

Abstract
The Hsp70 chaperones (Heat shock protein 70 kDa) are key to cellular protein homeostatis. However, they also have the ability to inhibit tumor apoptosis, and contribute to aberrant accumulation of hyperphosphorylated tau in neuronal cells affected by tauopathies, including Alzheimer’s disease. Hence, Hsp70 are increasingly been identified as targets for therapeutic intervention in these widely abundant diseases. Hsp70 proteins are allosteric machines and offer besides classical active site targets, also opportunities to target the mechanism of allostery. In this work, it is demonstrated that the action of the potent anti-cancer compound MKT-077, is through differential interaction with the Hsp70 allosteric states. MKT-077 (1-ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4- oxothiazolidin-2-ylidenemethyl] pyridinium chloride) is therefore an “allosteric drug”. Using NMR spectroscopy, the compound’s binding site on human HSPA8 (Hsc70) is identified. The binding pose is obtained from NMR-restrained docking calculations, subsequently scored by molecular dynamics-based energy and solvation computations. Suggestions for improvement of the compound’s properties are made on the basis of the binding location and pose.
The Hsp70 chaperones (Heat shock protein 70 kDa, systematic name: HSPA) assist in protein folding, protein refolding, protein transport and protein targetting1. The human genome contains 13 HSPA genes which encode 12 distinct proteins2. HspA5 (Bip), HSPA8 (Hsc70) and HSPA9 (mt-Hsp70 or Mortalin) are expressed constitutively. They are resident in the ER, cytosol and mitochondria, respectively. Cells respond to stress such as, but not limited to, elevated temperature, by upregulating the expression of the other HSPAs, which help reverse the stress-induced protein misfolding. The chaperones carry this out by unfolding the misfolded proteins in ATP-driven cycles of binding and release3.
HSPAs are significantly up-regulated in tumors4, and are required for the survival of these cells5,6; 7. Knockdown of HSPA2 by siRNA inhibits human tumor growth in nude mice8. Selective HSPA1 knockdown induces apoptosis in pancreatic cancer cells9. Enhanced expression of HSPAs in tumor cells is likely caused by conditions which mimic stress10; 11. The HSPAs are thought to attempt to neutralize the conformational changes in mutated proteins12,13 which are common in tumorogenic cells. In addition, HSPAs are found to specifically inhibit cell death pathways11; 14; 15; 16; 17; 18. Lastly, HSPA9 may directly inactivate p53 tumor suppressor protein19; 20. Hence, inhibition of HSPA has been recognized as a promising avenue for the prevention or therapy of a wide variety of cancers.
HSPAs are also involved in several CNS disorders. Diseases such as Alzheimer’s, Pick’s disease, progressive supranuclear palsy, corticobasal degeneration and argyrophilic grain disease are characterized by the aberrant accumulation of hyperphosphorylated tau, called tau-tangles21; 22; 23; 24. HSPAs participate in the clearance of tau-tangles through a mechanism that is not well understood as of yet25. Recently, we found that inhibitors of HSPA8 led to a rapid increase in tau ubiquitination and proteasome-dependent degradation, in tau-overexpressing HeLa cells26. Clearance of tau-tangles is being recognized as therapeutic to Alzheimer-affected neuronal cells27. Hence, inhibition of HSPA is also a promising avenue for the prevention or therapy of CNS disorders28.
HSPA proteins are becoming recognized as very druggable29 because they offer so many opportunities for interference. HSPA proteins consist of a nucleotide-binding domain30 (NBD) which competitively binds ATP and ADP31, and substrate-binding domain (SBD) which harbors the hydrophobic substrate-binding cleft32. Compounds that compete for binding in either of these sites would modulate HSPA function. Examples of such compounds are adenosine analogs dibenzyl-8-aminoadenosine33; 34 and VER-15500833, which compete for the ATP binding site. Compounds that modulate HSPAs by competing with substrate binding such as Gentamycin35, geranylgeranylacetone36 and several peptidomimetics37 have been described as well.
In addition, HSPA are regulated by the DnaJ co-chaperone family and by a diverse set of nucleotide exchange factors38. They also interact with HIP39, HOP40 and CHIP41, and some specialized factors such as ZIM (for HSPA9)42. Competing with or enhancing the interaction of these ancillary proteins with HSPA may also be an avenue for HSPA modulation. One of the first HSPA inhibitors, 15-deoxyspergualin, was long known to bind to the C-terminal EEVD sequence43; 44. This C-terminus is now also known to be the CHIP-HSPA interaction site45, so likely 15-deoxyspergualin functions by competition with CHIP. Recently, we discovered an inhibitor of the bacterial HSPA orthologue DnaK, that most likely functions by competing with the J-HSPA protein interaction46 .
Finally, HSPA are allosteric proteins. Substrate binding enhances ATP hydrolysis while ATP binding stimulates substrate release. Recently, much insight in the allosteric mechanism has been gained from comparing the structural properties of HSPA in the ADP-state, bound to substrate (referred to as HSPA-ADP-SUB), and in the HSPA-ATP-APO state. HSPA-ADP-SUB is best described as a dynamic ensemble in which NBD and SBD are loosely tethered47; 48, while the NBD and SBD in HSPA-ATP-APO are docked47. In the HSPA-ADP-SUB state, a protective LID docks to the SBD41, while the LID releases in the HSPA-ATP-APO state. The subdomains of the NBD rearrange and reorient between ADP and ATP state49. A surface cleft on the NBD opens and closes between these states40,49. Compounds that can recognize such allosteric differences and perturb the allosteric equilibrium between the states should also modulate HSPA function.
Recently, we identified a flavonoid called myricetin, which binds to the Hsp70 of E. coli, DnaK, at an non-canonical site on he nucleotide binding domain. This compound blocks the binding of DnaK to DnaJ.50 It is the first example of an allosteric HSPA drug.
Taken together, there are many opportunities for the manipulation of HSPA function. In principle, the protein could be targeted by a combination of compounds that are individually not very potent, but act in synergy. In analogy to tumor irradiation therapy, using several low-intensity beams reinforcing each other at the target, a molecular target-specific combination therapy may also have reduced toxicity. In addition, the potential of addressing several different HSPA functions with small compounds may provide an avenue to HSPA-directed personalized medicine.
In the current work, we investigate the interaction of a well-known HSPA interacting compound, MKT-077. MKT-077 is known to interact with HSPA951 and HSPA852. We show here that MKT-077 affects HSPA8 function through differential interaction with HSPA’s different allosteric states. Hence, MKT-077 is found to be an “allosteric drug”. MKT-077 is a Fuji dye compound with the systematic name (1-ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride) (see Figure 1). MKT-077 has an IC50 of 0.35-1.2 uM against several human cancer cell lines53. The IC50 of MKT-077 towards these tumor cell lines is more than 100 times lower than the IC50 against healthy cell lines. Because of these very favorable properties, MKT-077 has been evaluated as cancer chemo-therapeutic in a Phase I trial 54. The trial was aborted because of renal toxicity of MKT-077. Despite this finding, interest in MKT-077 and its derivatives have remained strong 55,56; 57.
Figure 1.
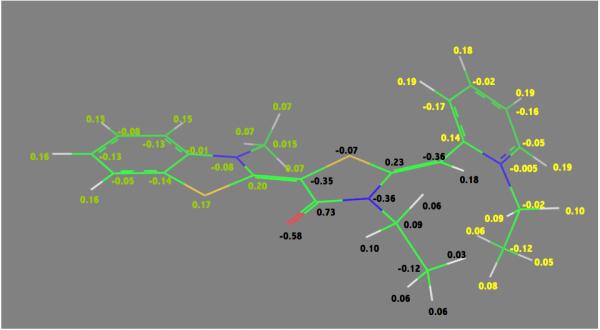
The quantum-mechanically optimized structure of MKT-077. Nitrogen is in blue, sulfur in yellow, oxygen in red. Double-bond assignments were made based on quantum mechanical calculations. AM1-BCC charges76 are shown. ∑Qgreen=+0.56; ∑Qblack=−0.29; ∑Qyellow=+0.73 (see Methods for details)
As mentioned above, we recently found that inhibitors of HSPA8 also lead to a rapid increase in tau ubiquitination and proteasome-dependent degradation, in tau-overexpressing HeLa cells26. We show here that MKT-077 also enhances tau clearance, which makes the compound also of interest for therapy of CNS disorders such as Alzheimer’s. We determine by NMR the binding site of MKT-077 to the ADP-state of HSPA8 (Hsc70). The drug locates itself in a negatively charged pocket close to, but not identical to, the nucleotide binding site. The identification of its binding pocket and binding pose should allow for the design of more potent, more selective, and less toxic MKT-077 derivatives.
Results
Inhibition of the HSPA8 by small molecules such as methylene blue or azure c causes clearance of tau-tangles in transfected HeLa cells26. It was hypothesized that the compounds interfere with the dissociation of HSPA8-tau complexes, leading to clearance through the ubiquitin-proteasome system26. Based on this, we wondered whether the known HSPA951 and HSPA852 inhibitor, MKT-077, would also lead to clearance of hyper-phosphorylated tau. Figure 2 shows that such indeed is the case, suggesting that MKT-077 also interacts and interferes with the function of HSPA8 in these cells. Inhibition of HSPA8 with MKT-077 therefore is a potential avenue for therapeutic intervention with tauopathic diseases such as Alzheimer’s.
Figure 2.
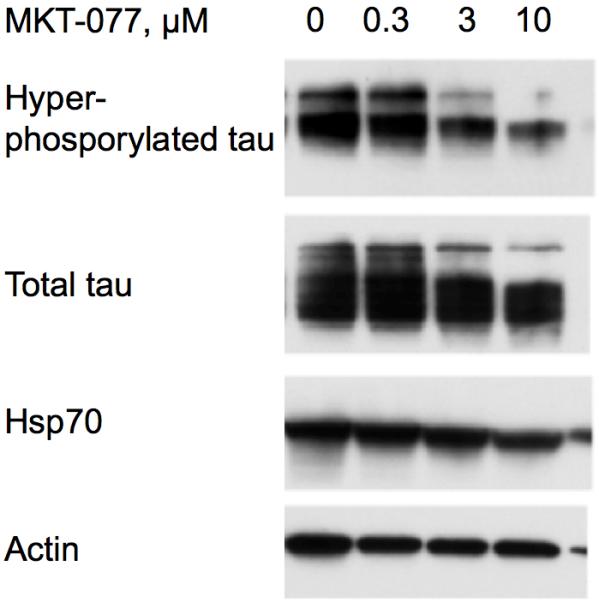
MKT-077 promotes the clearance of hyperphosphorylated tau (top line) in engineered HeLaC3 cells in a dose-dependent way (24-hour incubations).
In recent work, we have used NMR spectroscopy to locate the binding sites of several compounds to the bacterial Hsp70 chaperone, DnaK46; 50; 58. Here we use the same approach, combined with extensive computer modeling and molecular dynamics calculations, to decipher the binding location, pose and mechanism of MKT-077 with the nucleotide-binding domain (NBD) of human HSPA8.
The 15N-1H TROSY NMR spectrum of HSPA8 NBD in the ADP state is shown in the supplemental material. Many of the resonances in the spectrum of this 383-residue protein have been assigned by hand59 and double-checked by a computer algorithm60. Enlargements of the sections of the spectrum are shown in Figure 3. A select number of resonances show gradual chemical shift changes upon addition of MKT-077 up to a molar ratio of 1:1 (using a concentrated sample of 2H,15N,13C–labeled HSPA8-NBD) after which the chemical shifts do no change any more (using a more dilute sample of 15N–labeled HSPA8-NBD). The observed changes in chemical shifts for these two experiments are shown on the amino-acid sequence in the supplemental material.
Figure 3.






Details of some the chemical shift changes in the TROSY spectrum of HSPA8 NBD upon addition of MKT-077. Panels a and c: Blue, no MKT-077; Red, MKT-077: HSPA8 =1:1; Black, MKT-077: HSPA8=2:1 using a sample of 80 μM 15N-labeled HSPA8-NBD in the ADP state.
Ratios MKT-077/HSPA8 for all other panels: Blue, no MKT-077; Green, 0.25:1; Yellow, 0.5:1; Red, 0.75:1; Black, 1:1. For these, a sample of 260 μM 15N, 2H-labeled HSPA8-NBD in the ADP state was used.
Figures 4a and b show the composite chemical shifts observed depicted on a surface rendition of HSPA8 NBD. The majority of the shift changes take place in an area which turns out to be an otherwise unoccupied negatively charged pocket located on the interface of subdomains IA and IIA. As outlined in the introduction, HSPAs are allosteric proteins, which show global conformational and dynamical changes on the NBD between the ADP and ATP state59; 61. Hence, we wondered if MKT-077 would also bind to the ATP-state of the NBD. Figures 4c and d shows a similar display of changes in the TROSY spectrum of HSPA8 NBD in the AMP-PNP state. Significantly, none of the residues affected by MKT-077 binding in the ADP state are affected in AMPPNP state. We conclude that MKT-077 does not bind to Hsc70 NBD in the AMP-PNP state. We also monitored MKT-077 binding to HSPA8 NBD in the true ATP state. No chemical shift changes in the (largely unassigned) NMR spectrum could be discerned, indicating that MKT-077 does not bind to the ATP-state (not shown), indicating that the results for the AMP-PNP state are representative for the ATP state. We also monitored MKT-077 binding to HSPA8 NBD in the apo state. Many changes occurred in the NMR spectrum (not shown) but those were largely uninterpretable because a lack of NMR assignments for this state. Nevertheless, we may conclude that MKT-077 also binds to the apo-state.
Figure 4.
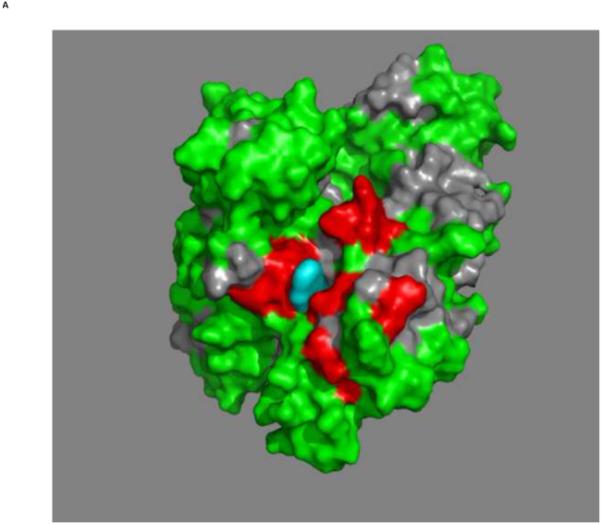
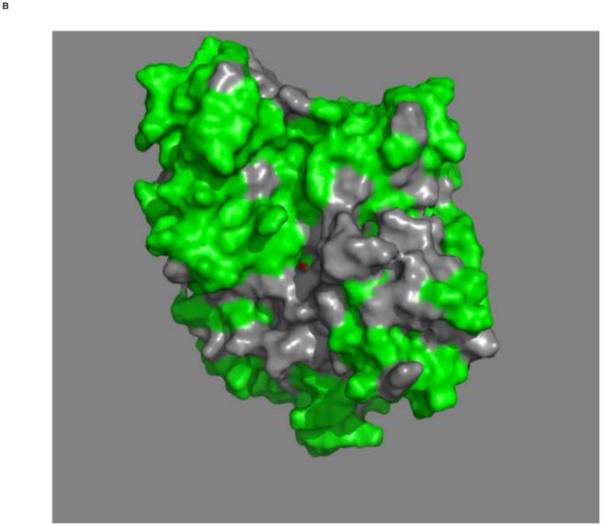

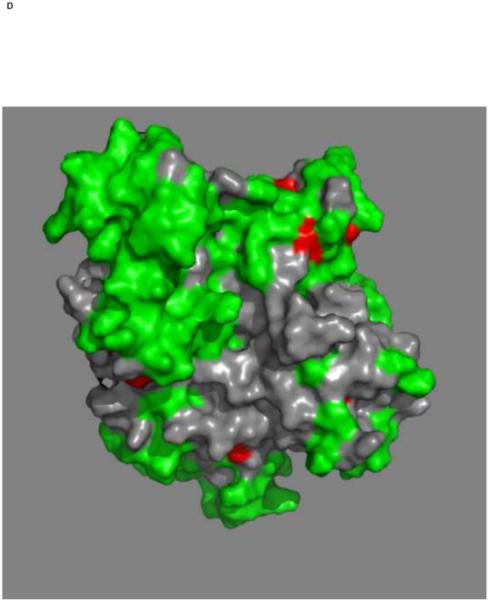
Mapping of the chemical shift changes of MKT-077 on the structure of HSPA8 NBD (3HSC.pdb). Panels a and b: ADP state. Left and right images are rotated 180 degrees along the vertical axis with respect to each other. Color coding: Red, sites detected using 250 μM 15N-2H labeled HSPA8-NBD; Green, no changes; grey, no assignments and cannot be decided. Cyan, approximate position of MKT-077 as determined using AUTODOCK.
Panels c and d: AMPPNP state. Panels and color scheme as above.
The figure was generated in Pymol84.
The MKT-077-induced changes to the spectrum of HSPA8 NBD in the ADP state are in the fast-exchange limit (see Figure 3), with little or no line broadening. The largest shifts in the spectrum are approximately 20 Hz, which sets a lower limit of 100 s−1 for MKT-077’s off-rate. This sets the limit KD > 1 uM, when assuming an upper limit on the diffusion-controlled on-rate of 108 M−1s−1. The shifts are linear over the titration interval (see Figure 5), and saturate abruptly at equivalence (see Figure 3a and 3c). This is compatible with a KD that is at least order of magnitude smaller than the protein concentration; with Hsc70 concentration of 200 uM, we obtain KD < 10 uM. In combination, thus, we estimate from the NMR titration 1 uM < KD < 10 uM. Attempts to determine the binding affinity of MKT-077 to Hsc70 by ITC have not been successful.
Figure 5.

Fractional shifts upon addition of MKT-077 in the 15N-2H-labeled HSPA8. Cyan: T222; Red: A223; Blue: D225; Green H227; Purple, L228; Black, Average. For each resonance, the shift at molar ratio 0.5 was defined as 50% completed.
HSPA8 NBD is a flexible molecule, which can rotate its subunits with respect to each other, especially in the ADP and apo states59; 61. Hence, chemical shift changes at the interface between sub-domains should be eyed with some skepticism. Indeed, there is an area of chemical shift changes (residues 140-144; see Fig 2 of supplemental material) which is not contiguous with the primary shift area shown in Figure 4a. The former area is completely buried, and the shifts must be caused by conformational changes due to MKT-077 binding at the primary and solvent-accessible pocket comprised by the residues shown in figure 5.
The entrance to pocket located on the interface of subdomains IA and IIA is hydrophobic, while the interior is negatively charged due to the presence of the sidechains of Glu175, Asp199 and Asp206. As matter of fact, the electrostatic potential of the entire binding area is strongly negative (see Figure 6). Hence, the chemistry of the binding area complements the chemical properties of MKT-077, which is positively charged, very well (see Fig 1). In support of the hypothesis that electrostatics is a important contributor to the binding energy of MKT-077 is the fact that an uncharged MKT-077 derivative does not perturb the NMR spectrum of HSPA8 (results not shown).
Figure 6.

Surface representation of the electrostatic charge on HSPA8 NBD (3C7N.pdb, Molecular Dynamics relaxed). The APBS module90 in Pymol84 was used. The color coding shows the electrostatic energy of a putative unit charge on the surface of the protein in units of kT. Red, negative potential; blue, positive potential.
Together, the evidence strongly suggests that MKT-077 binds preferentially to the ADP- and not the ATP-bound state of HSPA8. To test whether this compound could stabilize the ADP-like conformation, we performed limited proteolysis experiments. It has been previously shown that trypsin treatment of human Hsc70 yields digestion patterns diagnostic of the chaperone’s nucleotide state62. Consistent with those reports, we confirmed that Hsc70 saturated with ATP was primarily cleaved into three high molecular weight bands, including prominent bands at approximately 69 kDa (band 1) and 66 kDa (band 2) (Figure 7). Conversely, treatment with ADP strongly favors band 2. Addition of the J-domain of prokaryotic DnaJ, which stimulates ATP turnover in Hsc70, converted the ATP-like pattern to an ADP-like pattern. Next, we tested whether MKT-077 could stabilize the ADP-like configuration. Addition of MKT-077 (200 μM) largely blocked formation of band 1, consistent with the NMR data that this compound binds preferentially to the ADP-bound state of Hsc70.
Figure 7.
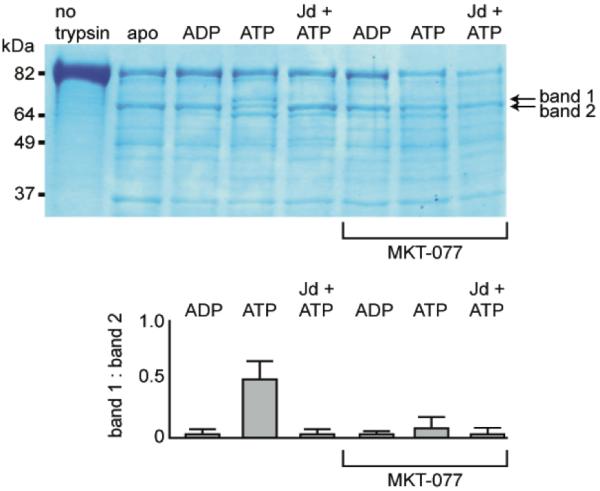
MKT-077 favors the ADP-bound conformation of HSPA8, as measured by limited trypsin proteolysis. Full-length HSPA8 was treated with trypsin in the presence of excess nucleotide and MKT-077. By this analysis, the ATP-bound state is characterized by the appearance of both bands 1 and 2, while the ADP-bound Hsc70 predominantly shows band 2. Addition of a J domain (Jd) converts the ATP- to the ADP-like pattern. MKT-077 (200 μM) suppressed the ATP-bound form and favored band 2. Bands 1 and 2 were quantified in an NIH Image J densitometer and the ratio of these bands (e.g. relative formation of the ATP-bound state) is shown in the bottom panel. These results are representative of experiments performed in duplicate.
Discussion
Our key finding is that MKT-077 binds to the ADP state of HSPA8, but not to the ATP state, which are the two most extreme allosteric states that an HSPA can adopt. The compound binds close to, but not at the natural nucleotide binding site. This qualifies MKT-077 as an allosteric effector, acting not unlike 2,3-DPG that modulates hemoglobin function by binding to a non-heme site that is exposed in the T-state and closed in the R-state63. MKT-077 binds to the interface of the four HSPA NBD subdomains. We have recently shown that the HSPA NBD subdomains change their relative orientations between the different allosteric states61 (see Fig 8). In detail, it was found (a) that the relative orientations of the subdomains of the HSPA ortholog DnaK of Thermus thermophilus in the ADP state resemble, in solution, those of HSPA8 complexed with nucleotide exchange factor as seen in a crystal, and (b) that the relative orientations of the subdomains of the HSPA ortholog DnaK of Thermus thermophilus in the AMP-PnP state resemble, in solution, those of HSPA8s NBD in a crystal, irrespective of the latter’s nucleotide state. We defined the ATP state as the “closed” state and the ADP state as the “open” state. In this context, the binding of MKT-077 to the ADP (open) state and not to the ATP (closed) state makes perfect sense: the binding pocket is (more) accessible in the ADP state but (less) not in the ATP state (see Figure 8).
Fig 8.
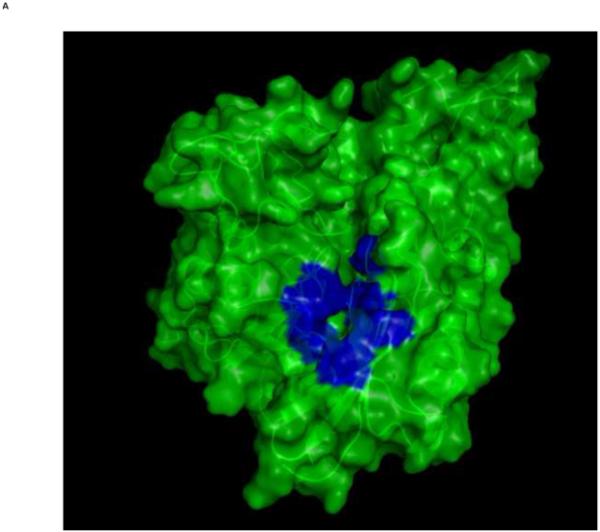
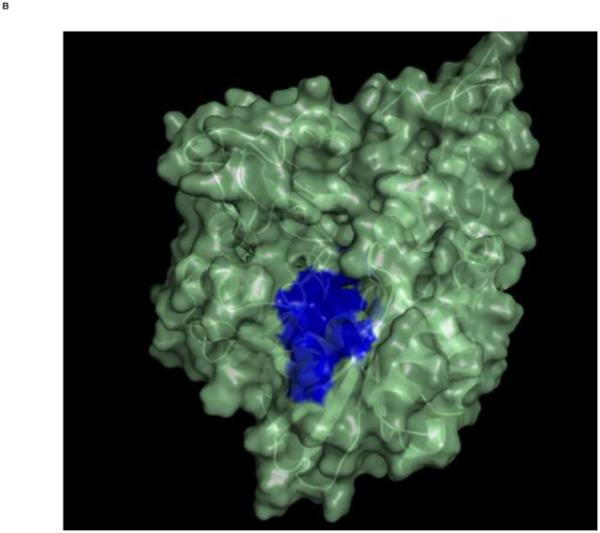
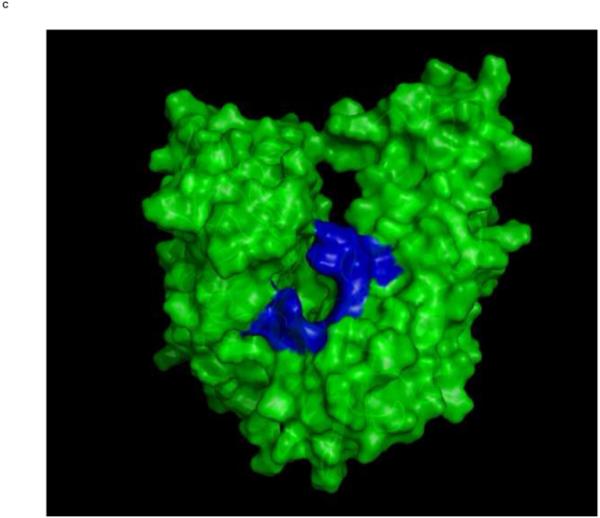
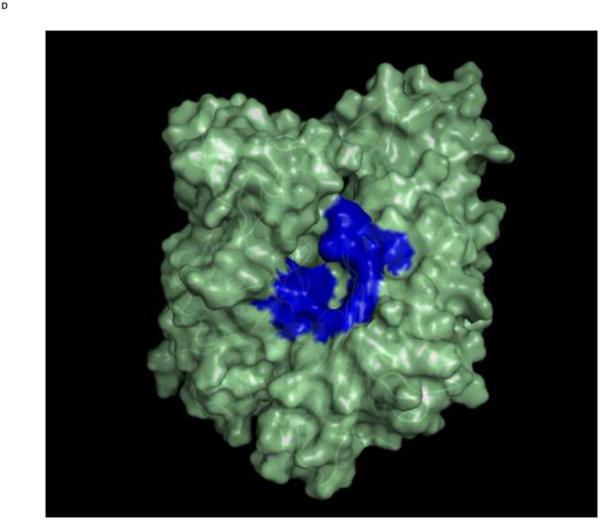
Top: Changes in the overall architecture of the NBD of DnaK of Thermus Thermophilius in solution. A: The open ADP state; B: the closed AMPPNP (ATP) state61. The MKT contact residues of Hsc70 NBD are projected on these conformations in blue.
Bottom: Changes in the overall architecture of the NBD of human HSC70 in the crystal. C: The (equilibrated) crystal structure of Human Hsc70 NBD.ADP in complex with yeast Hsp110 (3C7N) resembles the solution open state. D: The (equilibrated) crystal structure of isolated Hsc70 NBD.ADP resembles the solution closed (ATP) state. The figure was generated in Pymol84.
These observations lead us to formulate the following hypothesis of the mechanism of action of MKT-077 (see Figure 9). The figure summarizes a large body of structural, dynamical and interaction characteristics for the HSPA8, based on the work of our48; 59; 61 and other labs, especially those of L. Gierasch47; 64 and M. Mayer/B.Bukau65; 66; 67. Briefly, in the ADP-substrate-bound state, the nucleotide binding cleft is mostly open and the substrate binding domain is loosely tethered. The lid is docked to the substrate domain. In the ATP-apo state, the nucleotide binding cleft is closed, while the substrate binding domain is docked. In this state, the lid is dynamically tethered.
Figure 9.
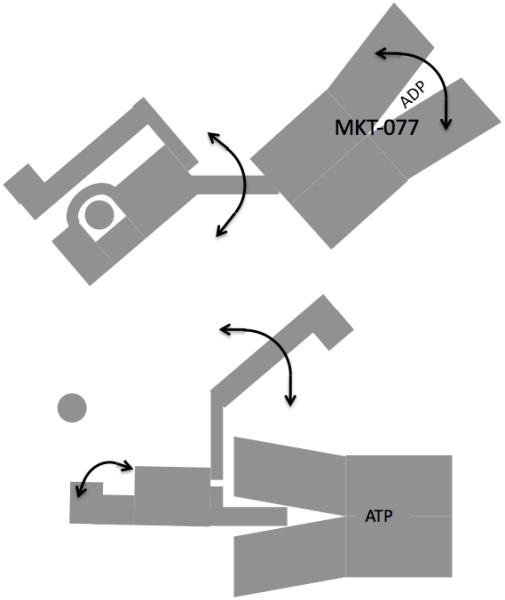
Cartoon showing the properties of HSPA in the ADP state (top) and ATP state (bottom). The SBD is on the left, the NBD is on the right. The substrate is shown as a circle. The double-headed arrows indicate dynamical processes. We hypothesize that MKT-077 stalls HSPA8 in the ADP state
In the current work, we observed that MKT-077 selectively binds to the ADP-allosteric state. By doing so, it likely stalls the HSPA protein-refolding cycle in that state. As a consequence, the HSPA cannot release its substrate. In the context of tauopathies, one hypothesizes68 that the lifetime of the tau-HSPA8 complex becomes long enough to engage in a triple complex with CHIP (C-terminal Hsp70 Interacting Protein). CHIP is an ubiquitin ligase69, which promotes the ubiquitination of the HSPA8-MKT trapped tau, and clearance by the proteasome.
Interference of MKT-077 with HSPA’s substrate binding and release cycle should also directly interfere with HSPA’s abilities to promote survival of tumor cells.
We used AUTODOCK70 to obtain insight in the MKT-077 binding geometry at this pocket. Based on the information that MKT-077 only binds to the ADP state, we used the crystal structure of HSPA8 NBD71 in complex with Hsp110 (3C7N) for the protein coordinates (see figure 8c). The selection of this particular HSPA-NEF complex (there are 5 different ones in the Protein Data Bank) is based on the fact that it is the only one that contains ADP. The crystal coordinates of the NBD were minimized, equilibrated and relaxed to 300 K using a Amber11 72 molecular dynamics protocol (see Methods). The coordinates and degrees of freedom for MKT-077 were optimized and determined from quantum mechanical calculations.
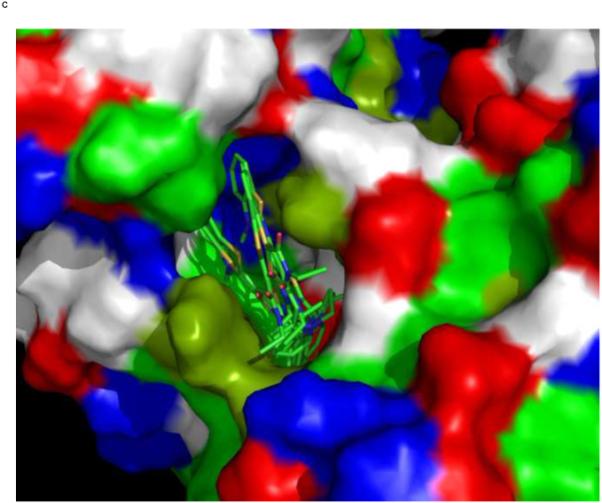
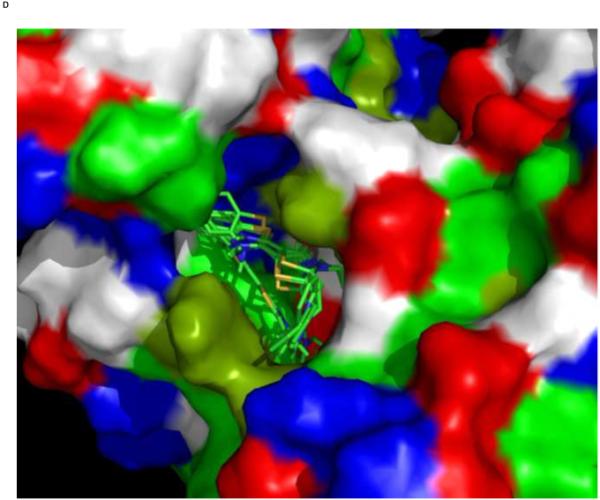
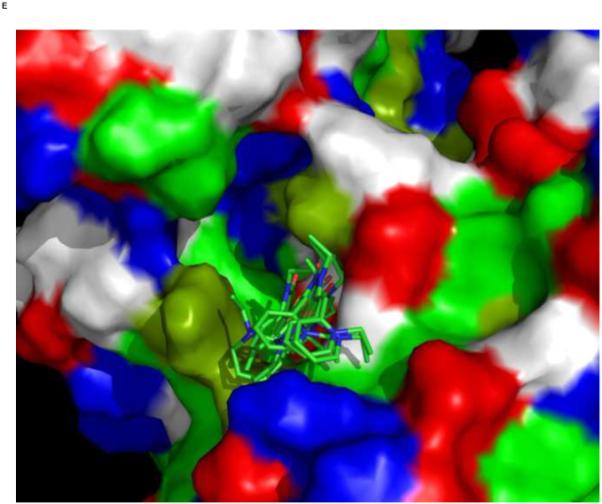
We used an AUTODOCK search box localized around the NMR-determined MKT-077 binding site. The results are shown in Figure 10a. On the one hand, there is satisfactory agreement between the AUTODOCK results and the NMR data: all good docks are located in the area identified by NMR chemical shifts. On the other hand, the AUTODOCK computations yield a wide variety of possible binding poses with AUTODOCK energies ranging between 4 and 7 kcal/mol. The electrostatic interaction does not exceed 0.4 kcal/mol in any of these calculations, which we feel is at odds with the prominent role of electrostatics in MKT-077 action. After culling those dockings which have positive MKT charges contacting positive protein residues, we subdivide the dockings in four families as shown in Figures 10 C-F.
Figure 10.
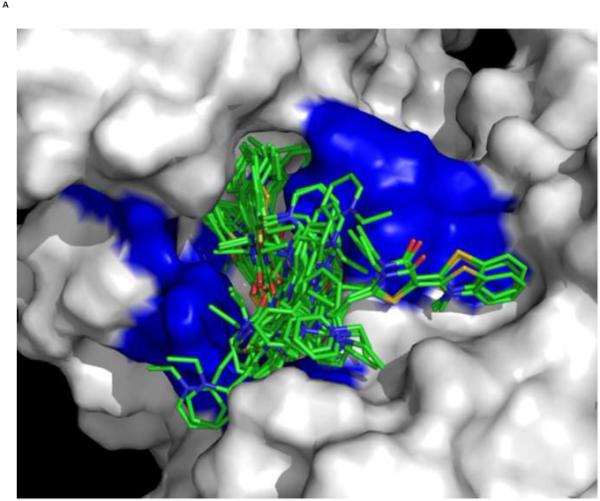
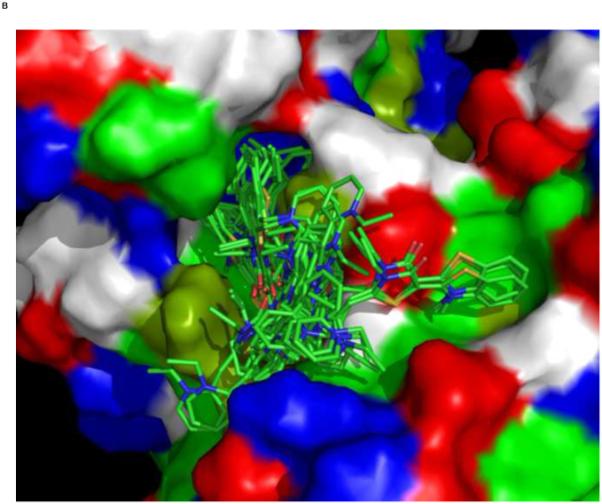
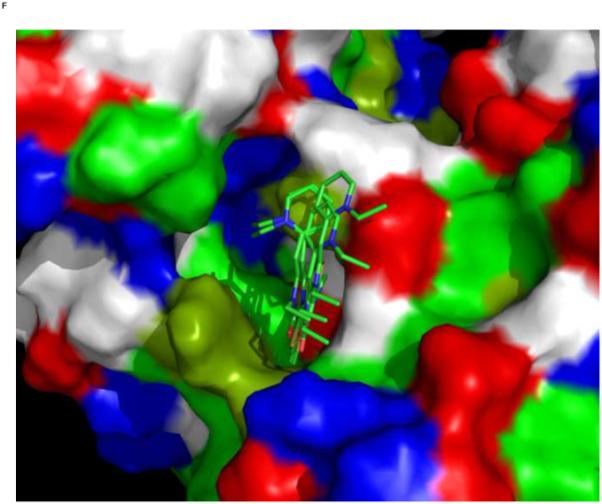
Results of AUTODOCK. The search box was restricted to the binding area as determined by the NMR chemical shift perturbations. Panel A: all dockings ranging from 4.5 – 7.0 kcal/mol in relation to the NMR shifts in blue. Panel B, the same ensemble in relation to the chemical properties of the protein (green: A,C,F,I,L,M,P,V,W; dark green: T,Y; blue: H,K,R; red:D,E; white:G,N,Q,S).
Panel C, a family of feasible dockings with an energy of 7.03 kcal/mol for its best member.
Panel D, a family of feasible dockings with an energy of 6.32 kcal/mol for its best member.
Panel E, a family of feasible dockings with an energy of 5.36 kcal/mol for its best member.
Panel F, a family of feasible dockings with an energy of 5.25 kcal/mol for its best member. Also see Table 2. The figures were generated in Pymol84.
All docked families are in close vicinity / contact with the residues that show chemical shift perturbations. All of them are also close to Glu 175 and Asp 206 that reside deep within the pocket and contribute much to the negative electrostatic potential of the pocket. These residues, as well as the other residues that are in contact with the MKT-077, are 100% conserved between the 13 human HSPA isoforms (see Table 1). Hence, it is more than likely that MKT-077 also binds at this site in the mitochondrial protein, HSPA9, which was earlier identified as prime target of the drug73; 74,51.
Table 1.
HSPA8 residues in contact with the five MKT-077 docking families, shown in the context of the human HSPA paralogs. The alignment was carried out in BLAST-P.
Red, contacts with 4 or 5 families; orange, contacts with 2 families; yellow, contacts with 1 family. These residues are also shown in Fig 11.
The residues that showed chemical shifts are shown in italic. Numbering is for HSPA8.
| 11 | 12 | 13 | 14 | 15 | 69 | 70 | 71 | 72 | 73 | 74 | 75 | 76 | 80 | 81 | 82 | 83 | 84 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HSPA8 | L | G | T | T | Y | D | A | K | R | L | I | G | R | D | A | V | V | Q | |||||
| HspA1A | L | G | T | T | Y | D | A | K | R | L | I | G | R | D | P | V | V | Q | |||||
| HspA1L | L | G | T | T | Y | D | A | K | R | L | I | G | R | D | P | V | V | Q | |||||
| Hspa2 | L | G | T | T | Y | D | A | K | R | L | I | G | R | D | A | T | V | Q | |||||
| HSPA5 | L | G | T | T | Y | D | A | K | R | L | I | G | R | D | P | S | V | Q | |||||
| HSPA6 | L | G | T | T | Y | D | A | K | R | L | I | G | R | D | T | T | V | Q | |||||
| HSPA9 | L | G | T | T | N | A | T | K | R | L | I | G | R | D | P | E | V | Q | |||||
| HSP12A | F | G | T | T | S | Y | A | A | R | D | F | Y | H | P | N | E | A | K | |||||
| HSPA12b | F | G | T | T | S | Y | T | A | R | D | Y | Y | H | P | E | E | A | R | |||||
| HSPA13 | L | G | T | T | Y | D | A | K | R | F | I | G | K | A | E | E | L | E | |||||
| HSPA14 | L | G | C | T | S | K | V | K | Q | I | L | G | R | D | P | Q | A | Q |
| 145 | 146 | 147 | 148 | 149 | 150 | 151 | 152 | 153 | 154 | 155 | 156 | 173 | 174 | 175 | 176 | 177 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HSPA8 | T | V | P | A | Y | F | N | D | S | Q | R | Q | I | N | E | P | T | ||||||
| HspA1A | T | V | P | A | Y | F | N | D | S | Q | R | Q | I | N | E | P | T | ||||||
| HspA1L | T | V | P | A | Y | F | N | D | S | Q | R | Q | I | N | E | P | T | ||||||
| Hspa2 | T | V | P | A | Y | F | N | D | S | Q | R | Q | I | N | E | P | T | ||||||
| HSPA5 | T | V | P | A | Y | F | N | D | A | Q | R | Q | I | N | E | P | T | ||||||
| HSPA6 | T | V | P | A | Y | F | N | D | S | Q | R | Q | I | N | E | P | T | ||||||
| HSPA9 | T | V | P | A | Y | F | N | D | S | Q | R | Q | I | N | E | P | T | ||||||
| HSP12A | T | V | P | A | I | W | K | Q | P | A | K | Q | A | L | E | P | E | ||||||
| HSPA12b | T | V | P | A | I | W | K | Q | P | A | K | Q | A | L | E | P | E | ||||||
| HSPA13 | S | V | P | A | E | F | D | L | K | Q | R | N | I | N | E | P | T | ||||||
| HSPA14 | T | V | P | F | D | F | G | E | K | Q | K | N | I | H | E | P | S |
| 202 | 203 | 204 | 205 | 206 | 207 | 208 | 220 | 221 | 222 | 223 | 224 | 225 | 226 | 227 | 228 | 229 | 230 | 318 | 319 | 320 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HSPA8 | G | G | T | F | D | V | S | K | S | T | A | G | D | T | H | L | G | G | E | K | A | ||
| HspA1A | G | G | T | F | D | V | S | K | A | T | A | G | D | T | H | L | G | G | E | K | A | ||
| HspA1L | G | G | T | F | D | V | S | K | A | T | A | G | D | T | H | L | G | G | E | K | A | ||
| Hspa2 | G | G | T | F | D | V | S | K | S | T | A | G | D | T | H | L | G | G | E | K | A | ||
| HSPA5 | G | G | T | F | D | V | S | V | A | T | N | G | D | T | H | L | G | G | Q | K | V | ||
| HSPA6 | G | G | T | F | D | V | S | K | A | T | A | G | D | T | H | L | G | G | E | K | A | ||
| HSPA9 | G | G | T | F | D | I | S | K | S | T | N | G | D | T | F | L | G | G | Q | K | A | ||
| HSP12A | G | G | T | V | D | L | T | K | A | T | G | G | P | Y | G | S | L | G | R | D | L | ||
| HSPA12b | G | G | T | V | D | L | T | K | A | S | G | G | P | Y | G | A | V | G | E | A | L | ||
| HSPA13 | G | G | T | L | D | V | S | R | A | M | S | G | N | N | K | L | G | G | Q | Q | V | ||
| HSPA14 | G | T | S | L | S | L | S | L | S | T | N | T | D | D | N | I | G | G | R | G | L |
One needs a detailed description of the interaction of MKT-077 with HSPA, to suggest where modifications can be made to improve MKT-077. However, our AUTODOCK computations cannot distinguish between different orientations of the compound. For instance, in the AUTODOCK family depicted in Fig 10C, the positive pyridinium ring is reaching into the pocket to contact Glu 175 and Asp 206. In the family depicted in Fig 10D, partial contact occurs. In the other families, the positive pyridinium ring is solvent-exposed. In family 10F, it is in contact with Asp 225, which lines the rim of the pocket (at 2 o’clock in Figure 10F). AUTODOCK slightly favors the family of Fig 10C (pyridinium ring buried), with an energy of −7.03 kcal/mol. However, experimental evidence strongly suggests that the pyridinium ring is not buried: MKT-077 derivatives with larger poly-aromatics in place of the pyridinium moiety are equally or more potent73. In addition, through derivitization of the pyridinium moiety, MKT-077 could be covalently linked to Sepharose and be used to bind to HSPA9 75.
Prima facie, there seems little reason for the methylbenzothiazolin moiety (left in Fig 1) to seek the negatively charged pocket, since it does not carry a formal net charge. However, when using the AM1-BCC method76 to compute the charge distribution on MKT-077, one obtains a sizeable positive charge on this moiety due to polarization (see Fig 1). The charges obtained by the AM1-BCC method are known76 to correspond closely to the “true” quantum mechanical charges. In addition, one may expect MKT-077 to become even more polarized when placed in the vicinity of negative charges.
We became interested if one could distinguish between the different AUTODOCK poses by using the MMGB/PBSA solvation / desolvation computational protocol77 on basis of snapshots of molecular dynamics simulations of the complex (see Materials). We first tested whether we could distinguish between pyridinium “in” and “out” conformations. As detailed in the Materials section, carefully equilibrated 250 ps molecular dynamics runs for the complexes as shown in Figure 10, Panel C, (pyridinium “in”) and in Figure 10, Panel F, (pyridinium “out”) were obtained using Amber 11. We used the Generalized Born / Poisson Boltzmann protocol to estimate the solvation enthalpy of ligand, protein and complex (MMGB/PBSA). For the pyridinium “in” conformation (7.03 kcal/mol binding energy by AUTODOCK) a total AMBER binding enthalpy of −12.1 +/− 4.1 kcal/mol was obtained, while for the pyridinium “out” conformation (5.25 kcal/mol binding energy by AUTODOCK) an AMBER binding enthalpy of −18.3 +/− 2.9 kcal/mol was obtained (Table 2). This indicates a marked preference for the correct pyridinium “out” conformation in AMBER. Moreover, since the pyridinium is the only freely rotatable moiety in MKT-077 which is still relatively free to move in the pyridinium “out” conformation (from checking the trajectories), the pyridinium “out” conformation should also have a substantial entropic advantage as compared to the pyridinium “in” conformation. Having passed this test, we proceeded to try to distinguish between the different pyridinium “out” poses as shown in Figs 10 D, E and F, which have indistinguishable AUTODOCK energies of 6.32, 5.36 and 5.25 kcal/mol, respectively. As is shown in Table 2, the results of the Amber calculations are also too close to make this call, but the family shown in Fig 10F has the best score.
Table 2.
MKT-HSPA8 docking energies
| Subfamily in Fig 10 | AUTODOCK Energy |
AMBER GB Energy |
AMBER PBSA Energy |
AMBER GB/PB average |
|---|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | kcal/mol | |
| Panel C | 7.03 | −16.6 ± 1.8 | −7.5 ± 3.7 | −12.1 ± 4.1 |
| Panel D | 6.32 | −18.6 ± 1.5 | −14.0 ± 2.1 | −16.3 ± 2.8 |
| Panel E | 5.36 | −18.1 ± 1.2 | −10.8 ± 2.0 | −14.5 ± 2.3 |
| Panel F | 5.25 | −22.8 ± 1.3 | −13.8 ± 2.6 | −18.3 ± 2.9 |
The AMBER results may be used to guide MKT-077 modifications that would allow tighter binding. No modification should be made on non-pyridinium moieties of the molecule since these parts are buried in the pocket. An obvious modification is to add positive charge to the compound, as it binds to an area that has overall strong negative electric potential as is shown in Figure 7. However, positively charged compounds are more toxic to the kidneys than neutral compounds78. Hence, adding extra charge to MKT-077 is likely to exacerbate the toxic side-effects79 which were cause for the termination of the Phase I trial54. MKT-077’s renal toxicity may potentially be reduced by converting MKT-077 to a zwitter-ion, with a negative functionality on the pyridinium moiety to complement the sidechain of His 227. His 227 is very close to the MKT pyridinium moiety in the best pose (see Tables 1, 2 and Figure 11). By trying to exploit such an interaction, the compound’s affinity may be enhanced as well. However, the negative charge would likely interfere with MKT-077’s ability to selectively target the strongly negatively charged mitochondria of tumor cells80. Hence, the suggested conversion of MKT-077 to a zwitter-ion will likely take MKT-077 out of contention as an anti-tumor drug, but may allow it to be used in the treatment of tauopathies such as Alzheimer’s, potentially without the toxic renal side-effects found in the phase I trial. In fact, neutralization of the overall charge of MKT-077 could help enhance the compound’s ability to cross the blood-brain barrier81.
Fig 11.

HSPA8 residues in contact with the five MKT-077 docking families.
Red, contacts with 4 or 5 families; orange, contacts with 2 families; yellow, contacts with 1 family. His 227 is shown in salmon (see text). Mg2+ is in blue, ADP in cyan. The docked MKT-077 structure of Fig 10 F is shown in green. Also compare Table 1. The figure was generated in Pymol84.
The pyridinium moiety is also close to an apolar surface area composed of residues V82 and T226. Modification of the pyridinium moiety to a larger poly-aromatic to exploit additional dispersion and hydrophobic interactions with these residues, may lead to a compound with a higher affinity. This may explain why the larger MKT-derivatives FJ-5744 and FJ-5826 are more potent HSPA inhibitors73.
Conclusion
We have determined that MKT-077 binds to a negatively charged pocket at the interface of the four HSPA8 NBD subdomains. All residues lining the pocket are conserved in all human HSPA isoforms. The compound binds selectively to the ADP state of the protein, and can hence be qualified as an allosteric drug. We suggest modifications that would potentially retain the compound’s binding affinity but which likely would diminish the compound’s renal toxicity.
Materials and Methods
Protein preparation
The plasmid for the expression of bovine HSPA8 NBD domain (100% identical to the human protein) was a generous donation by Dr. D. B. McKay (Stanford School of Medicine, Stanford, CA). It was expressed in Escherichia coli strain BL21(DE3). In order to increase the yield of the induction, the cells were inoculated from a freshly transformed Petri dish a 2ml LB culture, were grown until OD(600)=0.4, transferred to a 100ml flask of M9 media and were grown until OD(600)=0.4. Subsequently the cells were centrifuged and transferred to a 1L M9 medium containing 98% D20, 2g/l protonated [13C glucose], and 1g/l 15NH4Cl. The expression was induced by isopropylthio-B-galactoside to 0.5mM at OD(600)=0.8. Harvested cells were removed by centrifugation and disrupted mechanically by a French press. Cell debris was removed by centrifugation and the supernatant was loaded onto a DEAE52 column and eluted with 150mM KCl. The Hsc70 fractions were dialyzed against 20mM HEPES (pH 7.0)/25mM KCl/10mM EDTA. The EDTA was precipitated by the addition of 25mM MgCl2 in the dialyzed protein pool yielding 5mM free Mg+2. The precipitate of (Mg)EDTA was removed by centrifugation. The supernatant was loaded onto an ATP-agarose affinity column and was eluted with 3mM ADP. The protein was concentrated by using Amicon concentrators. The protein was at all times stabilized with protein inhibitors and was kept at 4C.
NMR Assignments
For the assignment of the protein’s NMR spectrum, we used a 15N, 13C, 2H labeled 280 μM HSPA8-NBD sample that contained 5mM MgCl2, 25mM KCl, 20mM Tris-HCl, 10mM ADP, 5 mM K3PO4, 0.005% sodium azide and 10% (vol/vol) D20 (pH 7.2). The experiments were performed at 26 °C on a Varian Inova 800 MHz NMR spectrometer equipped with a triple-resonance cold probe. The protein backbone resonances were assigned from 3D HNCA, HN(CO)CA, HNCO, HN(CA)CO, HNCACB, HN(CO)CACB TROSY data. All spectra were processed with NMRPIPE82 and converted into Sparky83. The Sparky program was used to peak-pick the spectra. The peak pick lists were manually curated to obtain consistent NH root labeling throughout the different spectra. Assignments were made from these curated lists using the automatic assignment program SAGA60. Alternative assignments as produced by SAGA were evaluated and further checked using SPARKY. Only assignments of stretches of more then 2 connected spin systems were retained. By enlarge, they corresponded to the previous manual assignments for this protein domain 59.
Synthesis of MKT-077
The dye, originally manufactured by Fuji, Inc, is no longer commercially available. It was synthesized by us (Y.M. and J.E.G). The synthetic protocol will be described elsewhere. The 1H spectrum of MKT-077 in H2O was assigned using DQF-COSY and ROESY acquired on a 500 MHz Bruker Avance spectrometer. The ROESY spectra confirmed the trans configuration of the pyridinium ring and the nitrogen in the oxothiazolidin moiety over the double bond as shown in Fig 1.
NMR Titrations
HSPA8 NBD samples of 80 – 240 μM in 5mM MgCl2, 25mM KCl, 20mM Tris-HCl,10mM ADP, 5 mM K3PO4, 0.005% sodium azide and 10% (vol/vol) D20 (pH 7.2) were used for the titrations, using 4 mM solutions of MKT-077 in water (neutral pH) as titrant. The results of two titrations are reported here. Using a sample of 80 μM 15N-labeled HSPA8-NBD, MKT-077 was added to ratios of 1:1 and 2:1. Duplicate TROSY spectra of 8 hours each were recorded for each titration step. Using a triple-labeled 270 μM sample of HSPA8 NBD, MKT was titrated to ratios of 0.25:1, 0.5:1, 0.75:1 and 1:1. Duplicate TROSY spectra of 4 hours each were recorded for each titration step. The shifts upon the addition of the drugs were manually recorded in Sparky and were mapped on the crystal structure coordinates of HspA8 NBD (3HSC.pdb) using Pymol84.
Partial Proteolysis
The partial proteolysis protocol was adapted from a previously described method85. Briefly, HSPA8 (6 μM) in 40 mM HEPES buffer (20 mM NaCl, 8 mM MgCl2, 20 mM KCl, 0.3 mM EDTA, pH 8.0) was incubated with 1 mM nucleotide (ADP or ATP), either a buffer control or MKT-077 (200 μM) and a J-domain (residues 2-108, 24 μM) when noted. Samples were incubated at room temperature for 30 minutes. The trypsin (SIGMA Ec 3.4.2.1.4) was added to a final concentration of 0.9 μM and the samples were incubated for another 30 minutes at room temperature. The reaction was then quenched with the addition of 25 μl of 3x SDS loading buffer (240 mM Tris, 6 % w/v SDS, 30 % v/v glycerol, and 16 % v/v β-mercaptoethanol, 0.6 mg/ml bromophenol blue, pH 6.8) and heated to 95 °C for 3 minutes. Bands were separated by 12 % SDS-PAGE and stained with Coomassie blue.
Docking computations
AUTODOCK-486 was used for the docking of MKT-077 to HSPA8-NBD. The initial structure for MKT-077 was minimized in Jaguar (Schrödinger, LLC) at the B3LYP/6-31G* level. Standard charging methods within AUTODOCK leave MKT-077 uncharged. To obtain better charging models, we used the Antechamber87 suite in AMBER72. In one round of computations, we use Gasteiger charging as afforded by Antechamber, and just added +1 to the charge of the pyridinium nitrogen. We also used AM1-BCC charges as computed in AMBER. AM1-BCC predicts considerable polarization over the remainder of the conjugated molecule (see Fig 1) which also seems more realistic than the charge pattern in Gasteiger in which polarization is absent. The AMBER-charged models were hand-edited into the AUTODOCK. pdbqt files.
All available crystal structures for the NBD of HSPA8 conform to the closed (ATP) state61. These structures cannot be used for docking because MKT-077 does not bind to this state. In absence of a “true” high-resolution structure for the open ADP state, we chose one of the available crystal structures of HSPA8 in complex with a nucleotide exchange factor. According to the NMR analysis of a bacterial Hsp70 in the ADP and ATP state, HSPA8 in complex with a nucleotide exchange factor is a reasonable representation of the ADP state61. We chose HSPA8 in complex with yeast Hsp110 (3C7N.pdb88) as our model, because it is the only complex that contains ADP and Mg++. We discarded the coordinates of Hsp110 in the docking computations. In order to relieve possible strain exerted by crystal and/or Hsp110, the extracted HSPA8-NBD coordinates were relaxed using restrained minimization and equilibration in AMBER (see below). The coordinates of the equilibrated protein, including ADP and Mg++ were used for the AUTODOCK runs.
The AUTODOCK gridbox was located on the interface of the four subdomains IA-IB-IIA-IIB with a 0.2 Å resolution. We used a Lamarckian genetic algorithm with the following parameters : the GA runs were set to 100, the size of the initial population to 1500, the maximum number of evaluations was set to long, the maximum number of top individuals that automatically survive was set to 1, the rate of gene mutation was set to 0.02, the rate of crossover was 0.8, the GA crossover mode was twopt, the mean of Cauchy distribution for gene mutation was set to 0, the variance of Cauchy distribution for gene mutation was set to 1 and the number of generations for picking worst individuals was set to 10. The calculations were performed on a MacIntosh computer. The docked structures were clustered and evaluated by hand using Pymol84.
Molecular dynamics
The crystal structure coordinates of HSPA8 in complex with yeast Hsp110, ADP, SO42− and Mg++ ions (3C7N.pdb) were used as a starting point. The coordinates of Hsp110 and SO42− ions were removed. The NBD, ADP and Mg coordinates were minimized in 1000 steps using a hybrid forcefield (ff99SB for the protein, gaff for metal ions and a gaff-derived forcefield89 for ADP) in the Amber 11 program suite72. The coordinates were restrained to the X-ray coordinates with a force constant of 5 kcal/mol/Å2. Subsequently we carried out a molecular Langevin dynamics equilibration run in implicit generalized Born solvent with 0.1 M ionic strength, running from 0 - 300 K over 10 ps. The same hybrid forcefield was used. The dynamics equilibration was restrained by 940 CA-CA distances in the 2-5 Å range, 2952 CA-CA distances in the 10-20 Å range, and 742 restraints between protein CA atoms and all atoms of ADP and Mg++ in the 1-15 Å range. The restraints had as lower bounds the actual distances as computed from the PDB file, and as upper bounds the actual distance + 1 Å, enforced by a parabolic potential of 20 kcal/mol/Å2. The calculations were performed on an Apple MacBookPro5.1 computer equipped with a 64-bit 2.4 GHz Inter Core 2 Duo processor, running MacOS X10.6.6.
Docking evaluation
The AUTODOCK solutions with “energies” of 7.03, 6.32, 5.36 and 5.25 kcal /mol were evaluated using AMBER (vs. 11) in the following way. Hydrogen-atoms were re-attached to the coordinates of the docked MKT-077 molecules and the double and aromatic bonds were reassigned in the Pymol Builder module84. These coordinates were converted to AMBER .prepi and forcefield modification files using the Amber Antechamber suite. We used AM1-BCC charging for MKT-077 as shown in Fig 1.
In order to carry out a proper MD-based binding energy evaluation, it is necessary to run the simulations using explicit solvation. For a molecule the size of HSPA8 NBD (380 residues) such computations become too time consuming for our labs, which do not have access to specialized super computers. Hence, we decided to carry out restrained MD simulations on a docking site consisting of HSPA8 residues 12-13+69-83+143-158+172-177+196-211+219-229+316-323 and the Mg++ ion, which was extracted from the equilibrated pdb files (see Figure S3). Four different docked MKT-077 poses, corresponding to the best members of each of the four families shown in Fig 10, were added the site (not simultaneously). Identical protocols were followed for the four computational series. The “complexes” were solvated with ~6500 TIP3P waters in a 12 Å3 periodic box. After a minimization step, a molecular dynamics temperature ramp running from 0 - 300 K over 50 ps was carried out. The same hybrid force field was used as described above. This run was restrained by 3790 restraints between N, HN, CA, C’ and O backbone atoms of the reduced binding site, 55 Mg++ - CA restraints, and ~2600 MKT - CA restraints based on the minimized structure (depending on family member). All restraints were defined within a 1.0 Å range and enforced with a 20 kcal/mol/Å2 gradient. Subsequently, a density equilibration (for 50 ps, with pressure relaxation time of 1 ps) and general equilization (for 100 ps, with pressure relaxation time of 2 ps) with the same restraints followed. These equilibration steps were followed by a production run of 250 ps restrained MD computations at 300 K. In the production runs, the MKT restraints were removed, while all others remained. The binding enthalpies were calculated using the MM-GB/PBSA protocol77 as implemented in the Amber 11 release, using 100 frames of the last 50 ps of the molecular dynamics production runs. The calculations were performed on an Apple MacBookPro5.1 computer equipped with a 64-bit 2.4 GHz Inter Core 2 Duo processor, running MacOS X10.6.6.
Supplementary Material
Acknowledgements
We thank Dr. Dan Weaver for the sample of triple-labeled HSPA8-NBD. We thank Mr. Peter Ung (lab of Dr. Heather Carlson) for tutorials on AMBER and AUTODOCK and Dr. Heather Carlson for the quantum mechanical calculations on isolated MKT-077. Mr Ung also provided parameters for ADP in AUTODOCK. We thank Dr. Daniel Roe (lab of Dr. David Case) for help with the AM1-BCC charging method in AMBER. Support from NIH grants GM63027-S02 (ERPZ), NS059690 (AR, ERPZ and JEG) and NS073899 and AG031291 (CAD) is gratefully acknowledged. CAD also thanks the Alzheimer’s Association for support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 2.Kampinga H, Hageman J, Vos M, Kubota H, Tanguay R, Bruford E, Cheetham M, Chen B, Hightower L. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chap. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schroder H, Langer T, Hartl FU, Bukau B. Dnak, Dnaj and Grpe Form a Cellular Chaperone Machinery Capable of Repairing Heat-Induced Protein Damage. Embo J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, Zhao R, Puravs E, Tra J, Michael CW, Misek DE, Hanash SM. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 5.Nylandsted J, Brand K, Jaattela M. Heat shock protein 70 is required for the survival of cancer cells. Ann N Y Acad Sci. 2000;926:122–5. doi: 10.1111/j.1749-6632.2000.tb05605.x. [DOI] [PubMed] [Google Scholar]
- 6.Kaul Z, Yaguchi T, Kaul SC, Hirano T, Wadhwa R, Taira K. Mortalin imaging in normal and cancer cells with quantum dot immuno-conjugates. Cell Res. 2003;13:503–7. doi: 10.1038/sj.cr.7290194. [DOI] [PubMed] [Google Scholar]
- 7.Kaul SC, Yaguchi T, Taira K, Reddel RR, Wadhwa R. Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp Cell Res. 2003;286:96–101. doi: 10.1016/s0014-4827(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 8.Garg M, Kanojia D, Seth A, Kumar R, Gupta A, Surolia A, Suri A. Heat-shock protein 70-2 (HSP70-2) expression in bladder urothelial carcinoma is associated with tumour progression and promotes migration and invasion. Eur J Cancer. 2010;46:207–15. doi: 10.1016/j.ejca.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–25. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- 10.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 12.Soti C, Csermely P. Chaperones and aging: role in neurodegeneration and in other civilizational diseases. Neurochem Int. 2002;41:383–9. doi: 10.1016/s0197-0186(02)00043-8. [DOI] [PubMed] [Google Scholar]
- 13.Soti C, Csermely P. Chaperones come of age. Cell Stress Chaperones. 2002;7:186–90. doi: 10.1379/1466-1268(2002)007<0186:ccoa>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abarzua F, Sakaguchi M, Tanimoto R, Sonegawa H, Li DW, Edamura K, Kobayashi T, Watanabe M, Kashiwakura Y, Kaku H, Saika T, Nakamura K, Nasu Y, Kumon H, Huh NH. Heat shock proteins play a crucial role in tumor-specific apoptosis by REIC/Dkk-3. Int J Mol Med. 2007;20:37–43. [PubMed] [Google Scholar]
- 15.Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. 2008;12:743–61. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calderwood SK, Ciocca DR. Heat shock proteins: stress proteins with Janus-like properties in cancer. Int J Hyperthermia. 2008;24:31–9. doi: 10.1080/02656730701858305. [DOI] [PubMed] [Google Scholar]
- 17.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 18.Garrido C, Schmitt E, cande C, Vahsen N, Parcellier A, Kroemer G. Hsp27 and Hsp70: potentially oncogenic apoptosis inhibitors. Cell Cycle. 2003;2:579–584. [PubMed] [Google Scholar]
- 19.Kaul SC, Duncan EL, Englezou A, Takano S, Reddel RR, Mitsui Y, Wadhwa R. Malignant transformation of NIH3T3 cells by overexpression of mot-2 protein. Oncogene. 1998;17:907–11. doi: 10.1038/sj.onc.1202017. [DOI] [PubMed] [Google Scholar]
- 20.Walker C, Bottger S, Low B. Mortalin-based cytoplasmic sequestration of p53 in a nonmammalian cancer model. Am J Pathol. 2006;168:1526–30. doi: 10.2353/ajpath.2006.050603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiological Reviews. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- 22.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VMY. ABNORMAL TAU-PHOSPHORYLATION AT SER(396) IN ALZHEIMERS-DISEASE RECAPITULATES DEVELOPMENT AND CONTRIBUTES TO REDUCED MICROTUBULE-BINDING. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 23.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Research Reviews. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 24.Geschwind DH. Tau phosphorylation, tangles, and neurodegeneration: The chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- 25.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–6. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, O’Leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE, Dickey CA. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29:12079–88. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy Targeting Pathological Tau Prevents Cognitive Decline in a New Tangle Mouse Model. Journal of Neuroscience. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong CX, Grundke-Iqbal I, Iqbal K. Targeting Tau Protein in Alzheimer’s Disease. Drugs & Aging. 2010;27:351–365. doi: 10.2165/11536110-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 29.Powers MV, Jones K, Barillari C, Westwood I, van Montfort RLM, Workman P. Targeting HSP70 The second potentially druggable heat shock protein and molecular chaperone? Cell Cycle. 2010;9:1542–1550. doi: 10.4161/cc.9.8.11204. [DOI] [PubMed] [Google Scholar]
- 30.Flaherty KM, Wilbanks SM, DeLuca-Flaherty C, McKay DB. Structural basis of the 70-kilodalton heat shock cognate protein ATP hydrolytic activity. II. Structure of the active site with ADP or ATP bound to wild type and mutant ATPase fragment. J Biol Chem. 1994;269:12899–907. [PubMed] [Google Scholar]
- 31.McCarty JS, Buchberger A, Reinstein J, Bukau B. The role of ATP in the functional cycle of the DnaK chaperone system. J. Mol. Biol. 1995;249:126–137. doi: 10.1006/jmbi.1995.0284. [DOI] [PubMed] [Google Scholar]
- 32.Zhu XT, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M, Lavan P, Matassova N, Francis GL, Graham CJ, Parsons R, Wang YK, Padfield A, Comer M, Drysdale MJ, Wood M. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemotherapy and Pharmacology. 2010;66:535–545. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- 34.Williamson DS, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Foloppe N, Francis GL, Graham CJ, Howes R, Macias AT, Murray JB, Parsons R, Shaw T, Surgenor AE, Terry L, Wang YK, Wood M, Massey AJ. Novel Adenosine-Derived Inhibitors of 70 kDa Heat Shock Protein, Discovered Through Structure-Based Design. Journal of Medicinal Chemistry. 2009;52:1510–1513. doi: 10.1021/jm801627a. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto S, Nakano S, Owari K, Fuziwara K, Ogawa N, Otaka M, Tamaki K, Watanabe S, Komatsuda A, Wakui H, Sawada K, Kubota H, Itoh H. Gentamicin inhibits HSP70-assisted protein folding by interfering with substrate recognition. FEBS letters. 2010;584:645–651. doi: 10.1016/j.febslet.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 36.Otaka M, Yamamoto S, Ogasawara K, Takaoka Y, Noguchi S, Miyazaki T, Nakai A, Odashima M, Matsuhashi T, Watanabe S, Itoh H. The induction mechanism of the molecular chaperone HSP70 in the gastric mucosa by Geranylgeranylacetone (HSP-inducer) Biochemical and Biophysical Research Communications. 2007;353:399–404. doi: 10.1016/j.bbrc.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 37.Rerole AL, Gobbo J, De Thonel A, Schmitt E, de Barros JPP, Hammann A, Lanneau D, Fourmaux E, Deminov O, Micheau O, Lagrost L, Colas P, Kroemer G, Garrido C. Peptides and Aptamers Targeting HSP70: A Novel Approach for Anticancer Chemotherapy. Cancer Research. 2011;71:484–495. doi: 10.1158/0008-5472.CAN-10-1443. [DOI] [PubMed] [Google Scholar]
- 38.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–51. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Hohfeld J, Minami Y, Hartl F-U. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. cell. 1995;83:589–598. doi: 10.1016/0092-8674(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 40.Johnson BD, Schumacher RJ, Ross ED, Toft DO. Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem. 1998;273:3679–3686. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- 41.Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 42.Burri L, Vascotto K, Fredersdorf S, Tiedt R, Hall MN, Lithgow T. Zim17, a novel zinc finger protein essential for protein import into mitochondria. J Biol Chem. 2004;279:50243–9. doi: 10.1074/jbc.M409194200. [DOI] [PubMed] [Google Scholar]
- 43.Nadler SG, Dischino DD, Malacko AR, Cleaveland JS, Fujihara SM, Marquardt H. Identification of a binding site on Hsc70 for the immunosuppressant 15-deoxyspergualin. Biochemical and Biophysical Research Communications. 1998;253:176–180. doi: 10.1006/bbrc.1998.9775. [DOI] [PubMed] [Google Scholar]
- 44.Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem. 2004;279:51131–40. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- 45.Graf C, Stankiewicz M, Nikolay R, Mayer MP. Insights into the Conformational Dynamics of the E3 Ubiquitin Ligase CHIP in Complex with Chaperones and E2 Enzymes. Biochemistry. 2010;49:2121–2129. doi: 10.1021/bi901829f. [DOI] [PubMed] [Google Scholar]
- 46.Wisen S, Bertelsen EB, Thompson AD, Patury S, Ung P, Chang L, Evans CG, Walter GM, Wipf P, Carlson HA, Brodsky JL, Zuiderweg ERP, Gestwicki JE. Binding of a Small Molecule at a Protein-Protein Interface Regulates the Chaperone Activity of Hsp70-Hsp40. Acs Chemical Biology. 2010;5:611–622. doi: 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertelsen EB, Zuiderweg ER. Solution conformation of E.Coli Hsp70 complexed with ADP and substrate. Proceedings of the National Academy of Sciences, USA. 2008 doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bork P, Sander C, Valencia A. AN ATPASE DOMAIN COMMON TO PROKARYOTIC CELL-CYCLE PROTEINS, SUGAR KINASES, ACTIN, AND HSP70 HEAT-SHOCK PROTEINS. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang L, Miyata Y, Ung PM, Bertelsen EB, McQuade TJ, Carlson HA, Zuiderweg ER, Gestwicki JE. Chemical Screens against a Reconstituted Multiprotein Complex: Myricetin Blocks DnaJ Regulation of DnaK through an Allosteric Mechanism. Chem Biol. 2011;18:210–21. doi: 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, Maruta H, Kaul SC. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60:6818–21. [PubMed] [Google Scholar]
- 52.Tikoo A, Shakri R, Connolly L, Hirokawa Y, Shishido T, Bowers B, Ye LH, Kohama K, Simpson RJ, Maruta H. Treatment of ras-induced cancers by the F-actin-bundling drug MKT-077. Cancer Journal. 2000;6:162–168. [PubMed] [Google Scholar]
- 53.Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido, Chen LB. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56:538–43. [PubMed] [Google Scholar]
- 54.Propper D, Braybrooke J, Taylor D, Lodi R, Styles P, Cramer J, Collins W, Levitt N, Talbot D, Ganesan T, Harris A. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo-resistant solid tumours. Annal of Oncology. 1999;10:923–927. doi: 10.1023/a:1008336904585. [DOI] [PubMed] [Google Scholar]
- 55.Pilzer D, Saar M, Koya K, Fishelson Z. Mortalin inhibitors sensitize K562 leukemia cells to complement-dependent cytotoxicity. Int J Cancer. 2010;126:1428–35. doi: 10.1002/ijc.24888. [DOI] [PubMed] [Google Scholar]
- 56.Modica-Napolitano JS, Nalbandian R, Kidd ME, Nalbandian A, Nguyen CC. The selective in vitro cytotoxicity of carcinoma cells by AZT is enhanced by concurrent treatment with delocalized lipophilic cations. Cancer Lett. 2003;198:59–68. doi: 10.1016/s0304-3835(03)00274-x. [DOI] [PubMed] [Google Scholar]
- 57.Abdul M, Hoosein N. Potentiation of the antiproliferative activity of MKT-077 by loperamide, diltiazem and tamoxifen. Oncol Rep. 2003;10:2023–6. doi: 10.3892/or.10.6.2023. [DOI] [PubMed] [Google Scholar]
- 58.Chang L, Bertelsen EB, Wisen S, Larsen EM, Zuiderweg ER, Gestwicki JE. High-throughput screen for small molecules that modulate the ATPase activity of the molecular chaperone DnaK. Anal Biochem. 2008;372:167–76. doi: 10.1016/j.ab.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Zuiderweg ER. The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci U S A. 2004;101:10272–7. doi: 10.1073/pnas.0401313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crippen GM, Rousaki A, Revington M, Zhang Y, Zuiderweg ER. SAGA: rapid automatic mainchain NMR assignment for large proteins. J Biomol NMR. 2010;46:281–98. doi: 10.1007/s10858-010-9403-2. [DOI] [PubMed] [Google Scholar]
- 61.Bhattacharya A, Kurochkin AV, Yip GN, Zhang Y, Bertelsen EB, Zuiderweg ER. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J Mol Biol. 2009;388:475–90. doi: 10.1016/j.jmb.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J. Biol. Chem. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- 63.Arnone A, Perutz MF. STRUCTURE OF INOSITOL HEXAPHOSPHATE-HUMAN DEOXYHEMOGLOBIN COMPLEX. Nature. 1974;249:34–36. doi: 10.1038/249034a0. [DOI] [PubMed] [Google Scholar]
- 64.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–8. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 65.Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281:16493–501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- 66.Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281:38705–11. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 67.Schlecht R, Erbse AH, Bukau B, Mayer MP. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol. 2011;18:345–51. doi: 10.1038/nsmb.2006. [DOI] [PubMed] [Google Scholar]
- 68.Jinwal UK, Miyata Y, Koren J, Jones JR, Trotter JH, Chang L, O’Leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ERP, Gestwicki JE, Dickey CA. Chemical Manipulation of Hsp70 ATPase Activity Regulates Tau Stability. Journal of Neuroscience. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Human Molecular Genetics. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 70.Morris G, Goodsell D, Halliday R, Huey R, Hart W, RK B, AJ O. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 71.Flaherty KM, Deluca-Flaherty C, McKay DB. 3-Dimensional Structure of the ATPase Fragment of a 70k Heat-Shock Cognate Protein. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 72.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr., Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, Maruta H, Kaul SC. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Research. 2000;60:6818–6821. [PubMed] [Google Scholar]
- 74.Deocaris CC, Widodo N, Shrestha BG, Kaur K, Ohtaka M, Yamasaki K, Kaul SC, Wadhwa R. Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer Lett. 2007;252:259–69. doi: 10.1016/j.canlet.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 75.Kawakami M, Suzuki N, Sudo Y, Shishido T, Maeda M. Development of an enzyme-linked immunosorbent assay (ELISA) for antitumor agent MKT 077. Analytica Chimica Acta. 1998;362:177–186. [Google Scholar]
- 76.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. Journal of Computational Chemistry. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 77.Fogolari F, Brigo A, Molinari H. Protocol for MM/PBSA molecular dynamics simulations of proteins. Biophys J. 2003;85:159–66. doi: 10.1016/S0006-3495(03)74462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vegt E, de Jong M, Wetzels JF, Masereeuw R, Melis M, Oyen WJ, Gotthardt M, Boerman OC. Renal toxicity of radiolabeled peptides and antibody fragments: mechanisms, impact on radionuclide therapy, and strategies for prevention. J Nucl Med. 2010;51:1049–58. doi: 10.2967/jnumed.110.075101. [DOI] [PubMed] [Google Scholar]
- 79.Somogyi A. Renal transport of drugs: specificity and molecular mechanisms. Clin Exp Pharmacol Physiol. 1996;23:986–9. doi: 10.1111/j.1440-1681.1996.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 80.Chiba Y, Kubota T, Watanabe M, Otani Y, Teramoto T, Matsumoto Y, Koya K, Kitajima M. Selective antitumor activity of MKT-077, a delocalized lipophilic cation, on normal cells and cancer cells in vitro. Journal of Surgical Oncology. 1998;69:105–110. doi: 10.1002/(sici)1096-9098(199810)69:2<105::aid-jso11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 81.Doniger S, Hofmann T, Yeh J. Predicting CNS permeability of drug molecules: comparison of neural network and support vector machine algorithms. Journal of Computational Biology. 2002;9:849–864. doi: 10.1089/10665270260518317. [DOI] [PubMed] [Google Scholar]
- 82.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 83.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 2000. [Google Scholar]
- 84.DeLano W. The PyMOL Molecular Graphics System DeLano Scientific. San Carlos; CA, USA: 2002. [Google Scholar]
- 85.Chang L, Thompson AD, Ung P, Carlson HA, Gestwicki JE. Mutagenesis Reveals the Complex Relationships between ATPase Rate and the Chaperone Activities of Escherichia coli Heat Shock Protein 70 (Hsp70/DnaK) Journal of Biological Chemistry. 2010;285:21282–21291. doi: 10.1074/jbc.M110.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morris G, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson A. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J. Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- 87.Wang JM, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. Journal of Molecular Graphics & Modelling. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 88.Schuermann JP, Jiang JW, Cuellar J, Llorca O, Wang LP, Gimenez LE, Jin SP, Taylor AB, Demeler B, Morano KA, Hart PJ, Valpuesta JM, Lafer EM, Sousa R. Structure of the Hsp110: Hsc70 nucleotide exchange machine. Molecular Cell. 2008;31:232–243. doi: 10.1016/j.molcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meagher K, Redman L, Carlson H. Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry. 2003;24:1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 90.Lerner M, Carlson H. Pymol APBS Tools. 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.