

Abstract
We investigated three families whose offspring had extreme microcephaly at birth and profound mental retardation. Brain scans and postmortem data showed that affected individuals had brains less than 10% of expected size (≤10 standard deviation) and that in addition to a massive reduction in neuron production they displayed partially deficient cortical lamination (microlissencephaly). Other body systems were apparently unaffected and overall growth was normal. We found two distinct homozygous mutations of NDE1, c.83+1G>T (p.Ala29GlnfsX114) in a Turkish family and c.684_685del (p.Pro229TrpfsX85) in two families of Pakistani origin. Using patient cells, we found that c.83+1G>T led to the use of a novel splice site and to a frameshift after NDE1 exon 2. Transfection of tagged NDE1 constructs showed that the c.684_685del mutation resulted in a NDE1 that was unable to localize to the centrosome. By staining a patient-derived cell line that carried the c.83+1G>T mutation, we found that this endogeneously expressed mutated protein equally failed to localize to the centrosome. By examining human and mouse embryonic brains, we determined that NDE1 is highly expressed in neuroepithelial cells of the developing cerebral cortex, particularly at the centrosome. We show that NDE1 accumulates on the mitotic spindle of apical neural precursors in early neurogenesis. Thus, NDE1 deficiency causes both a severe failure of neurogenesis and a deficiency in cortical lamination. Our data further highlight the importance of the centrosome in multiple aspects of neurodevelopment.
Introduction
The human brain has evolved rapidly in size and complexity; in the short evolutionary time scale of 5 million years it has tripled in relative weight compared to other higher apes.1,2 The principal explanation for this increase is an expansion of the cerebral cortex, a mammalian-specific brain structure.3,4 This process of encephalization is hypothesized to underpin our increased intellect and has resulted in both our environmental adaptability and our language development.5 Here, we present data suggesting that NDE1 is a major component facilitating encephalization and describing a disease characterized by a profound reduction in prenatal neuron production that results in affected brains less than 10% of expected size. The degree of reduction in brain and cerebral cortex size is far greater than that seen in primary autosomal-recessive microcephaly, caused by biallelic ASPM mutations (MCPH5 [MIM 608716]) among others, or in microcephalic osteodysplastic primordial dwarfism type 2, caused by biallelic PCNT mutations (MOPD II [MIM 210720]) that produce, in addition to microcephaly, significant reductions in whole-body growth.6–8 Both of these phenotypes have also been considered potential atavistic human diseases of encephalization, and in support of this, genes involved in MCPH have been shown to have undergone significant Darwinian selection in parallel with the increase in size that began with monkey and ape brains and culminates in human brains.9,10 NDE1, however, has a more complex role in human neurodevelopment because it affects not only neuron production but also cortical lamination. Complementary data are presented by Alkuraya et al. in this issue.11
Subjects and Methods
Clinical Studies
We investigated three families in which offspring had extreme congenital microcephaly (brains smaller than −10 standard deviation [SD]) and profound mental retardation (Figures 1A and 1B). The study received approval from the local research ethics committee (Yale human investigation committee protocol number 0908005592) and the informed written consent from the families involved was obtained by the referring physicians.
Figure 1.
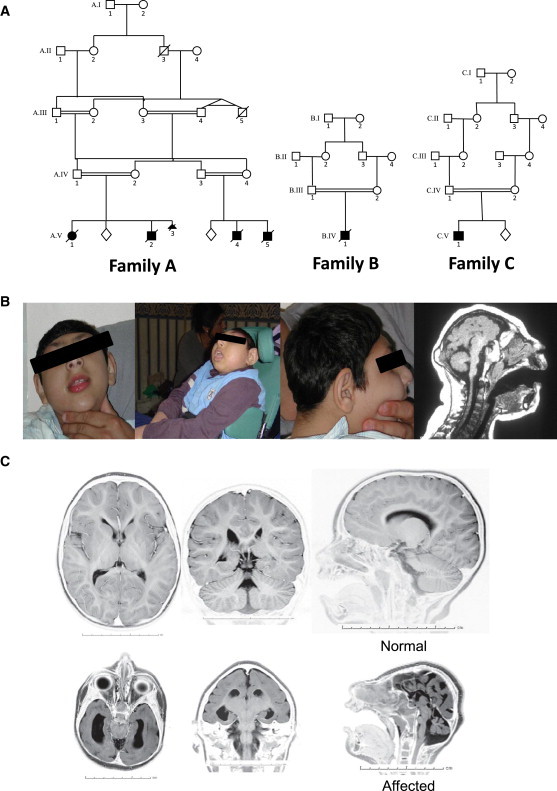
Clinical Details of Research Families
(A) The pedigrees of families A, B, and C. The closest consanguineous links between family members are shown; affected individuals are filled-in shapes and unaffected sibling existence is marked as a lozenge. Number and gender of unaffected siblings is undisclosed to protect privacy.
(B) Pictures of affected children. (Right) A sagittal plane image of the computed tomography brain scan of an affected child at 4 years old.
(C) Magnetic resonance images of an affected child at 2 years old. Axial, coronal, and sagittal MR images show severe microcephaly with cortical simplification, agenesis of the corpus callosum, and a hypoplastic cerebellum. The upper three panels show a control for comparison. Photographically inverted T2 images are shown.
Using Molecular Genetic Studies to Identify NDE1 Mutations
Families A and B were evaluated and analyzed independently of family C.
For families A and B, we used an autozygosity mapping approach to seek genetic linkage, whereas in family C the mutation was concomitantly identified by whole-exome sequencing. Standard protocols were used for blood and DNA preparation.
We used genomic DNA from two of the affected children in family A (individuals A.V-1 and A.V-2 from Figure 1) and a modified Weber panel of polymorphic microsatellite markers, as previously described.12 We used an ExcludeAR analysis, which is based on the principle that any chromosomal region that remains after excluding regions of nonlinkage must contain the locus of interest, to delineate five regions of concordant homozygosity shared between the two affected individuals.13 We then used the panel of microsatellite markers to examine the single affected individual from family B (individual B.IV-1 in Figure 1) and look for homozygosity at each of these five loci. This individual (B.IV-1) was only homozygous for 9 cM of chromosome 16. We designed further polymorphic microsatellite markers to confirm and augment this result, define the boundaries of the shared region, and seek a region of common haplotype between both families.12 We selected candidate genes on the basis of our knowledge of the primary microcephaly genes, whether the gene was expressed in the neuroepithelium of the prenatal brain, and whether the protein was associated with centrosomes.14 We designed primers to sequence all recorded NDE1 exons (NM_001143979.1, CCDS10564.1), including splice sites and polyadenylation signals; we used the Human Genome Browser, Primer3, and BLAST. Genomic DNA from affected individuals from each family was sequenced. We used the sequencing primers to test whether the mutation segregated as a recessive disease phenotype by sequencing the affected exon (exon 6) of NDE1 in all available family members. We also used the same primers to sequence DNA samples from 160 healthy, unrelated Pakistani adults.
In family C, we used a Sequence Capture Human Exome 2.1M Array (Roche NimbleGen) according to the manufacturer's protocol to capture the exome of the affected individual (individual C.V-1 in Figure 1). We performed sequencing of the library with a Genome Analyzer IIx (Illumina) by using a single-lane single-read sequencing at a read length of 74 base pairs. The Illumina pipeline version 1.5 was utilized for image analysis and base calling. Analysis of the sequencing data was performed according to the previously described data-analysis pipeline15 (see Tables S1–S4, available online). In brief, the reads were mapped to the human genome (NCBI36/hg18) by Maq and BWA software.16,17 Coverage rates and distributions, as well as error positions and frequencies, were detected with Perl scripts developed in house. Variants were identified with SAMtools18 and annotated for novelty as compared to the dbSNP build 131, nine personal genomes published, and 1000 Genomes database (August 4, 2010 release), as well as additional exome sequencing experiments that we have performed. Novel variants were further evaluated for their impact on the encoded protein; conservation across 44 vertebrate species, Caenorhabditis elegans, and Drosophila melanogaster; expression patterns; and potential overlap with known miRNAs.
mRNA NDE1 Studies of the c.83+1G>T Mutation
The total RNA was isolated from both the lymphoblastoid cell lines immortalized with Epstein-Barr virus and fresh blood from the patient and controls with RNeasy Mini Kit (QIAGEN) according to the manufacturer's instructions. After isolation, RNA samples were treated with DNase (Turbo DNase, Ambion) and quantified. mRNA was reverse transcribed into first-strand cDNA with Oligo dt primers (Invitrogen) and the Omniscript RT Kit (QIAGEN) according to the manufacturer's instructions. Multiple primer pairs spanning exon-exon junctions of NDE1 were designed with University of California Santa Cruz (UCSC) Genome Browser, BLAST, and Sequencher 4.8 (Gene Codes, Ann Arbor, MI, USA). PCR amplification of NDE1 cDNA from the first-strand cDNA libraries was performed. The PCR products were evaluated by gel electrophoresis for specificity, cycle sequenced on ABI's 9800 Fast Thermocyclers, and analyzed on 3730xl DNA Analyzer (Applied Biosystems).
NDE1 Cloning and Generation of Expression Plasmids
We amplified full-length NDE1 by PCR with IMAGE clone 3140369 (BC001421) as a template and cloned into the XhoI and BamHI restriction sites of pEGFP-TUB (Clontech) to get the final clone, NDE-WT (full-length NDE1 tagged at its N terminus with EGFP). The c.684_685del (p.Pro229TrpfsX85) mutation was inserted into NDE1-WT with the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer's instructions to give clone NDE1-M. A stop codon was introduced at the site of the c.684_685del mutation to give clone NDE1-MSTOP. We made this clone by using BC001421 as template and including a stop codon in the reverse PCR primer, then cloning this product into the XhoI and BamHI restriction sites of pEGFP-TUB. All constructs were sequenced to verify their identity. Cloning primers are available on request.
NDE1 mRNA In Situ Hybridization Studies
Human embryonic brain sections were obtained from the MRC-Wellcome Trust Human Developmental Biology Resource, Institute of Human Genetics, Newcastle University. The samples were collected with appropriate maternal consents and ethical approval by the Newcastle and North Tyneside Research Ethics Committee. We used sections of Carnegie stage (CS) 23 (∼58 days after conception). Sense and antisense probes were made as previously described.19 The riboprobes were designed so that they would specifically detect NDE1 mRNA and not the closely homologous NDEL1. The riboprobes used correspond to nucleotides 2767–3223 of NDE1 cDNA (NM_001143979.1). The results were compared to those we concurrently generated by using a known positive control to SHH (MIM 600725) where all cells of the notochord showed expression (data not shown).
Transfection of NDE1-GFP Fusion Constructs
Cell lines were cultured in DMEM supplemented with 10% FCS. HeLaM cells were transiently transfected with the GFP-NDE1 constructs with either Lipofectamine 2000 (Invitrogen) or TransIT-LT1 (Mirus). 24 hr after transfection, the cells were fixed with ice-cold methanol for 5 min, permeabilized with acetone, and blocked with 3% BSA for 40 min. GFP was directly visualized. Staining was carried out with mouse monoclonal anti-γ-tubulin (Abcam, ab27074), which was detected with the secondary Alexa Fluor 546 goat anti-mouse IgG (Invitrogen). Slides were counterstained with DAPI, mounted with Fluoromount-G (Southern Biotech), and examined with a Zeiss LSM510 META confocal laser-scanning inverted microscope (Carl Zeiss) equipped with an argon-krypton laser beam.
Immunocytochemistry
We studied endogenous expression of NDE1 in cell lines by fixing the cells with ice-cold methanol for 10 min and blocking them with 3% BSA for 40 min. We carried out staining by using rabbit polyclonal anti-NDE1 (a kind gift from Dr. Yuanyi Feng, Northwestern University, USA), which was detected with the secondary Alexa Fluor 555 goat anti-rabbit IgG (Invitrogen); mouse monoclonal anti-γ-tubulin (Abcam, ab27074), detected with the secondary Alexa Fluor 488 goat anti-mouse IgG (Invitrogen); and rat polyclonal anti-α-tubulin (AB-Direct, MCA77G), detected with the secondary Alexa Fluor 633 anti-rat IgG (Invitrogen). Slides were counterstained with DAPI, mounted with Fluoromount-G (Southern Biotech), and examined with a Zeiss LSM510 META confocal laser-scanning inverted microscope (Carl Zeiss) equipped with an argon-krypton laser beam.
Endogenous expression and subcellular localization of NDE1 were tested in cell lines derived from a control subject and from the affected patient from family C (family C, individual V-1 in Figure 1). Lymphoblastic cell lines were maintained in RPMI 1640 and supplemented with 2 mM L-glutamine and 15% fetal bovine serum. Cells were fixed in 4% paraformaldehyde for 20 min at 37°C and blocked with 3% BSA for 45 min at room temperature. Staining was performed overnight at 4°C with rabbit polyclonal anti-NDE1 (ProteinTech Group, 10233-1-AP) at a 1/200 dilution and anti-γ-tubulin (Abcam, ab27074) at a 1/400 dilution. FITC-conjugated secondary anti-rabbit IgG (Jackson ImmunoResearch, 626111) and Cy5-conjugated anti-mouse IgG (Jackson ImmunoResearch, 715175150) were used at a dilution of 1/400. Cells were mounted in Vectashield Mounting Medium containing DAPI (Vector Labs H-1200) and examined with a Zeiss LSM510 META confocal laser-scanning microscope.
Immunoblot
Cellular lysates were analyzed by immunoblot with rabbit polyclonal anti-NDE1 (Protein Tech Group, 10233-1-AP) at dilutions of 1/500 and 1/1000 and by a secondary anti-rabbit IgG (Amersham Biosciences, NA9340V) at a dilution of 1/10,000. Anti-GAPDH antibody (Santa Cruz Biotechnology, sc-25778) was used as loading control at a dilution of 1/200. Horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody was obtained from Amersham Biosciences (NA9340V).
Immunohistochemistry
Pregnant CD-1 mice were purchased from a Charles River (Margate, UK) supplier. Central nervous system embryonic tissues were harvested in accordance with the Animals (Scientific Procedures) Act 1986. Embryonic day (E) 11, 13, and 15 tissues were fixed with 4% paraformaldehyde overnight at 4°C and incubated in 30% sucrose before cryostat sectioning took place. Human CS22 paraffin-embedded sections fixed with 4% paraformaldehyde were obtained from the MRC-Wellcome Trust Human Developmental Biology Resource (see Web Resources). Mouse and human sections were boiled for 10 min with 10 mM sodium citrate buffer (pH 6) and then treated with a blocking buffer containing 0.3% Triton X-100 detergent and bovine serum albumin (3%) for 1 hr. Primary antibodies were incubated overnight in the blocking buffer. Appropriate Alexa-Fluor-conjugated secondary antibodies (Invitrogen) were incubated for 1 hr along with Hoescht 33342 for nuclei staining. Primary antibodies used on tissue sections were rabbit polyclonal anti-NDE1 (a kind gift from Dr. Yuanyi Feng, Northwestern University, USA), mouse monoclonal anti-SOX2 (R&D systems, MAB2018), mouse monoclonal anti-γ-tubulin (Abcam, ab11316), chicken anti-nestin (Neuromics, CH23001), and mouse monoclonal anti-β-tubulin III (Sigma, T8660).
Results
Clinical Data
We investigated three families in which six offspring had extreme congenital microcephaly (heads were more than 10 SD smaller than the mean) and profound mental retardation (Figure 1A). The clinical findings in the six affected children allow an unambiguous phenotype to be described. During pregnancy, a reduced head circumference was recorded by 18 weeks of gestation and thereafter gradually declined in comparison to expected norms. Otherwise, the pregnancy and delivery were uneventful. All affected offspring had head circumferences that were so small as to be difficult to measure accurately but were more than 10 SD smaller than the expected age and sex mean throughout their lives. Mental retardation was evident within the first few months of life; none of the children recognized their parents, and they showed little response to the outside world, even to painful stimuli. Epileptic seizures began by 3 months and evolved from occasional rhythmic jerks into complex partial and tonic-clonic seizures. The children gradually adopted a fetal position over the first 3 years of life and had a paucity of movements but were not hypotonic or spastic. Apart from microcephaly, the children grew normally, were not dysmorphic, and did not have any other congenital anomalies (Figure 1B). Health problems were thought to be related to their condition; they had seizures and difficulties with feeding and coping with chest infections. All but one died in the first 5 years of life because of chest infections and aspiration. There was no evidence of a neurological or cognitive decline or of immune deficiency. The clinical diagnosis was of an extreme form of autosomal-recessive primary microcephaly.
Extensive biochemical, hematologic, and cytogenetic investigations yielded normal results. Brain imaging was available from one individual from each family. The findings were consistent: severe microcephaly, a simplified gyral pattern of the cerebral cortex, a small cerebellum, normal gross brain architecture, and normal cortical ribbon thickness (Figure 1C and Figure S1). The child in family C (C.V-1) had agenesis of the corpus callosum. The radiological classification would be within the autosomal-recessive microcephaly with normal or minor short stature and very poor function phenotype.20 In four cases, prenatal ultrasound scans were performed at 18–20 weeks of gestation; all showed microcephaly but without structural defects and with a slow decrease in relative head circumference as pregnancy progressed.
One child died of aspiration during an episode of infective diarrhea at 10 months of age. The postmortem showed anomalies only within the central nervous system (Figure S2). Appearance and length were normal for the child's age, but the weight was −2 SD from the mean, and head circumference was 31.5 cm (the mean head circumference for a male fetus at 34 weeks of gestation). When the cranium was opened, the appearance was that of hydrocephalus ex vacuo with a small shrunken brain and abundant cerebro-spinal fluid surrounding it. The brain was 10% of the expected weight, 75 g versus 809 g (it was less than −10 SD from the mean). There was severe hypoplasia of the frontal lobes, which had an abnormal gyral pattern. The temporal and occipital lobes were almost smooth, and the only major sulcus visible was the Sylvian fissure. The cerebral cortex was reduced to 4 mm in thickness. The cerebral hemispheres weighed 67 g, the cerebellum 2.8 g (giving a ratio of 24:1 compared to the normal of 10:1). Histological examination confirmed a thin cerebral cortex. The cortical layering was abnormal; although the molecular layer appeared normal, the internal and external granular layers along with the external pyramidal layer were jumbled together and disorganized. There was a huge reduction in neuron number but no evidence of abnormal migration of neurons from the neuroepithelia to the cortical mantle and no ectopic collections of neurons. The cerebellum was grossly abnormal and had poorly formed folia. Purkinje cells were abundant, but the granular layer was very sparse. Gross and histological examination of all other organs was unremarkable. This disorder is best described as a microlissencephaly.
All three families were consanguineous; families A and B originated from Pakistan, and family C was Turkish. The inheritance pattern of the condition is autosomal recessive: Males and females were equally affected, parents were unaffected (no heterozygote effects), and there were unaffected siblings (represented by a lozenge on the pedigrees). No translocations or copy number variants were found on cytogenetic or on microarray analysis (Affymetrix Human SNP Array 6.0, data not shown).
Linkage and Gene Mutation Identification
For families A and B, linkage between the condition and chromosome 16p13 was achieved by use of autozygosity mapping (Figure 2A). Homozygosity mapping identified a 9 cM shared locus between the families, which contained many genes. The assumption is that a mutation in one of these genes within the autozygosity interval will cause the phenotype. The candidate gene is selected among the others based on our knowledge of primary microcephaly genes, among other factors; we selected NDE1 as a candidate gene. Bidirectional sequencing of all NDE1 exons and splice sites from genomic DNA revealed a homozygous 2 bp deletion, c.684-685delAC, in all affected individuals in families A and B (Figure S3 and data not shown). The c.684_685del mutation occurs within an exonic short dinucleotide repeat (AC)3 in exon 6 of the 9 exon NDE1. It is predicted to cause a frame shift in the NDE1 mRNA and thus results in the loss of amino acids 229 to 335 (C terminus) but with the addition of a novel sequence of 84 amino acids. The resultant novel open reading frame is predicted to terminate in a STOP codon at position 314. This mutation would be expected to give rise to a stable protein and not be subject to nonsense-mediated decay.21
Figure 2.
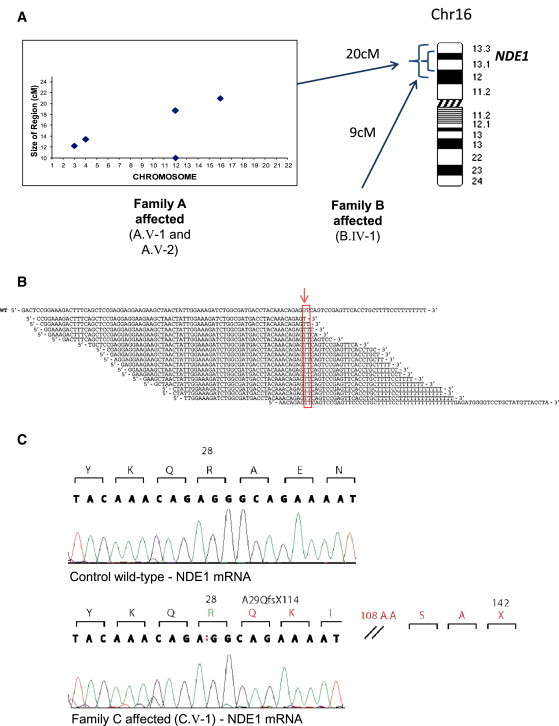
Discovery of Mutations in NDE1
(A) Linkage strategy employed for families A and B. As previously published,39 an ExcludeAR analysis is based on the principle that by excluding regions of nonlinkage, any chromosomal region that remains must contain the locus of interest. It detects heterozygous or homozygous but discordant results for each consecutive SNP and allows the detection of significant shared regions of concordant homozygosity in up to four affected family members. The output plot for family A is shown on the left and depicts the five concordant homozygous regions of significant size that were found. On the right, an illustration of Chromosome 16 shows the largest concordant region in family A and where it overlaps with a homozygous region found in family B. NDE1 is within this defined linkage region.
(B) Exome sequencing results. Exome sequencing demonstrates a G to T substitution in NDE1. Across the top of the figure is the wild-type (WT) sequence orientated 5′ to 3′ relative to NDE1. The wild-type G base mutated in the family is shown in green in the reference sequence (red arrow). Beneath this are the reads from the affected child from family C. The canonical splice donor site is shown in the red box. The depth of coverage across the variant was 17×, and all of the reads showed the substitution.
(C) Sequencing of mRNA extracted from family C lymphocytes. The two electropherograms depict the wild-type (top panel) and affected child (bottom panel) sequences as derived from mRNA sequencing. Called nucleotides are depicted above the electropherogram, and above this the codon is defined and translated into amino acids. The affected child in family C bears a homozygous c.83+1G>T mutation, and the red colon in the called nucleotides of this child shows that one base has been lost from the NDE1 mRNA. The codon amino acid track shows amino acid 28 of mutated NDE1 remains an arginine, but thereafter the frame shift causes the introduction of a novel sequence of 113 amino acids followed by a premature termination codon (p.Ala29GlnfsX114) (Figure S4).
In family C whole-genome genotyping confirmed consanguinity with an inbreeding coefficient (IBC) of 1.64, consistent with a second-cousin marriage, and allowed us to identify homozygous genomic segments (Table S1). Whole-exome capture and massively parallel sequencing were successfully performed (Tables S2 and S3). We identified two missense mutations in PRC1 (MIM 603484) and RRN3 (MIM 605121) and one homozygous splice-site mutation in NDE1; no coding insertion or deletion mutations were observed (data not shown). We prioritized the NDE1 variant because the other two were missense variants affecting residues that are not well conserved in orthologous proteins. Although the missense variant in PRC1 is observed in hedgehogs, the variant in RRN3 is present in cows, horses, and medaka (see Table S4 for phyloP scores). The splice-site NDE1 mutation c.83+1G>T (p.Ala29GlnfsX114) affected the second exon donor site (Figure 2B and Figure S3).
To confirm this mutation, we used a cell line established from the affected child in family C (individual C.V-1 in Figure 1). We extracted and sequenced NDE1 mRNA from a control and from the affected individual. We found that a novel splice donor site had been formed with the loss of a single nucleotide from the NDE1 mRNA (Figure 2C and Figure S4). The consequence of the c.83+1G>T mutation was a frame shift in NDE1, commencing at the first codon of exon 3, and the consequent addition of 113 nonnative novel amino acids before a premature stop codon p.Ala29GlnfsX114. We performed an immunoblot analysis on protein lysates extracted from the lymphoblastoid cell lines of the patient (C.V-1) and a control subject by using NDE1 antibody (ProteinTech Group, 10233-1-AP), the epitope of which is not preserved in the predicted mutant peptide. GAPDH antibody (Santa Cruz Biotechnology, sc-25778) was used as a loading control. As expected, NDE1 was detected in the control, as evidenced by a 38 kDa band in the blot, which was reduced to a significantly fainter band in the patient (C.V-1) (Figure S5).
All NDE1 mutations were found segregated as expected for an autosomal-recessive disorder in each family and were not present in public databases or in 320 ethnically matched control chromosomes.
The Origin of the c.684_685delAC Mutation and the Genetic Heterogeneity of the NDE1 Phenotype
Because the c.684_685del mutation had occurred in two Pakistani families who were not known to be related, we aimed to determine whether this mutation was recurrent or ancestral. We genotyped 11 SNPs from within the NDE1 genomic region in one affected child from each family A and B. The results were identical (data not shown); thus, the c.684_685del mutation was ancestral. We also investigated six further families with the same phenotype but found no linkage to NDE1 or NDE1 mutations. Therefore, the microlissencephaly phenotype we describe exhibits aethiological heterogeneity.
Cell Biology Studies of the c.83+1G>T and c.684_685del NDE1 Mutations
We first determined the mutational mechanism of the c.684_685del mutation by transfection of NDE1 constructs into HeLaM cells. Three fusion constructs were made, each with an in-frame 5′ green fluorescent protein (GFP) gene: wild-type (GFP-NDE1), the c.684_685del mutation (NDE1-M), and a stop codon introduced at position 228 of the wild-type protein (NDE1-MSTOP) (Figure 3). Wild-type GFP-NDE1 localized to the centrosome as expected,22,23 whereas both of the mutant NDE1 failed to do so at any stage during the cell cycle (Figure 4A and Figure S6). This agrees with mouse data that the NUDE_C domain of NDE1 is required for centrosome localization.24 The NDE1-M construct transfection leads to the accumulation of protein foci within the cytoplasm; these were rarely seen with the NDE-MSTOP construct despite similar levels of transfection. We concluded that when expressed in HeLaM cells, the NDE1-M protein, which lacks 107 C-terminal amino acids of NDE1, fails to localize to the centrosome. Kinetochore localization of GFP-NDE1 was normal, whereas NDE1-MSTOP failed to localize to kinetochores (data not shown).
Figure 3.
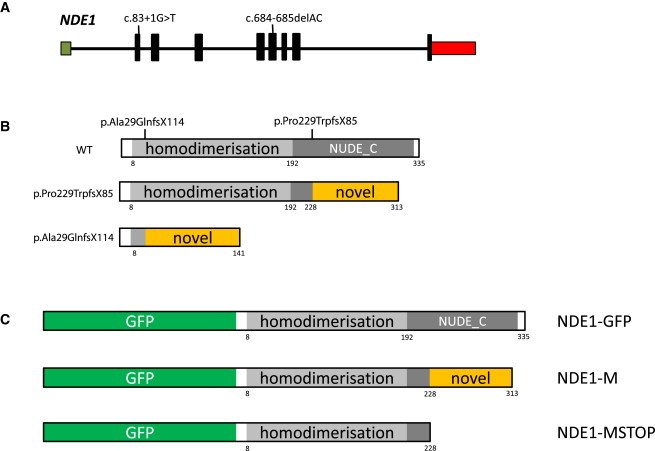
NDE1, NDE1, and Family Mutations
(A) NDE1 shown 5′ to 3′ with an untranslated exon 1 within a CpG island is shown in green and followed by eight coding exons (shown in black) and the 3′ UTR terminating in a single polyadenylation signal (shown in red). The size of the exons relative to one another is shown as is the position of the two homozygous mutations found at the donor site of exon 2 in family C (c.83+1G>T) and in exon 6 in families A and B (c.684_685del).
(B) An illustration of the NDE1 protein is shown with its two defined and evolutionarily conserved domains shown in shades of gray (top). In the center is the predicted protein deriving from the c.684_685del mutation, with loss of almost all of NUDE_C domain and the substitution with a novel peptide sequence of 84 amino acids. At the bottom is the predicted protein deriving from the c.83+1G>T mutation with loss of both known domains and the addition of 113 novel amino acids. The novel amino acid additions to NDE1 have no homology to known proteins or protein domains.
(C) NDE1 construct engineering. The three constructs made to investigate the c.684_685del mutation are shown. At the top, we have the wild-type protein tagged with GFP, designated GFP-NDE1. In the middle is the expected protein product of the mutation with the novel peptide sequence tagged with GFP, designated NDE1-M. At the bottom is a construct made with a stop codon at position 227, thus creating a protein truncated for the NUDE_C domain tagged with GFP, designated NDE1-MSTOP.
Figure 4.
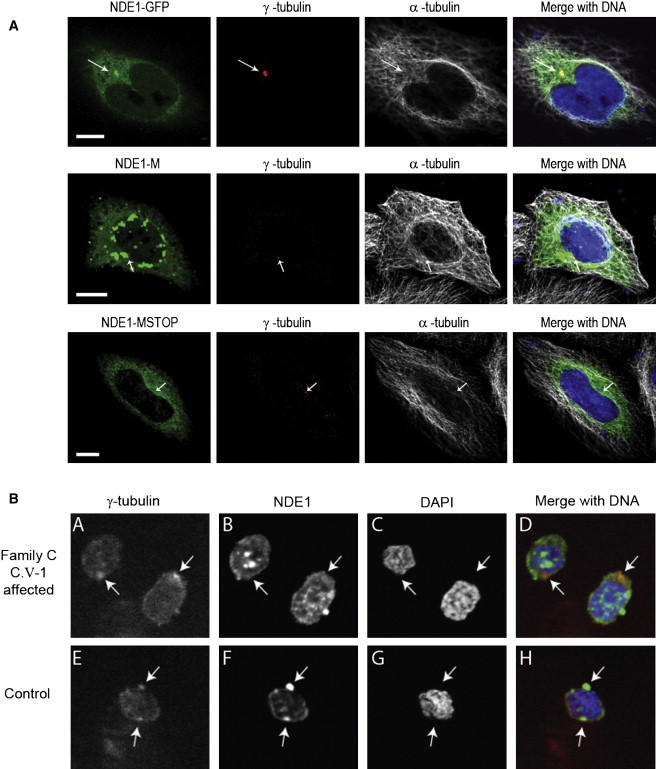
Determining the Consequence of the NDE1 Mutations
(A) The c.684_685del mutation prevents mutant NDE1 from localizing to the centrosome. The top four images of the panel show a representative HeLaM cell transfected with NDE1-GFP. On the left, NDE1 tagged with GFP is directly visualized in green. The two middle images show γ-tubulin in red, a constitutional marker of the centrosome, and α-tubulin in white. The merged image on the right shows the colocalization of NDE1 and γ-tubulin at the centrosome. The middle panel of four images shows the results after transfection with the NDE1-M construct. No centrosome localization could be found in any transfected cells (n > 200); instead aggregates formed within the more heavily transfected cells. In the bottom panel, we show the results obtained with NDE1-MSTOP construct; despite good levels of transfection, no NDE1 signal was seen at the centrosome, and aggregates were rarely seen. The scale bar indicates 10 μm.
(B) Subcellular localization of mutant NDE1 in patient from family C (individual C.V-1 in Figure 1) and wild-type control-derived lymphoblastoid cell line. We can see colocalization of NDE1 (green) with γ-tubulin (red) in the control but not in the patient cell line.
To determine the effect of the c.83+1G>T mutation, we stained cells from the lymphoblastoid cell line derived from the affected patient from family C (C.V-1) and a control individual (Figure 4). As expected, NDE1 colocalized with γ-tubulin in the control, but not in the patient sample.
Expression Pattern of NDE1 in Human Embryonic Brain
We examined the expression pattern of NDE1 in embryonic developing human brains during early neurogenesis, at CS23 (Figure 5). The mRNA expression pattern of NDE1 was determined by the use of antisense riboprobes specific to NDE1. Expression was seen in the neuroepithelium throughout the developing brain, including the cerebral cortex and cerebellum (Figures 5A and 5B and Figure S7). As a control, we used antisense riboprobes to SHH and sense riboprobes to NDE1. As expected, we observed no signal with the sense probes to NDE1, but we did observe strong positive expression of SHH.
Figure 5.

Expression of NDE1 in the Mammalian Brain
(A) NDE1 expression in a CS23 human embryo head. We performed in situ hybridization by using a riboprobe to NDE1 on a sagittal section of a human embryo. Darker red staining shows expression (see Figure S7 for control probe study). The black arrow indicates NDE1 expression in the neuroepithelium of the developing cerebral cortex, and the blue arrow indicates expression of the developing cerebellum in the neuroepithelium. The green star shows the mouth and hence the front of the face.
(B) A composite of two consecutively cut 10 μm sections through the developing cerebral cortex neuroepithelium. In the upper panel, dark red eosin and hematoxylin staining shows the structure of the neuroepithelium. The lower panel shows the results of NDE1 riboprobe in situ hybridization. Only cells within the neuroepithelium are stained. See Figure S7 for control probe study.
Embryonic Subcellular Localization of NDE1 and Nde1 in Human and Mouse Brain
We progressed to immunohistochemical analysis of embryonic human and mouse brains by examining a human embryonic brain at CS22 and a mouse embryonic brain at E11, E13 and E15. These times cover the periods of neurogenesis in the neuroepithelium, where apical neural precursors are proliferating through symmetric cell division and then commencing neuron production through asymmetric cell division.4,25–27 We found that NDE1 is strongly expressed in apical neuroepithelial cells in humans and mice (Figure 6). We found NDE1 to be strongly expressed in apical precursors in the ventricular zone and newborn neuronal population of the human embryonic brain but that it had a reduced expression in the subventricular zone as delineated by concomitant SOX2 antibody staining (Figure 6A). The subcellular localization of Nde1 included the centrosomes of all cells in the embryonic mouse brain. In apical neuroepithelial cells, the expression of centrosomal Nde1 was greatest in interphase and early mitosis and then reduced during metaphase (Figure 6B)—a pattern previously reported in other cell types.24 By measuring the intensity of the fluorescence signal of both γ-tubulin and Nde1 at the centrosome, we specifically proved colocalization of these proteins at the centrosome in apical neural precursor cells (Figure 6C).
Figure 6.
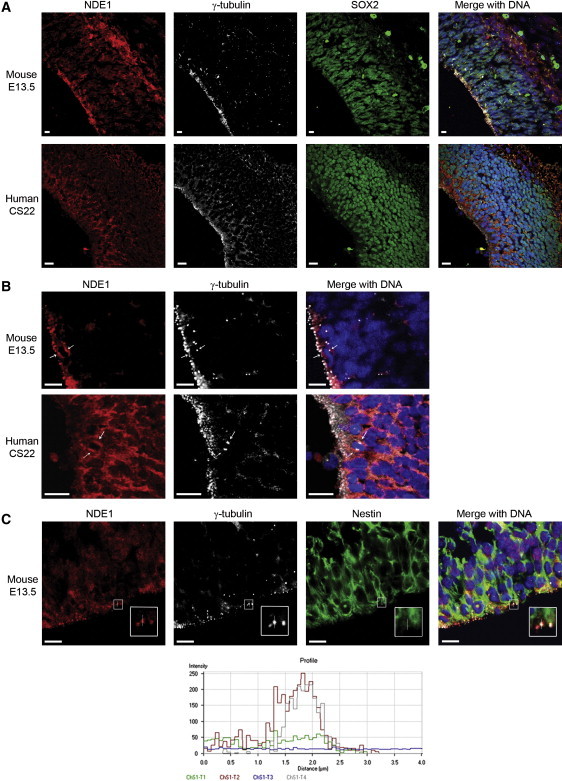
Immunohistochemical Analysis of E13 Mouse and CS22 Human Brains
(A) The upper set of four panels from a mouse indicate Nde1 (in red); Sox2 (in green) showing neuroepithelial cells; γ-tubulin (in white) showing centrosomes; and the merged image (on the right) including DAPI, showing DNA in blue. Nde1 is expressed at the apical rim (left and bottom) and more diffusely in the developing cortical plate (right and top). Immunohistochemical analysis of a CS22 human brain showing the cerebral cortex. This period of development is closest to E13 in mice. The color scheme is as above. NDE1 expression is marked in the neuroepithelium (left and bottom), and there is almost no expression in the subventricular zone (running left top to bottom right), whereas there is expression in the developing cortical plate (top right). Note that the subventricular zone is more developed in humans than in mice at the stages examined. Furthermore, the processing of the mouse embryonic brain can be better controlled to allow far greater clarity, particularly of microtubule-related proteins and structures, than in the human brain. Therefore, subcellular localization is likely to be less precise in our human studies than in mouse studies. The scale bar indicates 10 μm.
(B) Magnified image of mouse E13 and human CS22 neuroepithelium, clearly showing NDE1 cytoplasmic dots in apical mitotic precursors. Single, thin arrows indicate examples of apical neural precursor cells undergoing mitosis with a pair of centrosomes on either side of a DNA metaphase plate. The scale bar indicates 10 μm.
(C) Examination of Nde1 localization at the centrosome in apical neural epithelial cells. Nde1 is shown in red; DAPI shows DNA in blue; and Nestin, an apical progenitor cell marker, is shown in green; and γ-tubulin, a centrosome marker, is shown in white. The apical membrane of the neuroepithelial cells face the ventricle (the empty black space at the bottom). The white arrow dissects a representative centrosome for which we conducted an intensity profile analysis. The x-y graph below plots distance (defined by the path crossed by the arrow) on the x axis, and signal intensity on the y axis. The channels correspond respectively to Nestin (green, ChS1-T1), Nde1 (red, ChS1-T2), DNA (blue, ChS1-T3), and γ-tubulin (white, ChS1-T4). The intensity for both the white (γ-tubulin) and red (Nde1) channels reach a peak at the same point in the path defined by the arrow, whereas that for Nestin does not. We conclude that Nde1 is expressed at the centrosome of apical neural precursor cells where it colocalizes with γ-tubulin. The scale bar indicates 10 μm.
Subcellular Localization of Endogeneous NDE1 in HeLa and SK-N-SH and of Endogeneous Nde1 in Neuroepithelial Stem Cells
In order to further understand the role of NDE1 in neurogenesis, we investigated the subcellular colocalization of NDE1 in HeLa cells (human), SK-N-SH cells (human), and neuroepithelial stem cells ([NEP] considered to be fetal pluripotent neural stem cells and obtained from E10.5 mouse embryonic brains27,28). NDE1 was present in the cytoplasm and at the centrosome throughout the cell cycle in both HeLa and SK-N-SH cells. The centrosomal expression diminished during metaphase and anaphase (Figure S6). As previously reported,19,20 we also observed NDE1 at the metaphase kinetochores in these cell types (data not shown). In NEP cells, however, in addition to cytoplasmic and centrosomal sublocalization, we also found that Nde1 was highly enriched on the mitotic spindle (Figure 7). We did not see this spindle localization either endogeneously in HeLa or SK-N-SH or when we overexpressed NDE1 in HeLaM or SK-N-SH cells (data not shown).
Figure 7.
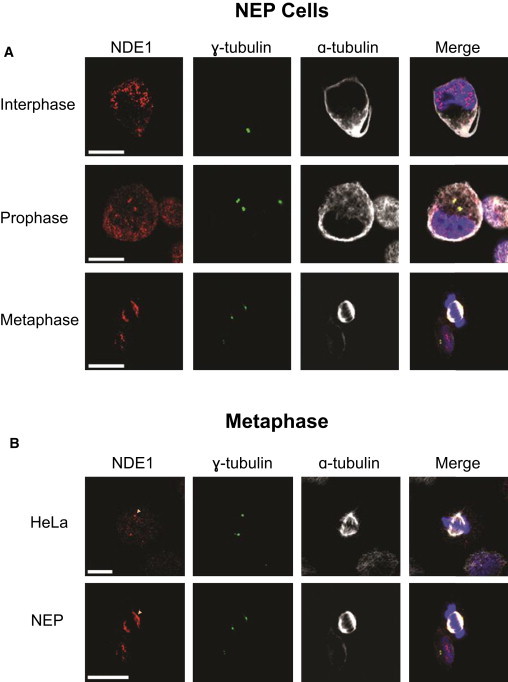
Subcellular Localization of NDE1 in NEP and HeLa Cells
(A) Representative NEP cells in interphase, prophase, and metaphase. Clear centrosomal staining is present during both interphase and prophase. In metaphase, NDE1 decorates the mitotic spindle. Cells were stained with antibodies against NDE1 (red); γ-tubulin (green), as a centrosome marker; α-tubulin (white), as a microtubule marker; and DNA (blue) stained with DAPI. The scale bars indicate 10 μm.
(B) NDE1 localization during metaphase in HeLa and NEP cells. NDE1 localizes to the centrosomes in HeLa cells, whereas it is present on the mitotic spindle in NEP cells. The scale bars indicate 10 μm.
Discussion
Here we have described a human developmental disease causing autosomal-recessive extreme primary microcephaly with disordered cortical lamination (microlissencephaly) caused by biallelic NDE1 mutations, the most severe deficit of prenatal neuron production described to date.29 Our data indicate that the dual pathogenesis of this disorder is a profound early failure of neuron production and a later, but still prenatal, deficiency of cortical lamination. It also suggests that loss of NDE1 at the centrosomes of apical neuroepithelial cells plays a critical role in these processes, further highlighting the importance of the so-far-unexplained involvement of the centrosome in neurogenesis.30
A mouse deficient for Nde1 has been described.31 Whether a mouse null for Nde1 is equivalent to the human mutations we describe here is unknown; however, the similarities in phenotype are striking, and the only major difference is the degree of brain size reduction. In Nde1−/− mice the brain was 33% smaller than in the wild-type; in humans, the brain was approximately 90% smaller. The fact that the human brain is so much more susceptible to NDE1 deficiency is a strong indication of the central importance of this gene in the cellular processes that make our brain, in some ways, unique. In both species, the cerebral cortex was most affected by the reduction in size (although we note that the human cerebellum was also reduced in size, whereas it was not in Nde1−/− mice). In addition, cerebral cortical layers II, III, and IV were particularly affected and had disorganized neurons. The relative preservation of layers V and VI (which predominantly contain intracerebral neurons or interneurons) compared to layers II, III, and IV (which provide an output to the cerebral cortex) is noteworthy. It could reflect a progressive failure in neuron production, the earlier born neurons that form layer VI being produced in more abundance than those neurons produced later, and destined for layers II, III, and IV. Or it could reflect a failure in long-range neuronal migration, whereby layer II–IV neurons having to transverse ever increasing distances.
NDE1 has a predominant 1005 nucleotide coding transcript, which consists of a single CpG island, 9 exons, and a consensus start codon in exon 2. Very unusually, the terminal exon is entirely contained within an exon of the neighboring MYH11 (MIM 160745). NDE1 has a homodimerization domain (the C-terminal part of which binds LIS1 when dimerized32) and a NUDE_C domain. The c.684_685del mutation is predicted to result in a novel open reading frame that terminates in a premature stop codon at position 313. This bisects the highly conserved C-terminal domain of NDE1 (NUDE_C) (Figure 3B). The NUDE_C domain is found exclusively in NDE1 and NDEL1 (MIM 607538).33,34 Studies of NDE1, predominantly in mammals, show that this domain allows interaction of NDE1 with CENPF (MIM 600236) and the dynein complex.19,35 Furthermore, loss of this domain leads to a failure of NDE1 to be present at the centrosome,24 a finding that we confirmed was also true of NDE1 in human cell lines. The mechanistic consequences of disruption to the NUDE_C domain in patients with microlissencephaly is explored in a sister paper by Alkuraya et al. in this issue.11 The c.684_685del mutations would be predicted to result in a protein maintaining its homodimerization domain and possibly also LIS1 (coded by PAFAH1B1, MIM 601545) binding.32,36 The predicted novel peptide resulting from the c.83+1G>T mutation maintains only the first 28 native amino acids of the wild-type protein on the N terminus followed by 113 novel amino acids. In addition to the complete loss of the highly conserved NUDE_C domain, the vast majority of the homodimerization domain is also lost. Even if the mutant NDE1 RNA detected in the cell line of the patient from family C (individual C.V-1 in Figure 1) is stable and possibly translated, it is not expected to retain the functions and interactions of native NDE1. It will be important to determine whether other types of mutations within NDE1 give rise to a phenotype different than that described here.
Although NDE1 has been shown to have multiple cellular functions, these seem either redundant (by expression of the homolog NDEL) or unnecessary in all tissues except the neuroepithelium of the central nervous system.20,30,35,37,38 We show that both the c.83+1G>T and c.684_685del mutations result in an inability of NDE1 to be targeted to the centrosome. In mouse NEP cells and human apical neural precursors, we observed Nde1 and NDE1, respectively, decorating the mitotic spindle adjacent to the spindle poles. We could not recapitulate this change in HeLaM or SK-N-SH cell lines by overexpression of NDE1. The principle determinant of neuron numbers in the mammalian brain is the ability of apical neural precursor cells to undergo symmetric mitotic cell divisions and thus exponentially generate further neural precursors; our data suggest that NDE1 is a crucial component of this process fundamental to mammalian encephalisation and human cerebral cortex growth.2,3,38 Human NDE1 mutations and mouse Nde1 loss lead to cortical-lamination deficits, which, together with a very reduced neuron production, explain the NDE1-associated phenotype of microlissencephaly.
Acknowledgments
The authors would like to thank the research families for their participation in this project, Ilias Kazanis and Veronique Marthiens for assistance with the project, and the Higher Education Commission of Pakistan, Action Research, and Wellcome Trust for funding (O.P.C., M.K., J.J.C., A.K.N., C.G.W.). The human embryonic and fetal material was provided by the MRC/Wellcome Trust-funded Human Developmental Biology Resource at the Institute of Human Genetics, Newcastle Upon Tyne, with funding from grant numbers G0700089 (MRC) and GR082557 (Wellcome Trust). We thank Cathy Camputaro for her help with the imaging studies. We acknowledge the use of Yale University Biomedical High Performance Computing Center for data analysis and storage. This study was supported by the Yale Program on Neurogenetics, the Yale Center for Human Genetics and Genomics, and National Institutes of Health grants RC2NS070477 (to M.G.).
Contributor Information
Murat Gunel, Email: murat.gunel@yale.edu.
C. Geoffrey Woods, Email: cw347@cam.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
HapMap, http://hapmap.ncbi.nlm.nih.gov/
Human Genome Browser, http://genome.ucsc.edu/
MRC-Wellcome Trust Human Developmental Biology Resource (HDBR), http://www.hdbr.org/
Personal Genome Variants track of UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=192378401&c=chr16&g=pgSnp
PFAM, http://pfam.sanger.ac.uk/search?tab=searchSequenceBlock
Primer3, http://frodo.wi.mit.edu/primer3/
Online Mendelian Inheritance of Man (OMIM), http://www.omim.org
References
- 1.Falk D. Hominid evolution. Science. 1986;234:11. doi: 10.1126/science.234.4772.11-a. [DOI] [PubMed] [Google Scholar]
- 2.Fish J.L., Dehay C., Kennedy H., Huttner W.B. Making bigger brains-the evolution of neural-progenitor-cell division. J. Cell Sci. 2008;121:2783–2793. doi: 10.1242/jcs.023465. [DOI] [PubMed] [Google Scholar]
- 3.Rakic P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009;10:724–735. doi: 10.1038/nrn2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Götz M., Huttner W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 5.Roth G., Dicke U. Evolution of the brain and intelligence. Trends Cogn. Sci. (Regul. Ed.) 2005;9:250–257. doi: 10.1016/j.tics.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Woods C.G., Bond J., Enard W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rauch A., Thiel C.T., Schindler D., Wick U., Crow Y.J., Ekici A.B., van Essen A.J., Goecke T.O., Al-Gazali L., Chrzanowska K.H. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 8.Griffith E., Walker S., Martin C.A., Vagnarelli P., Stiff T., Vernay B., Al Sanna N., Saggar A., Hamel B., Earnshaw W.C. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 2008;40:232–236. doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponting C., Jackson A.P. Evolution of primary microcephaly genes and the enlargement of primate brains. Curr. Opin. Genet. Dev. 2005;15:241–248. doi: 10.1016/j.gde.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Montgomery S.H., Capellini I., Venditti C., Barton R.A., Mundy N.I. Adaptive evolution of four microcephaly genes and the evolution of brain size in anthropoid primates. Mol. Biol. Evol. 2011;28:625–638. doi: 10.1093/molbev/msq237. [DOI] [PubMed] [Google Scholar]
- 11.Alkuraya F.S., Cai X., Emery C., Mochida G.H., Al-Dosari M.S., Felie J.M., Hill R.S., Barry B.J., Partlow J.N., Gascon G. Human mutations in the nde1 cause extreme microcephaly with lissencephaly. Am. J. Hum. Genet. 2011;88:536–547. doi: 10.1016/j.ajhg.2011.04.003. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bond J., Roberts E., Springell K., Lizarraga S.B., Scott S., Higgins J., Hampshire D.J., Morrison E.E., Leal G.F., Silva E.O. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 13.Woods C.G., Cox J., Springell K., Hampshire D.J., Mohamed M.D., McKibbin M., Stern R., Raymond F.L., Sandford R., Malik Sharif S. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thornton G.K., Woods C.G. Primary microcephaly: Do all roads lead to Rome? Trends Genet. 2009;25:501–510. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloğlu A., Ozen S., Sanjad S. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergnolle M.A., Taylor S.S. Cenp-F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr. Biol. 2007;17:1173–1179. doi: 10.1016/j.cub.2007.05.077. [DOI] [PubMed] [Google Scholar]
- 20.Stehman S.A., Chen Y., McKenney R.J., Vallee R.B. NudE and NudEL are required for mitotic progression and are involved in dynein recruitment to kinetochores. J. Cell Biol. 2007;178:583–594. doi: 10.1083/jcb.200610112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 22.Feng Y., Olson E.C., Stukenberg P.T., Flanagan L.A., Kirschner M.W., Walsh C.A. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 2000;28:665–679. doi: 10.1016/s0896-6273(00)00145-8. [DOI] [PubMed] [Google Scholar]
- 23.Bradshaw N.J., Ogawa F., Antolin-Fontes B., Chubb J.E., Carlyle B.C., Christie S., Claessens A., Porteous D.J., Millar J.K. DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem. Biophys. Res. Commun. 2008;377:1091–1096. doi: 10.1016/j.bbrc.2008.10.120. [DOI] [PubMed] [Google Scholar]
- 24.Hirohashi Y., Wang Q., Liu Q., Li B., Du X., Zhang H., Furuuchi K., Masuda K., Sato N., Greene M.I. Centrosomal proteins Nde1 and Su48 form a complex regulated by phosphorylation. Oncogene. 2006;25:6048–6055. doi: 10.1038/sj.onc.1209637. [DOI] [PubMed] [Google Scholar]
- 25.Uylings H.B. Development of the cerebral cortex in rodents and man. Eur. J. Morphol. 2000;38:309–312. doi: 10.1076/0924-3860(200012)38:5;1-h;ft309. [DOI] [PubMed] [Google Scholar]
- 26.Bystron I., Blakemore C., Rakic P. Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 2008;9:110–122. doi: 10.1038/nrn2252. [DOI] [PubMed] [Google Scholar]
- 27.Cai J., Wu Y., Mirua T., Pierce J.L., Lucero M.T., Albertine K.H., Spangrude G.J., Rao M.S. Properties of a fetal multipotent neural stem cell (NEP cell) Dev. Biol. 2002;251:221–240. doi: 10.1006/dbio.2002.0828. [DOI] [PubMed] [Google Scholar]
- 28.Rao M. Stem and precursor cells in the nervous system. J. Neurotrauma. 2004;21:415–427. doi: 10.1089/089771504323004566. [DOI] [PubMed] [Google Scholar]
- 29.Barkovich A.J., Kuzniecky R.I., Jackson G.D., Guerrini R., Dobyns W.B. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873–1887. doi: 10.1212/01.wnl.0000183747.05269.2d. [DOI] [PubMed] [Google Scholar]
- 30.Higginbotham H.R., Gleeson J.G. The centrosome in neuronal development. Trends Neurosci. 2007;30:276–283. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Feng Y., Walsh C.A. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Derewenda U., Tarricone C., Choi W.C., Cooper D.R., Lukasik S., Perrina F., Tripathy A., Kim M.H., Cafiso D.S., Musacchio A., Derewenda Z.S. The structure of the coiled-coil domain of Ndel1 and the basis of its interaction with Lis1, the causal protein of Miller-Dieker lissencephaly. Structure. 2007;15:1467–1481. doi: 10.1016/j.str.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Efimov V.P. Roles of NUDE and NUDF proteins of Aspergillus nidulans: Insights from intracellular localization and overexpression effects. Mol. Biol. Cell. 2003;14:871–888. doi: 10.1091/mbc.E02-06-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wainman A., Creque J., Williams B., Williams E.V., Bonaccorsi S., Gatti M., Goldberg M.L. Roles of the Drosophila NudE protein in kinetochore function and centrosome migration. J. Cell Sci. 2009;122:1747–1758. doi: 10.1242/jcs.041798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan X., Li F., Liang Y., Shen Y., Zhao X., Huang Q., Zhu X. Human Nudel and NudE as regulators of cytoplasmic dynein in poleward protein transport along the mitotic spindle. Mol. Cell. Biol. 2003;23:1239–1250. doi: 10.1128/MCB.23.4.1239-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenney R.J., Vershinin M., Kunwar A., Vallee R.B., Gross S.P. LIS1 and NudE induce a persistent dynein force-producing state. Cell. 2010;141:304–314. doi: 10.1016/j.cell.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niethammer M., Smith D.S., Ayala R., Peng J., Ko J., Lee M.S., Morabito M., Tsai L.H. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 38.Farkas L.M., Huttner W.B. The cell biology of neural stem and progenitor cells and its significance for their proliferation versus differentiation during mammalian brain development. Curr. Opin. Cell Biol. 2008;20:707–715. doi: 10.1016/j.ceb.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Woods C.G., Valente E.M., Bond J., Roberts E. A new method for autozygosity mapping using single nucleotide polymorphisms (SNPs) and EXCLUDEAR. J. Med. Genet. 2004;41:e101. doi: 10.1136/jmg.2003.016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.