculture. Survival was assessed using annexin V–FITC/PI staining on FACSCanto. *P < 0.05; **P < 0.01; n.s., not significant.
Secreted VEGF Is Essentially Involved in the Coculture-Mediated Survival Support
We intended to further elucidate whether VEGF is possibly one of the crucial secreted factors involved in the survival-support CLL cells gain from coculture with BMSCs. For that purpose, secreted VEGF was neutralized by the addition of an anti-VEGF monoclonal antibody (mAb293; R&D Systems). An isotype-matched antibody was used as control. Addition of 10 μg/mL neutralizing antibody significantly reduced the coculture-mediated survival support for CLL cells after 24 h (Figure 5A). Figure 5B displays a representative sample where neutralization of VEGF reduces the co-culture-mediated survival support by approximately 60% (survival advantage of 44.1% without VEGF neutralization versus survival advantage of 18.8% with VEGF neutralization). Interestingly, neutralization of VEGF in a monoculture setting did not influence CLL cell survival (Figure 5B, C).
Figure 5.

Blockage of secreted VEGF protein by addition of a VEGF-neutralizing monoclonal antibody (mAb). (A) Neutralization of secreted VEGF in a stromal cell/CLL cell coculture using the monoclonal antibody mAb293 (R&D Systems) (10 μg/mL) significantly reduced the coculture-mediated survival support for CLL cells (P = 0.030, paired two-tailed t test). Survival advantage was calculated by subtracting survival (%) in monoculture from survival (%) in coculture. (B) Annexin V–FITC/PI status of one representative sample is displayed as a flow cytometry dot blot. The left lower quadrant represents annexin V–FITC/PI–negative cells (alive), and the right upper quadrant represents annexin V–FITC/PI–positive cells (dead). (C) Addition of the VEGF-neutralizing antibody mAb293 (10 μg/mL) to CLL cells in a monoculture setting did not significantly influence CLL cell survival. Annexin V–FITC/PI status was analyzed on FACSCanto. *P < 0.05.
BMSC-Derived VEGF Is Crucial for Coculture-Mediated CLL Cell Survival
Besides blocking secreted VEGF in the coculture supernatant, which can be derived from both CLL cells or BMSCs, we reduced expression and secretion of VEGF by BMSCs using siRNA. At 24 h after transfection, siRNA was removed and CLL cells were added to confluent BMSCs with or without the addition of exogenous rhVEGF. siRNA treatment reduced amount of secreted VEGF in the supernatant of BMSCs compared with control siRNA (Figure 6A).
Figure 6.
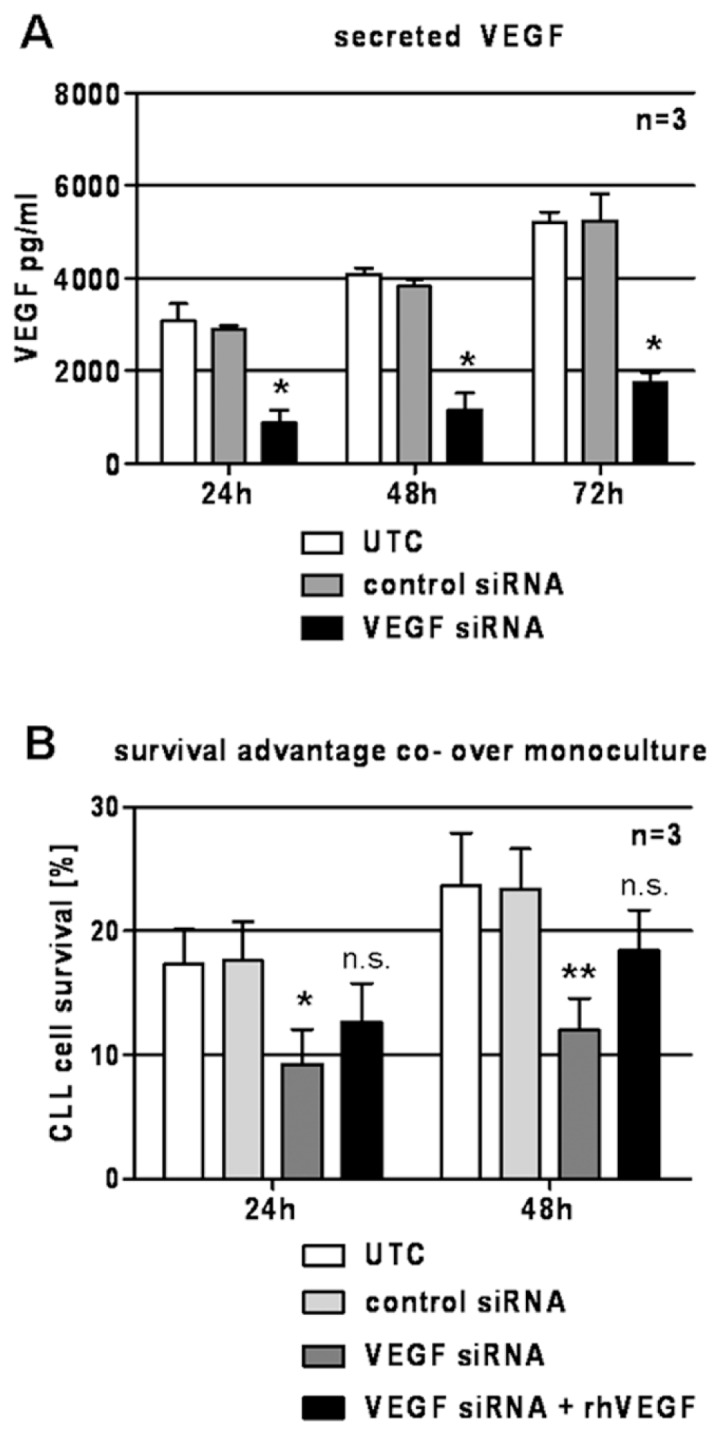
Downregulation of VEGF in BMSCs by siRNA. (A) The amount of secreted VEGF protein was reduced as a result of VEGF siRNA treatment. At 72 h, VEGF concentration in supernatant of control-treated stromal cells was 5,240.3 ± 478.3 pg/mL, whereas siRNA-treated stromal cells only secreted 1,742.8 ± 186.1 pg/mL VEGF (P = 0.013, paired two-tailed t test). (B) Stromal cells with reduced VEGF content provided significantly reduced survival support for CLL cells. The survival advantage of VEGF-reduced stromal cells was 9.3% ± 2.3% in comparison with a survival support of 17.6% ± 2.5% of control-treated stromal cells after 24 h (P = 0.028, paired two-tailed t test). At 48 h, the survival support of VEGF-reduced stromal cells was 12.0% ± 2.1%, whereas control-treated stromal cells led to an improved survival of 23.4% ± 2.7% compared with monoculture (P = 0.005, paired two-tailed t test). Addition of rhVEGF partly reestablished the survival support of VEGF-reduced BMSCs (survival advantage of 12.7% ± 2.5% and 18.4% ± 2.7% compared with control [P = 0.071 and P = 0.061, paired two-tailed t test] at 24 and 48 h, respectively). UTC, untreated control. *P < 0.05; **P < 0.01; n.s., not significant.
After 24 h, survival of CLL cells in co-culture with VEGF-knockdown BMSCs (VEGF siRNA) was significantly reduced compared to survival of CLL cells in co-culture with control siRNA-transfected BMSCs (Figure 6B). This effect became more obvious after 48 h of coculture. Addition of rhVEGF partly reconstituted the survival advantage. It is important to note that siRNA treatment did not affect BMSC survival (data not shown).
DISCUSSION
Until now, VEGF has commonly been accepted as a crucial prosurvival factor in CLL cells and was suggested to act in an autocrine fashion. However, detailed mechanisms underlying the VEGF- mediated survival support and the influence of the microenvironment are considered complex and are not yet resolved. From our data, we hypothesize that CLL cells profit from VEGF secreted from nonmalignant accessory cells such as BMSCs and not from autocrine VEGF.
In this study, we show secretion of VEGF by CLL cells and the presence of VEGFR2, which is in accordance with previous reports (5,9,10). This fact suggests possible autocrine stimulation mechanisms, which is further supported by the presence of VEGFR2 phosphorylation in CLL cells. We show phosphorylation on tyrosine 951, a major phosphorylation side in the kinase insert domain of the receptor, suggesting its signal transducing capacity. Interestingly, phosphorylated VEGFR2 was present intracellularly rather than on the plasma membrane. Whereas in the past, internalization of growth factor receptors was suggested as a mechanism solely to terminate signal transduction, it is now commonly accepted that internalization not only allows further signaling, but is even crucially involved in signaling regulation (26). Also in CLL, intracellular localization (perinuclear membrane) was demonstrated and was associated with active signaling (15). Active VEGF signaling was linked with improved survival. Because CLL cells die within a few days in culture, it is suggested that autocrine VEGF does not have a prosurvival impact on CLL cells, which was supported by this study, since blockage of secreted VEGF in CLL cell culture does not have any impact on CLL cell survival (compare Figure 5B, C). Because addition of rhVEGF to CLL cells in vitro resulted in upregulation of antiapoptotic proteins (15), it could be suggested that it is a solely quantitative problem. Nevertheless, others did not see prevention of spontaneous apoptosis when rhVEGF was added (13). Also in our group, addition of rhVEGF in concentrations up to 100 ng/mL (approximately 1,000-fold higher compared with VEGF secreted by CLL cells themselves) did not improve in vitro survival of CLL cells (data not shown). Hence, we conclude that VEGF acts in concert with other factors to yield efficient apoptosis protection. Farahani et al., for example, demonstrated that VEGF/VEGFR signaling and CD40/CD40L signaling collaborate to protect CLL cells from spontaneous apoptosis, since blocking secreted VEGF using a monoclonal antibody abolished the CD40L-induced prosurvival effect in vitro (16). In the same study, CLL cell–derived VEGF was in the absence of CD40L not sufficient to prevent spontaneous apoptosis of CLL cells in vitro, which is in-line with our results.
It is largely accepted that apoptotic resistance of CLL cells requires a complex interplay of several stimuli and components derived from the microenvironment (21). Besides several secreted factors, it should also be mentioned that direct cell–cell contacts are important as well. Nevertheless, Kay et al. (27) demonstrated that despite physical separation of CLL cells from a BMSC feeder layer (HS5 cell line and primary BMSCs), CLL cells still obtained a significant survival support, suggesting that secreted factors are crucially involved in the stromal cell–mediated survival protection.
Up-to-date VEGF was mainly suggested to act in an autocrine fashion, whereas its significance as a possible paracrine factor has largely been neglected. We therefore studied the influence of the microenvironment with regard to VEGF by monitoring the effect of the BMSC line HS5 on CLL cell survival in a coculture system. We show that BMSCs express and secrete high amounts of VEGF. In contrast to CLL cell monoculture, where phosphorylation of the VEGFR2 was lost over time, the phosphorylation status of VEGFR2 remained stable in cocultured CLL cells. As mentioned earlier, it can be concluded that this stability is not solely due to quantity (compare ~236 pg/mL VEGF secreted by CLL cells to ~4,300 pg/mL VEGF secreted by BMSCs), since addition of recombinant VEGF protein in three-digit ng/mL concentrations could not mimic the coculture effect. As expected, CLL cells cultured on BMSCs survived significantly better than in monoculture. This result is in agreement with several other studies where both the bone marrow stromal cell line HS5 as well as patient-derived bone marrow stromal elements effectively rescued CLL cells from spontaneous apoptosis in a cell culture setting (28–30). As already mentioned, a concerted interplay of secreted factors is believed to create a survival-supportive environment for CLL cells. We investigated whether these secreted factors are rather autocrine or paracrine and blocked protein secretion in either CLL cells or BMSCs by BFA treatment before coculture. Interestingly, prevention of CLL cell secretion did not significantly influence CLL cell survival in a coculture setting. On the other hand, if BMSCs were impaired of protein secretion, a significant reduction of CLL cell survival in coculture was observed. These results clearly underline the significance of paracrine-acting proteins for resistance of CLL cells toward apoptosis, hence their malignant phenotype.
Despite much secreted VEGF in BMSC/CLL coculture supernatants and phosphorylated VEGFR2, to what extent VEGF is involved in improving CLL cell survival in this coculture setting was not clear up to this point. We therefore investigated the impact of VEGF in BMSC-mediated survival support. Neutralization of secreted VEGF present in the supernatant of coculture using a monoclonal antibody noticeably reduced the coculture-mediated survival support for CLL cells, clearly identifying VEGF as an indispensable secreted prosurvival factor for CLL cells. To answer the question whether this VEGF is BMSC- or CLL cell–derived, VEGF was downregulated by siRNA in BMSCs. CLL cell survival in a coculture with downregulated BMSC-derived VEGF was considerably reduced compared with CLL cell survival in co-culture with unmodified BMSCs. The addition of recombinant human VEGF partly reconstituted the siRNA-mediated downregulation. It can be concluded that BMSCs are the source of prosurvival VEGF in the coculture system used in this study. Further, BMSCs may not only affect CLL cells but, vice versa, CLL cells may have an impact on BMSCs as well. Interestingly, it was recently demonstrated that CLL cell–derived microvesicles can induce VEGF production and secretion in BMSCs (HS5) via upregulation of hypoxia-inducible factor (HIF)-1α (31). Bidirectional interactions of CLL cells with their microenvironment have been demonstrated before. CLL cells, for example, actively recruit T cells by expression of T-cell-attracting chemokines (CCL17, CCL22) to profit from their CD40L production (32). Therefore, CLL cells seem to be capable of recruiting cells from the microenvironment to benfit their survival, which involves stimulation of BMSCs to produce VEGF.
In general, the present study was carried out in an in vitro model that does not fully represent the complexity of the actual in vivo situation; hence, its translational potential is limited. Nevertheless, this work adds substantial information toward understanding the complex interactions between CLL cells and BMSCs and suggests VEGF as an interesting target for a therapeutic approach, since it will contribute to disturbing the interactions of CLL cells and their natural microenvironment within the protective niches of the bone marrow.
ACKNOWLEDGMENTS
This work was supported by the José Carreras Leukemia Foundation, Munich, Germany.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Keating MJ. Chronic lymphocytic leukemia. Semin Oncol. 1999;26:107–14. [PubMed] [Google Scholar]
- 2.Reed JC. Molecular biology of chronic lymphocytic leukemia. Semin Oncol. 1998;25:11–8. [PubMed] [Google Scholar]
- 3.Meinhardt G, Wendtner CM, Hallek M. Molecular pathogenesis of chronic lymphocytic leukemia: factors and signalling pathways regulating cell growth and survival. J Mol Med. 1999;77:282–93. doi: 10.1007/s001090050351. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 5.Bairey O, et al. All three receptors for vascular endothelial growth factor (VEGF) are expressed on B-chronic lymphocytic leukemia (CLL) cells. Leuk Res. 2004;28:243–8. doi: 10.1016/s0145-2126(03)00256-x. [DOI] [PubMed] [Google Scholar]
- 6.Molica S, Vitelli G, Levato D, Gandolfo GM, Liso V. Increased serum levels of vascular endothelial growth factor predict risk of progression in early B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;107:605–10. doi: 10.1046/j.1365-2141.1999.01752.x. [DOI] [PubMed] [Google Scholar]
- 7.Gora-Tybor J, Blonski JZ, Robak T. Circulating vascular endothelial growth factor (VEGF) and its soluble receptors in patients with chronic lymphocytic leukemia. Eur Cytokine Netw. 2001;16:41–6. [PubMed] [Google Scholar]
- 8.Ferrajoli A, et al. High levels of vascular endothelial growth factor receptor-2 correlate with shortened survival in chronic lymphocytic leukemia. Clin Cancer Res. 2001;7:795–9. [PubMed] [Google Scholar]
- 9.Kini AR, Kay NE, Peterson LC. Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia. 2000;14:1414–8. doi: 10.1038/sj.leu.2401825. [DOI] [PubMed] [Google Scholar]
- 10.Gerber HP, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002;417:954–8. doi: 10.1038/nature00821. [DOI] [PubMed] [Google Scholar]
- 11.Masood R, et al. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood. 2001;98:1904–13. doi: 10.1182/blood.v98.6.1904. [DOI] [PubMed] [Google Scholar]
- 12.Lee YK, et al. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004;104:788–94. doi: 10.1182/blood-2003-08-2763. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Coad JE, Fortney JM, Gibson LF. VEGF-induced survival of chronic lymphocytic leukemia is independent of Bcl-2 phosphorylation. Leukemia. 2005;19:1486–7. doi: 10.1038/sj.leu.2403837. [DOI] [PubMed] [Google Scholar]
- 14.Paesler J, et al. The VEGF receptor tyrosine kinase inhibitors vatalanib and pazopanib potently induce apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Clin Can Res. 2010;16:3390–8. doi: 10.1158/1078-0432.CCR-10-0232. [DOI] [PubMed] [Google Scholar]
- 15.Lee YK, et al. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19:513–23. doi: 10.1038/sj.leu.2403667. [DOI] [PubMed] [Google Scholar]
- 16.Farahani M, et al. Autocrine VEGF mediates the antiapoptotic effect of CD154 on CLL cells. Leukemia. 2005;19:524–30. doi: 10.1038/sj.leu.2403631. [DOI] [PubMed] [Google Scholar]
- 17.Collins RJ, et al. Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br J Haematol. 1989;71:343–50. doi: 10.1111/j.1365-2141.1989.tb04290.x. [DOI] [PubMed] [Google Scholar]
- 18.Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43:461–6. doi: 10.1080/10428190290011921. [DOI] [PubMed] [Google Scholar]
- 19.Caligaris-Cappio F. Role of the microenvironment in chronic lymphocytic leukaemia. Br J Haematol. 2003;123:380–8. doi: 10.1046/j.1365-2141.2003.04679.x. [DOI] [PubMed] [Google Scholar]
- 20.Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052–7. doi: 10.1056/NEJM199510193331606. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen-Munk I, Reed J. Microenvironmental interactions and survival of CLL B-cells. Leukemia Lymphoma. 2004;45:2365–72. doi: 10.1080/10428190412331272703. [DOI] [PubMed] [Google Scholar]
- 22.Chilosi M, et al. Immunohistochemical demonstration of follicular dendritic cells in bone marrow involvement of B-cell chronic lymphocytic leukemia. Cancer. 1985;56:328–32. doi: 10.1002/1097-0142(19850715)56:2<328::aid-cncr2820560221>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 23.Jewell AP, Yong KL. Regulation and function of adhesion molecules in B-cell chronic lymphocytic leukaemia. Acta Haematol. 1997;97:67–72. doi: 10.1159/000203661. [DOI] [PubMed] [Google Scholar]
- 24.Ghia P, Caligaris-Cappio F. The indispensable role of microenvironment in the natural history of low-grade B-cell neoplasms. Adv Cancer Res. 2000;79:157–73. doi: 10.1016/s0065-230x(00)79005-1. [DOI] [PubMed] [Google Scholar]
- 25.Cheson BD, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87:4990–7. [PubMed] [Google Scholar]
- 26.Di Fiore PP, De Camilli P. Endocytosis and signaling. an inseparable partnership. Cell. 2001;13;106(1):1–4. doi: 10.1016/s0092-8674(01)00428-7. [DOI] [PubMed] [Google Scholar]
- 27.Kay NE, et al. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch.”. Leuk. Res. 2006;31:899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagneaux L, Delforge A, Bron D, De BC, Stryckmans P. Chronic lymphocytic leukemic B cells, but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91:2387–96. [PubMed] [Google Scholar]
- 29.Seiffert M, Stilgenbauer S, Dohner H, Lichter P. Efficient nucleofection of primary human B cells and B-CLL cells induces apoptosis, which depends on the microenvironment and on the structure of transfected nucleic acids. Leukemia. 2007;21:1977–83. doi: 10.1038/sj.leu.2404863. [DOI] [PubMed] [Google Scholar]
- 30.Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand AV. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol. 1996;92:97–103. doi: 10.1046/j.1365-2141.1996.00305.x. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh AK, et al. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010;115:1755–64. doi: 10.1182/blood-2009-09-242719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghia P, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32:1403–13. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]