

Abstract
The fact that hereditary hearing loss is the most common sensory disorder in humans is reflected by, among other things, an extraordinary allelic and nonallelic genetic heterogeneity. X-chromosomal hearing impairment represents only a minor fraction of all cases. In a study of a Spanish family the locus for one of the X-chromosomal forms was assigned to Xp22 (DFNX4). We mapped the disease locus in the same chromosomal region in a large German pedigree with X-chromosomal nonsyndromic hearing impairment by using genome-wide linkage analysis. Males presented with postlingual hearing loss and onset at ages 3–7, whereas onset in female carriers was in the second to third decades. Targeted DNA capture with high-throughput sequencing detected a nonsense mutation in the small muscle protein, X-linked (SMPX) of affected individuals. We identified another nonsense mutation in SMPX in patients from the Spanish family who were previously analyzed to map DFNX4. SMPX encodes an 88 amino acid, cytoskeleton-associated protein that is responsive to mechanical stress. The presence of Smpx in hair cells and supporting cells of the murine cochlea indicates its role in the inner ear. The nonsense mutations detected in the two families suggest a loss-of-function mechanism underlying this form of hearing impairment. Results obtained after heterologous overexpression of SMPX proteins were compatible with this assumption. Because responsivity to physical force is a characteristic feature of the protein, we propose that long-term maintenance of mechanically stressed inner-ear cells critically depends on SMPX function.
Main Text
As the most common sensory disorder in humans, hearing loss affects about 1 in 1000 newborns.1 It is assumed that at least half of the cases have a genetic basis, and more than two-thirds of this subset of cases are classified as nonsyndromic hearing loss (NSHL) because of the absence of additional symptoms. The vast majority of NSHL is caused by mutations in autosomal genes. X-chromosomal inheritance accounts for only 1%–5% of the cases.2 To date, four X-chromosomal NSHL loci (DFNX) have been mapped, and two genes have been implicated in this group of disorders (Hereditary Hearing Loss Homepage). X-linked DFNX1 (formerly DFN2 [MIM 304500]) is characterized by postlingual progressive hearing impairment and has a typical age at onset between 5 and 15 years for males and in the fifth decade for females. The gene mutated in DFNX1,3 PRPS1 (MIM 311850), encodes phosphoribosyl pyrophosphate synthetase 1, which participates in the nucleotide biosynthesis pathway by catalyzing the reaction of ribose-5-phosphate with ATP. Its function in the inner ear remains elusive. Apart from its contribution to NSHL, mutations in PRPS1 have also been implicated in PRS-I superactivity (MIM 300661), Charcot-Marie-Tooth disease type 5 (CMTX5 [MIM 311070]), and Arts syndrome (MIM 301835), all of which can be accompanied by hearing loss.4 POU3F4 (MIM 300039) encodes a member of the POU family of transcription factors and is mutated in DFNX2 (formerly DFN3 [MIM 304400]),5 which is characterized by prelingual sensorineural deafness accompanied by a conductive component because of stapedial fixation.
In the present study, we investigated a large German pedigree with a postlingual NSHL for which age at onset is 3–7 in males. Initially, there is a moderate hearing loss, especially for high frequencies, that progresses with age and affects all frequencies later. Onset of hearing loss in female carriers is in the second to third decades, and patients present with a severe hearing loss after 10–15 years (Figure 1). Vestibular function was normal, and tinnitus was not reported by the affected individuals. No signs of a conductive hearing loss-component (no air-bone gaps) were noticed in pure-tone audiometry; this finding indicates a normal middle-ear function. Computed tomography, magnetic resonance imaging and digital volume tomography showed a normal middle-ear cavity with regular ossicles, a normal mastoid, and no signs of malformation in the affected individuals. The regular inner-ear structures had fluid-filled, normally shaped cochlea that allowed later treatment by cochlear implantation.
Figure 1.
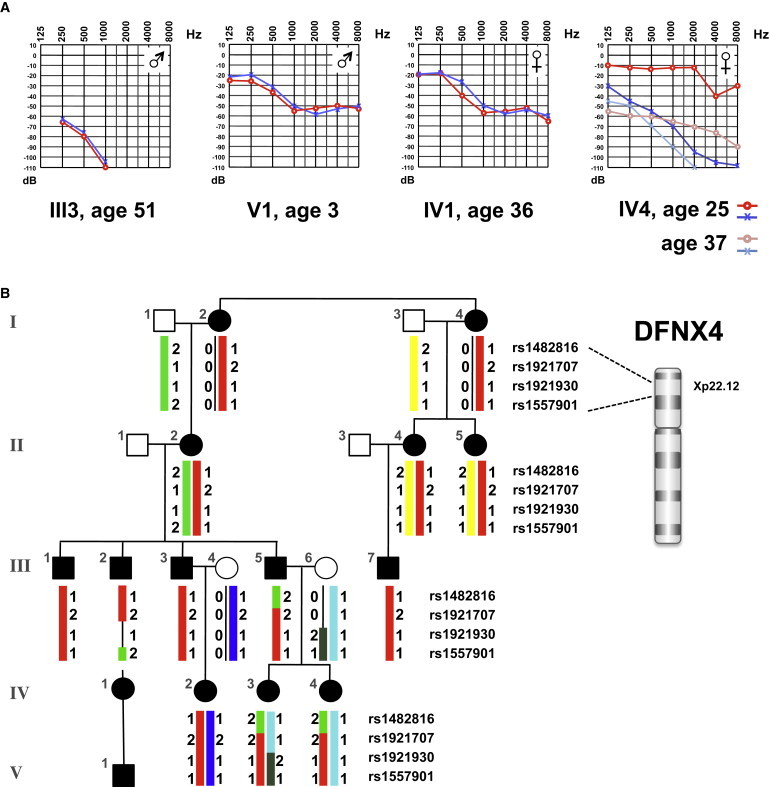
Clinical Phenotype and Segregation Analysis in a German Family Affected by Hereditary Hearing Loss
(A) Audiograms of selected family members (red indicates the right ear, and blue indicates the left ear). Hearing loss in males typically starts between ages 3–7, whereas hearing loss in females begins in the second to third decades.
(B) Haplotypes constructed by four Xp22.12 SNP markers mapped the disease locus between rs1482816 (hg19 X: 5746140) and rs1557901 (hg 19 X: 23222925). Individuals I-1–I-4, III-4, III-6, IV-1, and V-1 were not subjected to the 10K GeneChip linkage analysis.
Genome-wide linkage analysis (GeneChip Human Mapping 10K Array, Affymetrix, Santa Clara, CA, USA) on 11 affected family members revealed linkage to a 17.5 Mb interval in Xp22.12 and maximum LOD scores of 2.23 (Figure S1, available online). Analysis of multiple informative meioses assigned the disease locus between rs1482816 (hg19 X: 5746140) and rs1557901 (hg19 X: 23222925) (Figure 1). We calculated LOD score by using the ALLEGRO6 program and assumed dominant inheritance with full penetrance and a disease allele frequency of 0.0001. Haplotypes were reconstructed with MERLIN7 and presented graphically with HaploPainter.8
To identify the gene mutated in the family, we performed targeted enrichment of all exons and 1 kb of the promoter regions of the 88 protein-coding genes and known miRNAs within the critical interval (GRCh37/hg19) by using the Roche NimbleGen 385K custom sequence capture array. Final coverage of the design was 96.3% and included an offset of 100 bp. DNA of two affected males from the family (individuals III-2 and III-5, see Figure 1B) was subjected to target enrichment and sequencing. We prepared the sequencing library by using standard Illumina protocols including end repair; A-tailing, paired-end adaptor ligation; and amplification by PCR. Enrichment factors (284-fold and 280-fold) were determined by quantitative PCR of four control loci in the array in a comparison of enriched versus nonenriched DNA. The two libraries were subjected to massively parallel sequencing (Illumina GA IIx), resulting in approximately 2828.6 Mb and 2606.0 Mb of raw sequences for the two individuals analyzed. For primary data analysis, a semiautomated data-processing pipeline was established. We first mapped reads to version hg19 of the human reference genome by using the MAQ short-read alignment software9 on a large-scale compute cluster. We called single-nucleotide variants (SNVs) by using the MAQ downstream analysis tools.9 For indel calling, we repeated the alignment procedure by using the BWA aligner10 and the SAMtools11 software for downstream analysis. A total of 3858 and 3443 X-chromosomal variants were called for individuals III-2 and III-5 (Table S1).
At the same time, we collected DNA samples from additional affected males and genotyped them for highly polymorphic microsatellite markers; by doing this we reduced the linkage region to ∼8.5 Mb flanked by DXS987 (hg19 X: 14709303) at the telomeric end. When the refined interval (UCSC Genome Browser hg19 X:14709303-23223175) was taken into account, the number of SNVs remaining was 398 and 347 for the respective patients (Tables S1 and S2). Six variants located to exons and were not annotated as SNPs. We considered these variants for their impact on protein synthesis and the degree of evolutionary conservation by using GERP and PolyPhen.12,13 The nonsense mutation c.109G>T (p.Glu37X) in small muscle protein, X-linked (SMPX) (NM_014332.1, hg19 X:21761891) was considered the best candidate for NSHL in view of the character of the mutation and the GeneRIF entries and was confirmed by Sanger sequencing (Figure 2A, left panel).
Figure 2.
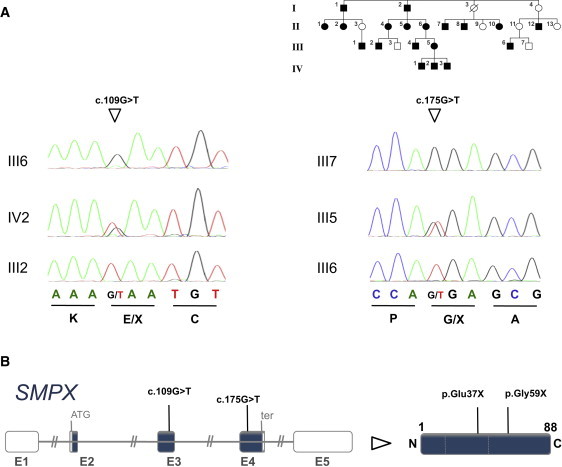
Identification of Nonsense Mutations in SMPX
(A) Sequencing of genomic DNA revealed the SMPX nonsense mutations c.109G>T in exon 3 in the German family (left) and c.175G>T in exon 4 in the Spanish family14 (right). Electropherograms of a respective heterozygous female carrier and a hemizygous male are shown in comparison to a reference sequence. No additional mutations have been identified in a cohort of 34 GJB2-negative individuals with early-onset hearing loss.
(B) SMPX is composed of five exons (left, the coding sequence is given in blue) and encodes an 88 amino acid protein (right) without known functional domains.
Remarkably, the candidate interval included DFNX4 (MIM 300066, formerly DFN6), the locus for one form of X-chromosomal NSHL mapped in a Spanish family.14 The NSHL in this family was bilateral, symmetric, sensorineural, postlingual, and progressive. In affected males, a high-frequency hearing loss was detected at age 5–7; by adulthood, it had become severe or profound and involved all frequencies. Females manifested a moderate hearing impairment for the high frequencies, and onset was in the fourth decade of life. Subsequent investigation of SMPX in this family identified the nonsense mutation c.175G>T (p.Gly59X) (hg19 X:21755773), which segregated with the hearing loss (Figure 2A, right panel). Both the c.109G>T and c.175G>T SMPX mutations (Figure 2B) result in transcripts with premature stop codons and are likely to undergo nonsense-mediated mRNA decay (NMD), suggesting loss-of-function as the underlying mechanism of the hearing impairment. These data demonstrate that SMPX is the gene mutated in the DFNX4-associated disease. In an independent concurrent study of two additional families with hearing loss, Schraders et al. (in this issue) also identified mutations in SMPX and their study provides additional support for our findings.15 Written informed consent was obtained from all the study participants after approval from the Institutional Review Boards at the participating institutions.
Human SMPX encodes a small 88-amino acid protein (NP_055147.1) that has no known functional domains and was initially cloned from muscle.16 SMPX was also identified in a screening for stretch-responsive skeletal muscle genes and shown to be highly upregulated in response to passive stretch in vivo.17 In adult striated myocytes, Smpx localizes to costamere structures,18 muscle-specific protein networks that couple the force-generating sarcomeres with the sarcolemma and the surrounding extracellular matrix. This functional unit acts as a buffer to protect the sarcolemmal plasma membrane from damage generated by the mechanical stress of contracting muscle cells. Costameres are believed to represent a striated-muscle-specific elaboration of focal adhesions in nonmuscle cells.19 These dynamic protein complexes, through which the cytoskeleton of a cell connects to the extracellular matrix, are mechanosensitive in that they respond to force by changing their size, dynamics, and signaling activity.20 In line with this, Smpx has been shown to partially colocalize with focal adhesion complexes upon heterologous expression and precipitate with the focal adhesion marker vinculin.21 In HeLa cells, heterologously expressed, C-terminally Myc-tagged SMPX (SMPX_myc) showed a predominant intracellular staining and enrichment in lamellipodia (Figure 3). A partial overlap with vinculin was observed, especially in adhesion complexes of the cell periphery. Of note, SMPX_myc did not reveal a substantial overlap with the mature focal adhesions that serve as anchor points for actin stress fibers, and this implies a nonexclusive role for the protein in adhesion processes. Staining of the truncated variant SMPX_59X_myc (p.Gly59X in the Spanish family) with the Myc antibody showed an intracellular signal comparable to wild-type; however, absence of the protein from the cell membrane and thus the vinculin-positive cell periphery indicated partial mistargeting (Figure 3). No specific staining was observed with the Myc antibody after expression of the sequence-verified SMPX_37X_myc variant (p.Glu37X, German family), indicating very low abundance or rapid degradation of newly synthesized polypeptides. These results suggest that the SMPX transcripts with early stop codons are degraded by NMD and represent a functional null allele. Alternatively, transcripts can escape NMD and produce largely truncated and misrouted, that is nonfunctional, polypeptides.
Figure 3.
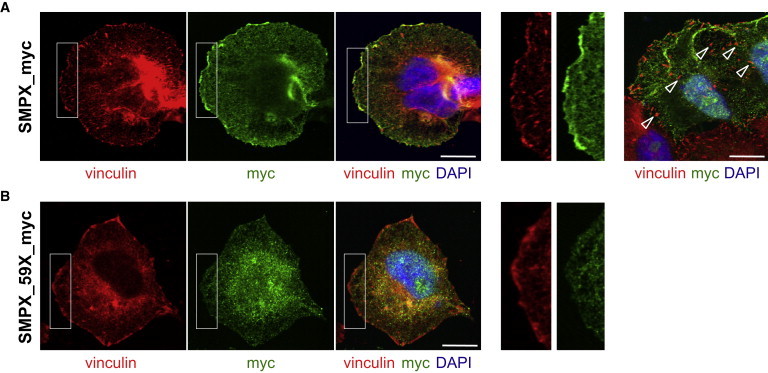
Heterologous Expression of the SMPX Mutant Proteins
(A) C-terminally Myc-tagged human SMPX (SMPX_myc) was overexpressed in HeLa cells. Costaining with Myc (Santa Cruz, A14 sc-789) and vinculin antibodies (Chemikon MAB3574) revealed overlap, especially in the focal adhesion sites of the cell periphery. Enlargements of the cell membrane (white rectangles) are given. Note that SMPX_myc was largely absent from the mature focal adhesions that serve as anchor points for actin stress fibers (right, arrowheads). The scale bar indicates 5μm.
(B) Truncated SMPX (SMPX_59X_myc) revealed an intracellular staining in HeLa cells; the signal was largely absent from adhesion sites at the cell membrane, indicating partial mislocalization of the protein. Enlargements of the cell membrane (white rectangles) are given on the right. SMPX_37X_myc, corresponding to the mutation identified in the German family, was not detectable with Myc antibodies after heterologous expression in HeLa cells (not shown). The scale bar indicates 5μm.
Immunolocalization studies by an Smpx antibody and longitudinal cross-sections of the mouse cochlea revealed staining in different cell types, including Böttcher cells, root cells, pillar cells, and interdental cells of the limbus spiralis. Smpx immunoreactivity was also detected at low levels in hair cells (Figure 4).
Figure 4.
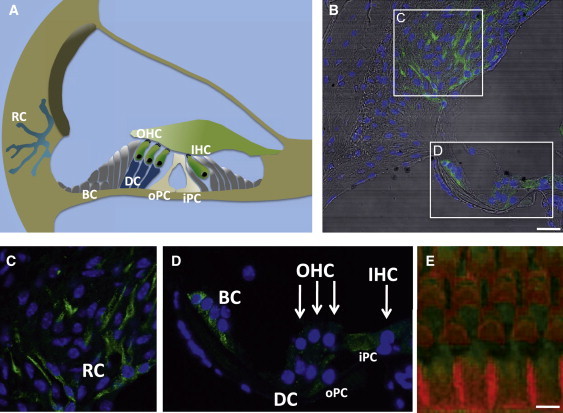
Localization of Smpx in the Mouse Inner Ear
(A) A schematic representation of the cochlea. The sensory epithelium is composed of inner hair cells (IHC) and outer hair cells (OHC). Nonsensory supporting cells include Deiters cells (DC), Böttcher cells (BC), and inner and outer pillar cells (iPC, oPC). Root cells (RC) build up a cellular network in the lateral wall of the cochlea.
(B) Cryosections (7-μm thickness) from the cochlea of adult C57BL/6J mice were stained with an Smpx antibody (Sigma Aldrich #AV41597, 1:500, green), and DAPI (blue). The overlay with differential interference contrast is shown. The scale bar represents 25μm. Stainings of SMPX_myc overexpressing HeLa cells with the Smpx rabbit-antiserum gave the same signal as with the mouse Myc-antibody (data not shown).
(C and D) Enlargements of spiral ligament cells and outer sulcus cells together with root cells (C) and the sensory epithelium with supporting cells (D). Note the Smpx localization in root cells, Böttcher cells, inner and outer pillar cell, and weaker signals in Deiters cells and hair cells (marked by arrows).
(E) Apical basilar membranes of fixed cochleae were dissected and immunostained with an Smpx antibody (green) and conjugated phalloidin (red). Weak Smpx immunoreactivity was detected in inner and outer hair cells. The scale bar indicates 5μm.
In the hearing process, the induction of sound causes fluid to move through the cochlear duct and thereby deflects the basilar membrane with the sensory epithelium against the tectorial membrane. Thus, chronic mechanical stress is characteristic for the inner ear. The mechanical force on hair cells leads to the transduction of sound into an electrical signal that underlies the hearing process. The tips of hair cells are equipped with mechanical-vibration-sensitive stereocilia that enable mechanosensory transduction. Stereocilia are arranged as a staircase and connected with lateral and tip links on the basis of elaborate actin-based cellular protrusions. Indeed, a significant number of genes in which variation is associated with deafness encode actin or actin-binding proteins, motor proteins of the myosin family, or proteins that are otherwise linked to the cytoskeleton.22,23 Given the association of SMPX with the cytoskeleton, the responsivity to mechanical force, and the detection of Smpx in hair cells in the mouse, it is tempting to speculate that SMPX might play a role in the maintenance of stereocilia, which are permanently exposed to physical forces. SMPX might also contribute to actin turnover and length regulation in stereocilia because these features are tightly regulated by extrinsic biomechanical forces.24 However, the antibody stainings failed to show a selective labeling of the stereociliar bundles of the inner and outer hair cells, which argues against high abundance of Smpx in stereocilia.
Mechanotransduction is not limited to hair cells and is crucial in the maintenance of many mechanically stressed tissues.25 Hence, any changes in normal intracellular-force transmission can result in altered mechanosensitive signals and cellular dysfunction. In addition to hair cells, physical force is also applied to other cells of the organ of Corti. SMPX might play a protective role against mechanical stress in different cell types of the organ of Corti, in line with the observed expression pattern, and functional loss of SMPX could lead to the progressive hearing impairment in the affected individuals.
Despite its strong expression in muscle cells, SMPX appears to be largely dispensable for muscle function because patients from the families studied do not show obvious signs of muscular dysfunction. Interestingly, elevated creatine kinase levels (400 U/l) and myalgias have been repeatedly reported in a 71-year-old male patient from the German family; however, the relation to the SMPX mutation is currently unclear. Mice with a targeted disruption of Smpx (named Csl) did not exhibit an overt muscle phenotype.18 Breeding of the Csl knockout-mouse line has been discontinued, and thus it is not available for hearing tests (R.P. Harvey, personal communication).
Remarkably, defects in the Rac1/p38 pathway, which is a target of Smpx,21 have also been shown to be associated with inner-ear dysfunction. Rac1 belongs to the family of small GTPases. It is activated by biomechanical stress, upon which it is recruited to sites of actin reorganization and integrin-mediated cell adhesions. Intriguingly, conditional ablation of Rac1 in the otic epithelium of mice resulted in defective morphogenesis of the auditory sensory epithelium and stereociliary bundle.26 p38 has also been shown to be of relevance for inner-ear function; inhibition of p38 MAP kinase phosphorylation suspended gentamicin-induced ototoxicity, a side effect of aminoglycoside therapy leading to permanent hair-cell loss and hearing impairment.27 Smpx is also stimulated by insulin-like growth factor-1 (IGF-1).18 IGF-1 mutations in humans are associated with syndromic sensorineural deafness28 and Igf-1-null mice exhibit hearing loss.29
In conclusion, our study identified mutations in SMPX in patients with X-chromosomal hearing impairment and suggested that the stress response of mechanically challenged inner-ear cells might critically depend on SMPX function.
Acknowledgments
We sincerely thank all of the family members for their participation and their constant interest in and support of this study. The authors also wish to thank Ingelore Bäßmann, Christian Kluck, and Christian Becker for expert technical assistance. Florian Wagner is an employee of ATLAS Biolabs GmbH. Peter Nürnberg is a founder, CEO, and shareholder of ATLAS Biolabs GmbH. ATLAS Biolabs GmbH is a service provider for genomic analyses. This work was supported by grants from the Werner-Otto-Stiftung (to I.K.), Fondo de Investigaciones Sanitarias (PI 08/0818 to I.d.C.), Fundación Ramón Areces (to I.d.C.), and Spanish Ministerio de Ciencia e Innovación (SAF2008-03216 to F.M.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ENSEMBL, http://www.ensembl.org
GeneDistiller, http://www.genedistiller.org/
Hereditary Hearing Loss, http://hereditaryhearingloss.org/
Online Mendelian Inheritance in Man (OMIM), www.omim.org
Primer Design, http://portal.ccg.uni-koeln.de/geneexplorer/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Morton C.C., Nance W.E. Newborn hearing screening—a silent revolution. N. Engl. J. Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Petersen M.B., Wang Q., Willems P.J. Sex-linked deafness. Clin. Genet. 2008;73:14–23. doi: 10.1111/j.1399-0004.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 3.Liu X., Han D., Li J., Han B., Ouyang X., Cheng J., Li X., Jin Z., Wang Y., Bitner-Glindzicz M. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am. J. Hum. Genet. 2010;86:65–71. doi: 10.1016/j.ajhg.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Brouwer A.P., van Bokhoven H., Nabuurs S.B., Arts W.F., Christodoulou J., Duley J. PRPS1 mutations: Four distinct syndromes and potential treatment. Am. J. Hum. Genet. 2010;86:506–518. doi: 10.1016/j.ajhg.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Kok Y.J., van der Maarel S.M., Bitner-Glindzicz M., Huber I., Monaco A.P., Malcolm S., Pembrey M.E., Ropers H.H., Cremers F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685–688. doi: 10.1126/science.7839145. [DOI] [PubMed] [Google Scholar]
- 6.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 8.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 9.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.del Castillo I., Villamar M., Sarduy M., Romero L., Herraiz C., Hernández F.J., Rodríguez M., Borrás I., Montero A., Bellón J. A novel locus for non-syndromic sensorineural deafness (DFN6) maps to chromosome Xp22. Hum. Mol. Genet. 1996;5:1383–1387. doi: 10.1093/hmg/5.9.1383. [DOI] [PubMed] [Google Scholar]
- 15.Schraders M., Haas S.A., Weegerink N.J.D., Oostrik J., Hu H., Hoefsloot L.H., Kannan S., Huygen P.L.M., Pennings R.J.E., Admiraal R.J.C. Next-Generation sequencing identifies mutations of SMPX which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am. J. Hum. Genet. 2011;88:628–634. doi: 10.1016/j.ajhg.2011.04.012. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patzak D., Zhuchenko O., Lee C.C., Wehnert M. Identification, mapping, and genomic structure of a novel X-chromosomal human gene (SMPX) encoding a small muscular protein. Hum. Genet. 1999;105:506–512. doi: 10.1007/s004390051138. [DOI] [PubMed] [Google Scholar]
- 17.Kemp T.J., Sadusky T.J., Simon M., Brown R., Eastwood M., Sassoon D.A., Coulton G.R. Identification of a novel stretch-responsive skeletal muscle gene (Smpx) Genomics. 2001;72:260–271. doi: 10.1006/geno.2000.6461. [DOI] [PubMed] [Google Scholar]
- 18.Palmer S., Groves N., Schindeler A., Yeoh T., Biben C., Wang C.C., Sparrow D.B., Barnett L., Jenkins N.A., Copeland N.G. The small muscle-specific protein Csl modifies cell shape and promotes myocyte fusion in an insulin-like growth factor 1-dependent manner. J. Cell Biol. 2001;153:985–998. doi: 10.1083/jcb.153.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ervasti J.M. Costameres: The Achilles' heel of Herculean muscle. J. Biol. Chem. 2003;278:13591–13594. doi: 10.1074/jbc.R200021200. [DOI] [PubMed] [Google Scholar]
- 20.Geiger B., Bershadsky A. Exploring the neighborhood: Adhesion-coupled cell mechanosensors. Cell. 2002;110:139–142. doi: 10.1016/s0092-8674(02)00831-0. [DOI] [PubMed] [Google Scholar]
- 21.Schindeler A., Lavulo L., Harvey R.P. Muscle costameric protein, Chisel/Smpx, associates with focal adhesion complexes and modulates cell spreading in vitro via a Rac1/p38 pathway. Exp. Cell Res. 2005;307:367–380. doi: 10.1016/j.yexcr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Petit C., Richardson G.P. Linking genes underlying deafness to hair-bundle development and function. Nat. Neurosci. 2009;12:703–710. doi: 10.1038/nn.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dror A.A., Avraham K.B. Hearing loss: Mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 2009;43:411–437. doi: 10.1146/annurev-genet-102108-134135. [DOI] [PubMed] [Google Scholar]
- 24.Manor U., Kachar B. Dynamic length regulation of sensory stereocilia. Semin. Cell Dev. Biol. 2008;19:502–510. doi: 10.1016/j.semcdb.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaalouk D.E., Lammerding J. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimsley-Myers C.M., Sipe C.W., Géléoc G.S., Lu X. The small GTPase Rac1 regulates auditory hair cell morphogenesis. J. Neurosci. 2009;29:15859–15869. doi: 10.1523/JNEUROSCI.3998-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei X., Zhao L., Liu J., Dodel R.C., Farlow M.R., Du Y. Minocycline prevents gentamicin-induced ototoxicity by inhibiting p38 MAP kinase phosphorylation and caspase 3 activation. Neuroscience. 2005;131:513–521. doi: 10.1016/j.neuroscience.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 28.Woods K.A., Camacho-Hübner C., Savage M.O., Clark A.J. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996;335:1363–1367. doi: 10.1056/NEJM199610313351805. [DOI] [PubMed] [Google Scholar]
- 29.Cediel R., Riquelme R., Contreras J., Díaz A., Varela-Nieto I. Sensorineural hearing loss in insulin-like growth factor I-null mice: A new model of human deafness. Eur. J. Neurosci. 2006;23:587–590. doi: 10.1111/j.1460-9568.2005.04584.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.