

Abstract
Infantile cardiomyopathies are devastating fatal disorders of the neonatal period or the first year of life. Mitochondrial dysfunction is a common cause of this group of diseases, but the underlying gene defects have been characterized in only a minority of cases, because tissue specificity of the manifestation hampers functional cloning and the heterogeneity of causative factors hinders collection of informative family materials. We sequenced the exome of a patient who died at the age of 10 months of hypertrophic mitochondrial cardiomyopathy with combined cardiac respiratory chain complex I and IV deficiency. Rigorous data analysis allowed us to identify a homozygous missense mutation in AARS2, which we showed to encode the mitochondrial alanyl-tRNA synthetase (mtAlaRS). Two siblings from another family, both of whom died perinatally of hypertrophic cardiomyopathy, had the same mutation, compound heterozygous with another missense mutation. Protein structure modeling of mtAlaRS suggested that one of the mutations affected a unique tRNA recognition site in the editing domain, leading to incorrect tRNA aminoacylation, whereas the second mutation severely disturbed the catalytic function, preventing tRNA aminoacylation. We show here that mutations in AARS2 cause perinatal or infantile cardiomyopathy with near-total combined mitochondrial respiratory chain deficiency in the heart. Our results indicate that exome sequencing is a powerful tool for identifying mutations in single patients and allows recognition of the genetic background in single-gene disorders of variable clinical manifestation and tissue-specific disease. Furthermore, we show that mitochondrial disorders extend to prenatal life and are an important cause of early infantile cardiac failure.
Main Text
Mitochondrial dysfunction is a major cause of metabolic disorders in adults and children and presents a wide variability of organ manifestations. Mitochondrial DNA (mtDNA) mutations explain 10%–30% of these disorders, the rest being caused by defects in nuclear-encoded mitochondrial proteins.1 The genetic causes of primary mitochondrial cardiomyopathies (CMPs) are, however, poorly known, although CMP is an important manifestation among children with mitochondrial disease.2–4 In addition to mutations in the mitochondrial tRNA for isoleucine (MT-TI [MIM 590045]),5 nuclear mutations in SCO2 (MIM 604272),6 TAZ (MIM 300394),7 and TMEM70 (MIM 612418)8 have previously been identified in disorders with early-onset mitochondrial CMP. The prognosis of children with mitochondrial CMP is particularly poor, with an 18% survival rate at 16 years of age, whereas patients with neuromuscular symptoms but without CMP have a 95% survival rate at the same age.3 Infantile CMPs typically lead to early death. In the present study, we set out to identify the genetic causes of mitochondrial CMPs by using whole-exome sequencing.
All patient samples were taken according to the Declaration of Helsinki, with informed consent given prior to sample collection. The project was approved by the review board of the Helsinki University Central Hospital. Our index patient was a girl with infantile mitochondrial hypertrophic CMP (patient II-1, family 1; Figure 1A). She was born healthy but somewhat small (2945 g) to nonconsanguineous parents after an uneventful pregnancy. At 3.5 months she was admitted to the hospital because of poor feeding, failure to thrive, delayed motor development, and severe generalized muscle weakness. She was alert, had normal eye movements without ptosis, but had hypoactive deep tendon reflexes. Ophthalmological examination did not reveal retinopathy or optic atrophy. Urinary organic acids showed marginal increase in ethylmalonic acid. Chest X-ray showed cardiomegaly, and cardiac ultrasound showed a severely hypertrophic left ventricle with decreased contractility (ejection fraction 40%). Brain magnetic resonance imaging was normal, but electroencephalogram showed mild background abnormality with isolated or multifocal spikes on the left hemisphere. She had lactic acidosis (up to 7 U/l; normal < 2.3 mmol/l) but normal plasma creatine kinase (108 U/l; normal 50–270 U/l) and alanine aminotransferase (36 U/l; normal < 50 U/l). At 4 months of age, her muscle sample showed scattered cytochrome c oxidase (COX, mitochondrial respiratory chain complex IV)-deficient muscle fibers, which suggested generalized muscle dysfunction and was considered a contraindication for heart transplantation. Other organs, including the retina and liver, showed no signs of disease. Her disorder progressed despite intensive medication for heart failure and supplementation of carnitine, CoQ, riboflavin, or medium chain triglycerides. She died at the age of 10 months of cardiac insufficiency. Autopsy showed a severely enlarged, dilated, and hypertrophic heart, which compressed the lung and caused mild pulmonary hypoplasia. Light microscopic examination of cardiac muscle showed scattered lymphocyte infiltration, ischemic myocytes, perinuclear vacuolization, and fat accumulation, consistent with histiocytoid CMP. Postmortem, 80% of cardiomyocytes and 60% of skeletal muscle fibers were COX deficient and succinate dehydrogenase (SDH) positive (Figure 1B). The cardiac COX deficiency manifested in the whole organ and was particularly prominent in the papillary muscles and underneath the endocardium. The liver histology showed mild fat infiltration. The skeletal muscle showed fat accumulation and moderate fiber size variation, including small atrophic and few enlarged, mostly type 1 fibers (Figure 1B). Neuropathologic findings were mild and unspecific, but some vacuolization of the neuropil combined with capillary congestion was detected, especially in the pontine tegmentum. Blue native electrophoresis (BN-PAGE) of the mitochondrial respiratory chain (RC) complexes9 revealed a near-total lack of COX and complex I (CI) in the heart, severe COX deficiency and reduction of CI in the brain, and partial complex III (CIII) deficiency in both tissues, whereas all complexes were unaffected in the liver (Figure 1C). For the exclusion of pathogenic mtDNA mutations, total DNA from frozen muscle tissue was extracted by standard methods, and mtDNA was amplified by PCR in two fragments, followed by DNA sequencing with several internal primers (Table S2 available online).
Figure 1.

The Mitochondrial Cardiomyopathy Patients and Their Families
(A) Pedigrees of families 1 and 2.
(B) Cytochrome c oxidase (COX) activity (shown in brown) of heart and skeletal muscle of patients 1 (II-1, family 1) and 2 (II-6, family 2). Simultaneous histochemical analysis for COX and succinate dehydrogenase (SDH, shown in blue) activities (Cox/Sdh) on frozen cryostat sections revealed mitochondrial COX deficiency with mitochondrial proliferation.
(C) Blue native electrophoresis analyses of mitochondrial respiratory chain complexes in the heart, brain, and liver of patient 1 (P1), compared to control samples (c). Ten micrograms of sample protein was loaded onto a gel. For protein detection, monoclonal antibodies (Mitosciences) against the 39 kDa subunit of Complex I (CI), the subunits core 2 or Rieske of Complex III (CIII), the cox1p and cox4 subunits of Complex IV (CIV), and the 70 kDa-Ip subunit of Complex II (CII) were used.
To identify the causative nuclear gene mutation, we sequenced the exome of patient II-1 in family 1. The patient's genomic DNA was isolated from cultured fibroblasts, and exome targets were enriched with the SureSelect Human All Exon Kit (Agilent Technologies), and sequenced with the use of the Illumina Genome Analyzer-IIx platform. The 2×82 bp paired-end sequence reads were aligned to the hg19 reference genome with the Burrows-Wheeler Alignment tool.10 The Pileup utility from the SAMTOOLS package was used for variant calling.11 The procedure yielded 5.3 Gb of mappable sequence, of which 3.1 Gb (58%) was mapped on SureSelect exome coordinates, resulting in 83× mean coverage for target bases. From the 65,849 identified SNPs, the pathogenic variant was identified by the following scheme: (1) The known dbSNP130 variants were excluded, under the assumption that the pathogenic variant was too rare to yet exist in the database. (2) Nongenic variants were excluded. (3) Homozygous changes were selected, because autosomal infantile-onset mitochondrial disorders are generally recessively inherited and homozygosity of the pathogenic variant was considered possible given that the patient originated from Finland, a genetic isolate. (4) The consequence of the remaining variants was assessed by the SIFT Genome tool.12 (5) Genes encoding proteins with a mitochondrial function were predicted by MitoProt and TargetP (Figure 2). The serial filtering of the data led us to identify a single change affecting a putative mitochondrial protein: a homozygous missense mutation (c.1774C>T [p.Arg592Trp], RefSeq accession number NM_020745.2) on chromosome 6 in exon 13 of AARS2 (MIM 612035), encoding a putative mitochondrial alanyl-tRNA synthetase (mtAlaRS) (Figure 3A). We independently carried out homozygosity mapping on the patient's DNA with the Infinium Human660W SNP Array (Illumina) and, using the runs of homozygosity screen option in PLINK,13 identified the largest continuous homozygosity (3.3 Mb, 716 consecutive SNPs) on the region of chromosome 6 that contained AARS2. Both parents were heterozygous for the mutation.
Figure 2.

Schematic Representation of the Exome Data Analysis and Data Filtering
(1) The known dbSNP130 variants were excluded assuming the pathogenic variant to be too rare to (yet) exist in the database. (2) Nongenic variants were excluded. (3) Homozygous changes were selected: autosomal infantile-onset mitochondrial disorders are generally recessively inherited and homozygosity of the pathogenic variant was considered possible as the patient originated from Finland, a genetic isolate. (4) The consequence of the remaining variants was assessed by SIFT Genome tool (5) Genes encoding proteins with a mitochondrial function were predicted by MitoProt and TargetP.
Figure 3.

AARS2 and Mutations
(A) AARS2 mutation sequences in patients 1 (II-1, family 1) and 2 (II-6, family 2).
(B) Cross-species protein conservation of mtAlaRS, flanking the altered amino acids p.Leu155Arg and p.Arg592Trp in mammals. The corresponding gene has not been fully characterized in other vertebrate species.
(C) Schematic representation of AARS2. Boxes represent exons 1–22. Two main functional domains, the aminoacylation (pink) and editing (green) domains, are indicated. M denotes the mitochondrial targeting signal. The p.Leu155Arg mutation is located in the aminoacylation domain, p.Arg592Trp in the editing domain.
(D) The AARS2-GFP fusion protein (green) colocalizes with Mitotracker Red (Invitrogen), indicating mitochondrial localization for mtAlaRS in HEK293T cells (overlay in yellow). The cells were examined and imaged with an Olympus IX8 fluorescence microscope.
AARS2 consists of 22 exons and encodes a protein of 985 amino acids (Figure 3C). We sequenced AARS2 in 14 patients with primary mitochondrial CMP (age at onset: 1 day–15 yrs) from our pediatric patient material (∼150 patients), all of whom have a suspected mitochondrial RC deficiency but no molecular diagnosis. The exons of AARS2 were sequenced from total DNA with primers designed to flank introns and untranslated regions (Table S3). We identified the c.1774C>T (p.Arg592Trp) mutation in a female, who was compound heterozygous for another missense mutation in exon 3, c.464T>G (p.Leu155Arg) (patient II-6, family 2; Figure 1A). She was from a family of healthy, nonconsanguineous parents with four healthy children and was born at week 40+6, weighing 3220 g. Toward the end of the pregnancy term, cardiac recordings showed extrasystolia every ten beats, but otherwise the pregnancy was uneventful. Her condition was poor immediately after birth; her Apgar score was 5/5. She required mechanical ventilation, but oxygen saturation was only 70%. Postnatal ultrasound showed a large hypertrophic heart with poor myocardial contractility, and she developed severe metabolic acidosis (lactacidosis; base excess −25). CNS affision was suspected because of staring gaze and periodical stiffness. EEG showed decreased basic activity and suspicion of bursts, with asymmetry between the hemispheres. Vitamin supplementations or carnitine had no effect on the progressive disease. She died at postnatal day 3. Autopsy showed an enlarged, hypertrophic heart and pulmonary hypoplasia but no pathological changes in the brain and liver, as well as severe COX deficiency in the heart and skeletal muscle (Figure 1B). Table S1 shows RC activity measurements,14 consistent with CI+CIV deficiency in the heart, brain, and skeletal muscle but not in the liver. Her brother (patient II-3, family 2; Figure 1A) had died in utero at pregnancy week 40+1, with the autopsy showing hypertrophic heart with cardiomyopathic histology and pulmonary hypoplasia. The parents of the patients were heterozygous carriers of the mutations. Neither mutation was found in 400 Finnish control chromosomes, which were screened by solid-phase minisequencing15 with mutation-specific detection primers (Table S2). Other CMP patients in our cohort did not have AARS2 mutations.
Our multiple sequence alignment of the putative mtAlaRSs with known bacterial alanyl-tRNA synthetases, obtained via the PROMALS3D server, showed that the human enzyme has 31% identity and 45% similarity with E. coli AlaRS, suggesting it to be the enzyme responsible for the charging of tRNAAla with alanine during mitochondrial translation. MitoProt and TargetP prediction algorithms suggested a likely N-terminal mitochondrial targeting signal for mtAlaRS. We cloned the full coding-region cDNA of AARS2 (excluding the stop codon) into pEGFP-N1 (Clontech) and used the Neon transfection system (Invitrogen) to express the AARS2-GFP fusion protein in HEK293T cells according to the manufacturer's protocol. As a result, AARS2-GFP fusion protein was efficiently targeted to mitochondria in the cells (Figure 3D).
The Leu155 and Arg592 residues are highly conserved in mammalian mtAlaRSs, except for the ortholog of Arg592 in mouse (Figure 3B). To assess the consequences of p.Leu155Arg and p.Arg592Trp substitutions on the enzyme function, we generated a homology model for mtAlaRS by using SWISS-Model Server (Figure 4A). For modeling of the fragment encompassing residues 36–491, the structure of the corresponding domain from the homologous E. coli AlaRS16 (residues 2–441) (PDB 3HXU, chain A) sharing 37.6% sequence identity was used. The domain of P. horikoshii AlaRS17 encompassing 540–752 residues (PDB 2ZZF, chain A) was used as a template for modeling of the human mtAlaRS aa 556–784 fragment. Sequence identity between these fragments is 30.2%. A structure model of the mtAlaRS molecule without 233 C-terminal residues was attained by superimposition of the obtained domains with the structure of the P. horikoshii AlaRs17 (PDB 2ZZF) (Figure 4A). Modeling of the tRNAThr (PDB 1QF6) into the editing domain of the modeled mtAlaRS was performed as previously described.17,18 Superimposition of the structures, docking of the tRNA, and structure analysis was performed with VMD software19 and Discovery Studio 2.5.5 (Accelrys). Sequence analysis and the attained structure predict that human mtAlaRS, similar to its bacterial orthologs, has an N-terminal aminoacylation domain (aa 36–481) for charging tRNAAla, an editing domain (aa 484–782) for deacylation, if tRNAAla is mischarged with serine or glycine,20,21 and a structural C-terminal domain (aa 883–985) linking the aminoacylation and editing functions.22 The residue Leu155 was found to be located in the aminoacylation domain and Arg592 within the editing domain (Figure 3C). Our model shows that Leu155 is one of the architectural residues surrounding the catalytic aminoacylation site (Figure 4B). Substitution of Leu155 for positively charged arginine is predicted to severely affect the aminoacyl adenylate formation by altering the position of the neighboring Trp241 and consequently Asn242, the analog of which was shown in bacterial AlaRSs (Asn194 in A. aeolicus and Asn212 in E.coli) to be one of the key catalytic residues.16,23
Figure 4.
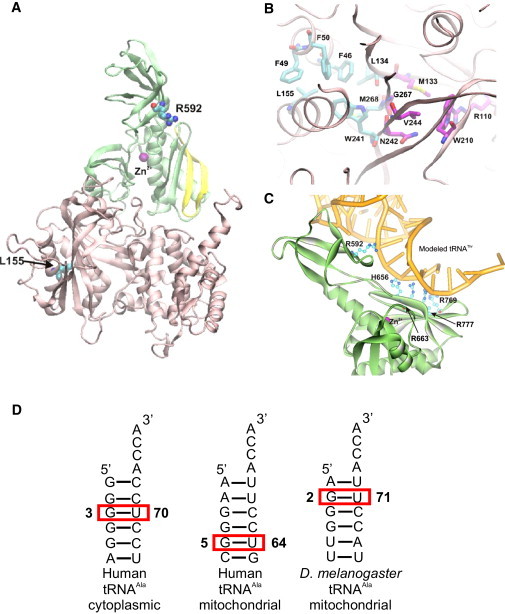
Modeled Human mtAlaRS
(A) The location of Leu155 within the aminoacylation domain (pink) and of Arg592 within the editing domain (green). The strand-loop-strand motif (aa 764–783) in the editing domain is shown in yellow.
(B) Hydrophobic architectural residues surrounding the catalytic aminoacylation site. Carbon atoms of the catalytic residues involved in amino acid binding and aminoacyl adenylate formation are shown in magenta.
(C) Conserved positively charged residues, predicted to be involved in the recognition and binding of tRNA (yellow) in the editing domain of human mtAlaRS.
(D) Comparative analysis of the predicted secondary structures of human cytoplasmic and mitochondrial tRNAAla revealed that the identity base pair of the latter is likely to be G5:U64, implying a structurally unique tRNA recognition site in the human mtAlaRS.
Arg592 is predicted to be located in the editing domain of mtAlaRS at a distance of 21.15 Å from the editing site at the same plane with an anti-parallel four-stranded β sheet of the domain. The strand-loop-strand motif of this β sheet formed by aa 64–783 (shown in yellow in Figure 4A) corresponds to the tRNAAla recognition motif of E. coli AlaRS (aa 680–699).17,18,24 The cytosolic alanyl-tRNA synthetases identify cytoplasmic tRNAs on the basis of a single conserved wobble G3:U70 base pair in the acceptor stem.25 However, in fruit fly mitochondrial tRNAAla, the position of the critical G:U base pair is shifted to 2:71.26 Using the DINAMelt server, comparative analysis of the predicted secondary structures of human cytoplasmic and mitochondrial tRNAAla (GenBank NC_012920.1, complement 5587–5655 nt) revealed that the identity base pair of the latter is likely to be G5:U64, which implies a structurally unique tRNA recognition site in the human mtAlaRS (Figure 4D). Analysis of the AlaRSs sequence alignment and the mtAlaRS structural model revealed that the surface-exposed basic amino acid residues Arg592, His656, and Arg769 are unique for the mitochondrial enzyme (Figure S1). Docking of bacterial tRNAThr as a template (tRNAAla has not been resolved) into the structural model showed that these residues together with invariant Arg663 and Arg777 form a positively charged patch that contacts the acceptor stem of tRNA from the minor groove in the same manner proposed for bacterial AlaRSs17,18,24 (Figure 4C). Given that Arg592 approaches the tRNA at the level of the G5:U64 base pair, we propose that it is involved in specific recognition and/or binding of mischarged mitochondrial tRNAAla in the editing domain. Therefore, p.Arg592Trp substitution likely impairs the editing activity of mtAlaRS, leading to increased mistranslation of alanine by serine or glycine. Homozygosity for the editing domain mutation was compatible with life for several months after birth, whereas aminoacylation domain defect in association with the editing dysfunction led to perinatal death.
Both AARS2 mutations caused a near-total deficiency of RC complexes in the heart of the patients, leading to primary manifestation as a CMP, and caused partial deficiency of RC complexes in the skeletal muscle and brain but not in the liver. The severe CMP led to early death of the patients, but the reduction of RC complexes in the other tissues indicated multiorgan manifestation. Next, we investigated the effects of the mutations on RC by BN-PAGE (data not shown) and on mitochondrial translation by translation assays in fibroblasts and in myoblasts or myotubes from the patients. For translation assays, primary cells were pulse-labeled for mitochondrial translation products with 35S methionine in the presence of emitine, a cytosolic translation inhibitor.14 No defects were, however, observed in any cell types through the use of standard culture conditions. If galactose is replaced by glucose as the carbon source for cultured cells, the slow metabolism of galactose to glucose-1-phosphate renders the cells dependent on glutamine and pyruvate for ATP production.27 Therefore, galactose promotes a metabolic shift from glycolysis to oxidative energy metabolism, and cells with a dysfunctional RC fail to grow.28 However, even in galactose culture, the mitochondrial translation and cell growth of the patient cells was similar to that of control cells (Figure S2A). Proofreading-deficient AlaRSs have been shown to allow misincorporation of serine, with profound functional consequences:16 cytoplasmic AlaRS defect has been shown to lead to accumulation of misfolded proteins and sensitivity to serine.29 However, we found no defects in mitochondrial translation, even when the proofreading-deficient mtAlaRS patient cells were grown in galactose medium with 5 μM L-Serine (Sigma-Aldrich) for 24 hr (Figure S2A). These results demonstrate that the manifestation of AARS2 mutations is restricted to certain postmitotic tissues, the mechanism of which is presently unclear.
We found the uncharged mitochondrial tRNA alanine (MT-TA) levels to be unaltered in the heart or muscle of patient 1 (II-1, family 1; Figure S2B). For the RNA blot, total RNA from heart and skeletal muscle postmortem tissues of the patient and of an age-matched control was extracted with Trizol reagent (Invitrogen) in a FastPrep 24 Lysing Matrix D tube (MP Biomedicals). Ten micrograms of RNA was run on a 15% 7M Urea polyacrylamide gel and blotted onto a positively charged nylon membrane (Amersham Hybond-XL nylon). The RNA was hybridized with γ-[32P] ATP-labeled oligonucleotide probes against: MT-TA 5′-ACTGAACGCAAATCAGCCAC-3′, and U6 small nuclear 1 (RNU6-1) RNA 5′-GGAACGCTTCACGAATTTGC-3′ was used as a loading control.
We hypothesized that the tissue-specific manifestation of the CMP could be explained by variable amino acid concentrations in different tissues, especially those of glycine and serine, which in the case of proofreading-deficient mtAlaRS could influence the misincorporation rate of serine and glycine in RC complexes. Therefore, we also quantified free amino acid levels in patient tissues by metabolomic analysis using comprehensive two-dimensional gas chromatography, combined with time-of-flight mass spectrometry.30 The available postmortem samples for the quantification of amino acids were from the heart, muscle, and liver of patient 1 (II-1, family 1), the heart and muscle of a 10-month-old child with a nonmitochondrial dilated familial CMP, and the heart and liver of a 10-year-old girl with an isolated CI deficiency and mitochondrial encephalopathy. The tissue samples were homogenized with Covaris cryohomogenizer and extracted with methanol after the addition of internal standard mixture (C17:0, valine-d and succinic acid-d4). After centrifugation, the supernatant was evaporated to dryness and the samples were trimethylsilylated (25 μl MOX, 45°C, 60 min; 25 μl MSTFA, 45°C, 60 min). For the analysis, a Leco Pegasus 4D GC × GC-TOFMS instrument (Leco, St. Joseph, MI, USA) equipped with a cryogenic modulator was used. For the quantitative analysis of amino acids, calibration curves were constructed at the concentration range at which the method was linear (20–1500 ng) and repeatable for all of the studied amino acids, with relative standard deviation below 10%. We did not detect considerable differences in glycine or serine concentrations between tissues, but we did find increased levels of alanine in the patient heart and muscle, but not in the liver (Figure 5). Alanine can be secreted from RC-deficient skeletal muscle for gluconeogenesis in the liver, and therefore the high alanine may be an unspecific response to RC defect. However, it could also be a compensatory feedback mechanism occurring upon deficient tRNAAla aminoacylation, which remains to be studied.
Figure 5.

Metabolomic Analysis: Postmortem Heart and Skeletal Muscle of Patient Shows Increased Alanine Levels
Patient 1 (II-1, family 1) heart, muscle, and liver samples were compared to those of an age-matched patient with nonmitochondrial dilated CMP (heart and skeletal muscle) and those of a 10-year-old patient with mitochondrial encephalopathy (heart and liver).
Mutations in the mitochondrial aminoacyl-tRNA synthetases are emerging as an important cause of disease, but fascinatingly, they underlie a spectrum of different phenotypes. DARS2 (MIM 610956) and RARS2 (MIM 611524) mutations have been shown to affect the CNS, causing childhood- or juvenile-onset leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation31 and infantile encephalopathy and pontocerebellar hypoplasia,32 respectively. A YARS2 (MIM 610957) mutation has been shown to affect the skeletal muscle and cause sideroblastic anemia,33 whereas very recently, SARS2 mutations were reported to lead to hyperuricemia, pulmonary hypertension, renal failure, and alkalosis.34 Our findings add mutations in another mitochondrial aminoacyl-tRNA synthetase as a cause of disease and indicate that these defects can underlie severe CMPs. We conclude that mitochondrial aminoacyl-tRNA synthetase defects mimic mitochondrial tRNA mutations, having a wide range of tissue-specific manifestations. However, defects in a mitochondrial aminoacyl-tRNA synthetase and its corresponding mitochondrial tRNA do not necessarily lead to similar disease phenotypes: for example, MT-TA mutations have been found to cause adult-onset ophthalmoplegia and dysphagia or pure myopathy (MIM 590000),35–37 not CMP.
Childhood CMPs often remain without molecular diagnosis because of small family materials, tissue-specific manifestation, and lack of a cell culture phenotype, which has left few tools for identification of gene defects by the means of linkage analysis or functional complementation. We demonstrate here that whole-exome sequencing is a powerful tool for use in finding a molecular diagnosis for such patients, providing clues that allow us to understand pathogenesis and a means of providing genetic counseling to the families. Our results show that the spectrum of mitochondrial CMPs extends to perinatal-manifesting lethal disorders.
Acknowledgments
The authors wish to thank Tuula Manninen and Anu Harju for technical assistance and Sanna Marjavaara, Pirjo Isohanni, Uwe Richter, and Brendan Battersby for discussions. This study was supported by the Jane and Aatos Erkko Foundation (to A.S. and T.T.), the Academy of Finland (to H.T. and A.S.), the Sigrid Juselius Foundation, the University of Helsinki (to A.S.), the Graduate School of Biotechnology and Molecular Biology, and the Finnish Cultural Foundation (to A.G.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
DINAMelt Server, http://dinamelt.bioinfo.rpi.edu/
MitoProt, http://ihg.gsf.de/ihg/mitoprot.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PROMALS3D Server, http://prodata.swmed.edu/promals3d/promals3d.php
SIFT Genome, http://sift.jcvi.org/
SWISS-Model Server, http://swissmodel.expasy.org/
UniProt, http://www.uniprot.org/
References
- 1.Thorburn D.R. Mitochondrial disorders: prevalence, myths and advances. J. Inherit. Metab. Dis. 2004;27:349–362. doi: 10.1023/B:BOLI.0000031098.41409.55. [DOI] [PubMed] [Google Scholar]
- 2.Holmgren D., Wåhlander H., Eriksson B.O., Oldfors A., Holme E., Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur. Heart J. 2003;24:280–288. doi: 10.1016/s0195-668x(02)00387-1. [DOI] [PubMed] [Google Scholar]
- 3.Scaglia F., Towbin J.A., Craigen W.J., Belmont J.W., Smith E.O., Neish S.R., Ware S.M., Hunter J.V., Fernbach S.D., Vladutiu G.D. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–931. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- 4.Yaplito-Lee J., Weintraub R., Jamsen K., Chow C.W., Thorburn D.R., Boneh A. Cardiac manifestations in oxidative phosphorylation disorders of childhood. J. Pediatr. 2007;150:407–411. doi: 10.1016/j.jpeds.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka M., Ino H., Ohno K., Hattori K., Sato W., Ozawa T., Tanaka T., Itoyama S. Mitochondrial mutation in fatal infantile cardiomyopathy. Lancet. 1990;336:1452. doi: 10.1016/0140-6736(90)93162-i. [DOI] [PubMed] [Google Scholar]
- 6.Papadopoulou L.C., Sue C.M., Davidson M.M., Tanji K., Nishino I., Sadlock J.E., Krishna S., Walker W., Selby J., Glerum D.M. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 7.Bione S., D'Adamo P., Maestrini E., Gedeon A.K., Bolhuis P.A., Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996;12:385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 8.Cízková A., Stránecký V., Mayr J.A., Tesarová M., Havlícková V., Paul J., Ivánek R., Kuss A.W., Hansíková H., Kaplanová V. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008;40:1288–1290. doi: 10.1038/ng.246. [DOI] [PubMed] [Google Scholar]
- 9.Wittig I., Braun H.P., Schägger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 13.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leary S.C., Sasarman F. Oxidative phosphorylation: synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol. Biol. 2009;554:143–162. doi: 10.1007/978-1-59745-521-3_10. [DOI] [PubMed] [Google Scholar]
- 15.Suomalainen A., Syvänen A.C. Quantitative analysis of human DNA sequences by PCR and solid-phase minisequencing. Mol. Biotechnol. 2000;15:123–131. doi: 10.1385/MB:15:2:123. [DOI] [PubMed] [Google Scholar]
- 16.Guo M., Chong Y.E., Shapiro R., Beebe K., Yang X.L., Schimmel P. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature. 2009;462:808–812. doi: 10.1038/nature08612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokabe M., Ose T., Nakamura A., Tokunaga K., Nureki O., Yao M., Tanaka I. The structure of alanyl-tRNA synthetase with editing domain. Proc. Natl. Acad. Sci. USA. 2009;106:11028–11033. doi: 10.1073/pnas.0904645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sokabe M., Okada A., Yao M., Nakashima T., Tanaka I. Molecular basis of alanine discrimination in editing site. Proc. Natl. Acad. Sci. USA. 2005;102:11669–11674. doi: 10.1073/pnas.0502119102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 20.Schimmel P., Ripmaster T. Modular design of components of the operational RNA code for alanine in evolution. Trends Biochem. Sci. 1995;20:333–334. doi: 10.1016/s0968-0004(00)89067-2. [DOI] [PubMed] [Google Scholar]
- 21.Swairjo M.A., Otero F.J., Yang X.L., Lovato M.A., Skene R.J., McRee D.E., Ribas de Pouplana L., Schimmel P. Alanyl-tRNA synthetase crystal structure and design for acceptor-stem recognition. Mol. Cell. 2004;13:829–841. doi: 10.1016/s1097-2765(04)00126-1. [DOI] [PubMed] [Google Scholar]
- 22.Guo M., Chong Y.E., Beebe K., Shapiro R., Yang X.L., Schimmel P. The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science. 2009;325:744–747. doi: 10.1126/science.1174343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swairjo M.A., Schimmel P.R. Breaking sieve for steric exclusion of a noncognate amino acid from active site of a tRNA synthetase. Proc. Natl. Acad. Sci. USA. 2005;102:988–993. doi: 10.1073/pnas.0409024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beebe K., Mock M., Merriman E., Schimmel P. Distinct domains of tRNA synthetase recognize the same base pair. Nature. 2008;451:90–93. doi: 10.1038/nature06454. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe K. Unique features of animal mitochondrial translation systems. The non-universal genetic code, unusual features of the translational apparatus and their relevance to human mitochondrial diseases. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 2010;86:11–39. doi: 10.2183/pjab.86.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lovato M.A., Chihade J.W., Schimmel P. Translocation within the acceptor helix of a major tRNA identity determinant. EMBO J. 2001;20:4846–4853. doi: 10.1093/emboj/20.17.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reitzer L.J., Wice B.M., Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- 28.Robinson B.H., Petrova-Benedict R., Buncic J.R., Wallace D.C. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- 29.Lee J.W., Beebe K., Nangle L.A., Jang J., Longo-Guess C.M., Cook S.A., Davisson M.T., Sundberg J.P., Schimmel P., Ackerman S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 30.Castillo S., Mattila I., Miettinen J., Orešič M., Hyötyläinen T. Data Analysis Tool for Comprehensive Two-Dimensional Gas Chromatography/Time-of-Flight Mass Spectrometry. Anal. Chem. 2011;83:3058–3067. doi: 10.1021/ac103308x. [DOI] [PubMed] [Google Scholar]
- 31.Scheper G.C., van der Klok T., van Andel R.J., van Berkel C.G., Sissler M., Smet J., Muravina T.I., Serkov S.V., Uziel G., Bugiani M. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 32.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giegé R. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., Segel R., Elpeleg O., Nassar S., Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McFarland R., Swalwell H., Blakely E.L., He L., Groen E.J., Turnbull D.M., Bushby K.M., Taylor R.W. The m.5650G>A mitochondrial tRNAAla mutation is pathogenic and causes a phenotype of pure myopathy. Neuromuscul. Disord. 2008;18:63–67. doi: 10.1016/j.nmd.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Spagnolo M., Tomelleri G., Vattemi G., Filosto M., Rizzuto N., Tonin P. A new mutation in the mitochondrial tRNA(Ala) gene in a patient with ophthalmoplegia and dysphagia. Neuromuscul. Disord. 2001;11:481–484. doi: 10.1016/s0960-8966(01)00195-x. [DOI] [PubMed] [Google Scholar]
- 37.Swalwell H., Deschauer M., Hartl H., Strauss M., Turnbull D.M., Zierz S., Taylor R.W. Pure myopathy associated with a novel mitochondrial tRNA gene mutation. Neurology. 2006;66:447–449. doi: 10.1212/01.wnl.0000196490.36349.83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.