

Abstract
In primary ciliary dyskinesia (PCD), genetic defects affecting motility of cilia and flagella cause chronic destructive airway disease, randomization of left-right body asymmetry, and, frequently, male infertility. The most frequent defects involve outer and inner dynein arms (ODAs and IDAs) that are large multiprotein complexes responsible for cilia-beat generation and regulation, respectively. Although it has long been suspected that mutations in DNAL1 encoding the ODA light chain1 might cause PCD such mutations were not found. We demonstrate here that a homozygous point mutation in this gene is associated with PCD with absent or markedly shortened ODA. The mutation (NM_031427.3: c.449A>G; p.Asn150Ser) changes the Asn at position150, which is critical for the proper tight turn between the β strand and the α helix of the leucine-rich repeat in the hydrophobic face that connects to the dynein heavy chain. The mutation reduces the stability of the axonemal dynein light chain 1 and damages its interactions with dynein heavy chain and with tubulin. This study adds another important component to understanding the types of mutations that cause PCD and provides clinical information regarding a specific mutation in a gene not yet known to be associated with PCD.
Main Text
Cilia are ancient, evolutionarily conserved organelles that project from the surfaces of most cells to perform diverse biological roles, including whole-cell locomotion; movement of fluid; chemo-, mechano-, and photosensation; and paracrine signal transduction. These organelles can be classified according to the arrangement of their microtubule cytoskeleton core, called an axoneme.1 The axoneme consists of nine outer-doublet microtubules, which are connected by nexin links and surround a central pair of microtubules (i.e., 9 + 2 pattern). In some cilia, the central pair of microtubules is absent (i.e., 9 + 0 pattern). The 9 + 0 primary cilia are immotile, except in the embryonic node, where they are involved in left-right asymmetry.2 The 9 + 2 motile cilia, structurally identical to spermatozoan flagella, are involved in the transport of extracellular fluids, as in the respiratory tract, where they propel mucus and therefore represent the first line of airway defenses.1 Motile cilia are powered by outer dynein arms (ODAs) and inner dynein arms (IDAs), multiprotein ATPase complexes that are attached to the peripheral doublets and are essential for ciliary motion.1 For most motile cilia, additional accessory structures, for example radial spokes and central pair projections, are involved in regulating dynein-mediated motility.
Primary ciliary dyskinesia (PCD [MIM 242650]) refers to a heterogeneous group of genetic disorders characterized by ultrastructural defects in the axonemal structure of the 9 + 2 motile cilia and sperm flagella.3 The incidence is estimated at 1:15,000–30,000,4 and there is a higher incidence in certain consanguineous and isolated populations.5,6 Clinical features reflect the distribution of immotile cilia in the body and include neonatal respiratory distress, chronic respiratory infections, sinusitis, and bronchiectasis because of deficient function of motile cilia in the upper and lower airways. Male and female subfertility occur as a result of defective sperm flagella and oviduct cilia, respectively. Occasionally, hydrocephalus occurs as a result of deficient ependymal cilia.7,8 In most families, there is an apparent randomization of left-right axis development; this randomization is proposed to result from the defective function of the embryonic node. This manifests in about half of patients as situs inversus or more severe laterality defects, such as cardiovascular abnormalities. The association of PCD and situs inversus is also referred to as Kartagener's syndrome (MIM 244400).9,10 There is a large variation in the severity of the clinical phenotype. Clinical features of PCD can also mimic those of other diseases, such as cystic fibrosis, allergies or immunologic disorders. Studying the ciliary-beat pattern and frequency by direct microscopy on transnasal brushings and examining the axonemal structure of respiratory cilia by transmission electron microscopy allows diagnosis of PCD. Typical ultrastructural defects in PCD consist of total or partial absence of dynein arms, absence or dislocation of central tubules, defects of the radial spoke, and peripheral microtubule anomalies. However, in a subset of patients, no ultrastructural defect are observed.11,12 Additionally, in patients with PCD, nasal nitric oxide (nNO) levels are often extremely low in comparison to controls. A nNO concentration of <200 ppb could indicate PCD.13
PCD is usually recessively inherited, and mutations that cause PCD have been identified in several genes and chromosomal loci.9 Most of the genes encode axonemal dyneins and are associated with reduction or loss of axonemal ODAs, which are the multisubunit axonemal ATPase complexes that generate the force for cilia motility and govern beat frequency. Mutations in DNAH5 (MIM 603335) and DNAI1 (MIM 604366) are a relatively more common cause of disease, underlying an estimated 28% (DNAH5)14 and 2%–10% (DNAI1)15,16 of total cases. The other genes are so far only associated with single or rare PCD cases; therefore, the genetic basis of at least 60% of PCD cases is not yet known. These genes include additional components of dynein arms: DNAI2, DNAH11, and TXNDC3.17 Mutations in genes encoding cytoplasmic proteins such as KTU and LRRC50 affect the assembly of both IDA and ODA complexes in the cytoplasm by a process that is still poorly understood.18–21 Mutations in RSPH9 and RSPH4A, encoding two radial spoke head proteins, have been recognized in Pakistanis and Bedouins.22 These mutations cause primary ciliary dyskinesia with central-microtubular-pair abnormalities involved in central-pair defects. Lastly, it was recently reported that mutations in CCDC3923 and CCDC40,24 two proteins for coiled-coil domains, cause a PCD variant that is characterized by misplacement of the central pair of microtubules and defective assembly of IDAs and dynein regulatory complexes. CCDC40 localizes to motile cilia at the apical cytoplasm and is required for axonemal recruitment of CCDC39;24 the latter underlies a substantial fraction of PCD cases with axonemal disorganization and abnormal ciliary beating.23
We have identified two consanguineous Bedouin families with three PCD patients (Figure 1A). The patients had the classical presentation of PCD, the clinical data and laboratory findings that established the PCD diagnosis of the patients are presented in Table 1. Example transmission electron microscope images demonstrating the absence of eight out of the nine ODAs and occasional marked shortening of the ninth is shown in Figure 1B in comparison to the healthy homozygous normal sibling IV-3. The same ODA defect was observed in a total of 37 cilia documented from the two patients of family A. Additional images of the 2 patients are presented in Figure S1, available online.
Figure 1.

PCD Bedouin Pedigrees, Fine Mapping of the Chromosome 14 Locus in Pedigree A, and Absence of the ODAs in Patient Cilia
(A) The haplotype analysis based on microsatellite markers from 14q24.2-q31.3 revealed a founder haplotype (gray bar) for which the patients are homozygous in the critical region harboring DNAL1. The numbers of the patients correspond to those presented in Table 1. Patient IV-1 of family B was identified by analysis of the mutation. Filled symbols represent the homozygote for the mutation and a double line indicates a consanguineous union. The two lines indicate the crossing events delimiting the homozygous interval shared by the two patients. Markers rs17176306 and rs41471545 define the minimal homozygosity locus associated with the disease.
(B) Transmission electron micrograph of nasal ciliary epithelium from patient IV-1 of family A in comparison to a control (sibling IV-3 of family A). The arrows in the micrographs point to the ODA that is missing in the patient.
Table 1.
Clinical Data and Laboratory Findings of the PCD Patients
|
Family A |
Family B |
||
|---|---|---|---|
| Patient IV-1 | Patient IV-2 | Patient IV-1 | |
| Patient characteristics | |||
| Age/gender | 17.5 years/male | 14.5 years /female | 6.6 years/male |
| Complete situs inversus | yes | yes | yes |
| Neonatal period | neonatal pneumonia | tachypnea, oxygen requirement for 12 days | tachypnea, oxygen requirement for 18 days |
| Reactive airway disease | yes | yes | yes |
| Chronic nasal discharge | yes | yes | yes |
| Chronic serous otitis media | yes | yes | yes |
| Bronchiectasis | yes | yes | no |
| Surgery | no | yes: adenoidectomy at 5.5 years, lymphangioma in right forearm | ventilation tubes |
| Cilia movementa | weak movement | weak movement | no movement |
| Electron microscopy | absence of ODAs | absence of ODAs | absence of ODAs |
| nNO | 5 ppb | 3 ppb | not done |
| Pulmonary function tests | |||
| Forced vital capacity (FVC)b | 105% | 103% | not done |
| Forced expiratory volume in 1 s (FEV1)c | 61% | 76% | not done |
| FEV1/FVC | 54% | 70% | not done |
| Forced midexpiratory flow rated | 25% | 40% | not done |
The following abbreviations are used: ODAs, outer dynein arms; nNO, nasal nitric oxide; and ppb, parts per billion.
Ciliary movement was measured by direct light microscopy.
The volume of air that can forcibly be blown out after full inspiration, measured in liters.
The average values for FEV1 in healthy people depend mainly on sex and age, and values between 80% and 120% of the average value are considered normal.
The rate of flow between 25% and 75% of the FVC. Its value is determined from the FVC curve, and it is reduced in early obstruction involving the smaller airways.
The study was approved by the institutional review board of the Soroka Medical Center, and informed consent was obtained from all participants or their legal guardians. Genomic DNA was isolated by standard methods directly from blood samples. Assuming disease by homozygosity of a mutation from a common ancestor, we excluded linkage to the DNAH5 and DNAI1 by the finding of heterozygosity of alleles at known polymorphic markers adjacent to these genes.25 We further searched for homozygous regions consistent with linkage by genotyping the patients and both parents from pedigree A with the Affymetrix (Santa Clara, CA) GeneChip Human Mapping 250K Sty arrays. We determined the genotype calls by using Affymetrix GeneChip Genotyping Analysis Software (GTYPE) and dedicated software KinSNP developed in house to automatically perform autozygosity analysis of the microarray results.26 A single large homozygous region between rs17176306 and rs41471545 (>16 cM and 14.4 Mb) on chromosome 14 was found to be shared by the two affected patients. To confirm linkage to this region, we tested all family members with both known polymorphic microsatellite markers and with additional markers developed for this purpose.27 Linkage was identified to chromosomal locus 14q24.2-q31.3, chr14: 70,128,727–84,524,979 (NCBI Build 36.1) (Figure 1A). We calculated the multipoint Lod score by using the Pedtool server and assuming recessive inheritance with 99% penetrance and an incidence of 0.01 or 0.001 for the disease allele in the population, was 2.49.
The linkage interval contains 84 genes; by using the ciliaproteome server, we found 22 of them. Among these, we considered DNAL1 to be the strongest candidate for PCD because it encodes the ortholog of the Chlamydomonas axonemal dynein light chain 1 (LC1), an ODA component that contains the molecular motors for ATP-dependent cilia movement. Therefore, we analyzed it first. Total RNA was extracted from lymphoblastoid cells of patient IV-1 with the EZ-RNA Reagent (Biologic Industries, Israel). Reverse transcriptase reactions were performed with the SuperScript II Reverse Transcriptase Synthesis Kit (Invitrogen). The whole cDNA coding region for DNAL1 was amplified in overlapping segments by PCR. No splice variants were detected. The PCR products were directly sequenced on an ABI PRISM 3100 DNA Analyzer with the BigDye Terminator v. 1.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA) according to the manufacturer's protocol. The primers that revealed the mutation were primer 5′-CCCTAGCAACCAGAGCAGTGA-3′ (forward) and primer 5′-AGGTTGGCATTACCAGTTTTG-3′ (reverse). We identified a homozygous nonsynonymous variation (NM_031427.3: c.449A>G; NP_113615.2: p.Asn150Ser) (Figure 2A). Because the patient in family B is from the same population, although not known to be related to family A, we evaluated him for the p.Asn150Ser mutation by sequencing of a PCR product amplified from his genomic DNA (see below) and found to be homozygous for the mutation. His parents, who were similarly analyzed, are heterozygous for this variant. The patient's siblings are either heterozygous (IV-2) or homozygous normal (IV-3). Testing for the variation in all members of both families and controls was performed by restriction analysis because the variation creates a BsmFI restriction site that does not exist in the wild-type sequence. PCR amplification of genomic DNA, around exon 7 of the gene, was performed with the use of the primers 5′- CCTCCCATCCTGTACTGTCTTC-3′ (forward) and 5′- GCTTTTGGTCTAGGGAGAATCTT-3′ (reverse). BsmFI restriction analysis of the PCR amplicons generated differential cleavage products of the wild-type (fragments were 370 bp and, uncut, 495 bp) versus variation (fragments were 370 bp and, cut, 148 bp and 347 bp) allele. Fragments were separated by electrophoresis on 2% agarose gel (Figure 2B). This variation segregates as expected in the families. The mutation was found in the heterozygous state in only one individual and was not found in the homozygous state in any individuals out of 124 healthy Bedouin controls.
Figure 2.
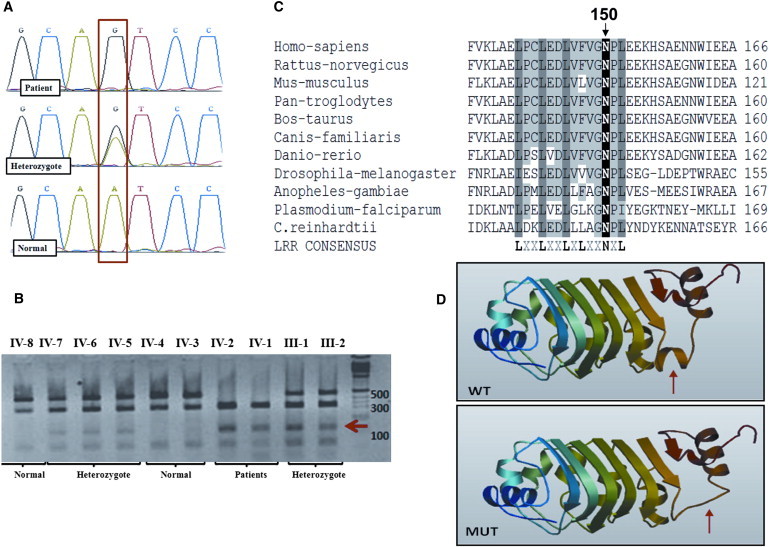
Identification of the DNAL1 Mutation
(A) Sequence of the genomic DNA corresponding to the c.449A>G mutation resulting in p.Asn150Ser. Patients were homozygous for the mutation; parents and siblings carrying the founder haplotype were heterozygous, and the healthy siblings without the founder haplotype were normal.
(B) Restriction analysis for family A with BsmFI enzyme. The 148 bp band (marked with a red arrow) is apparent only in the mutated allele. The wild-type (fragments were 370 bp and, uncut, 495 bp) versus mutation (fragments were 370 bp and, cut, 148 bp and 347 bp) alleles.
(C) Evolutionary conservation of the sixth LRR domain (conserved residues defining the LRR consensus are in bold). Position 150 of the mutated Asn is shown on top.
(D) Structural prediction of the mutated LC1 protein (MUT) in comparison to a normal (WT) structure performed by the SWISS-MODEL tool. The change in structure is marked with a red arrow.
We sought evidence for the pathogenic relevance of this variation by analyzing the conservation of the protein sequence. Sequence alignment of the human DNAL1 with other species orthologs demonstrated complete conservation of the leucine-rich-repeat (LRR) consensus domain. Asn at position150 in human DNAL1 is one of the LRR consensus residues (Figure 2C); it aligns with the Asn located between the β10 and α7 folds of the sixth LRR domain of the corresponding Chlamydomonas (LC1) ortholog.28 Human DNAL1 is highly conserved: alignment of its predicted 3D structure to the orthologous Chlamydomonas LC1 show minor differences, a loop instead of α7 helix and shortened C terminus.29 We verified the effect of the change on the protein conformation by using the SWISS-MODEL tool and found that the replacement of Asn by Ser at position 150 indeed has a major effect on the structure of the protein, in that it disturbs the folding of the sixth LRR domain (Figure 2D).
Next, we studied the effect of the mutation on the protein. We subcloned the mutated and normal protein sequences into the pc3myc plasmid to produce the DNAL1 protein fused at its N terminal to a Myc tag. In brief, we used RNA from lymphoblastoid cells of patient IV-1 in family A and lymphoblastoid cells of a healthy control to reverse transcribe (by using SuperScript II Reverse Transcriptase Synthesis kit [Invitrogen]) and amplify the whole coding sequence of DNAL1 by PCR with the primers EcoRI-F, 5′-GAATTCATGGCGAAAGCAACAACAATCAA-3′, and XhoI-R, 5′-CTCGAGGTTGTCTTCTTCCTCATCCCC-3′, by using the DreamTaq DNA Polymerase (Fermentas). The PCR product was restricted with EcoRI and Xho-I and subcloned into these sites of the plasmid. We sequenced the inserts with the flanking regions with the T7 and SP6 primers to ascertain insertion in frame with the Myc tag and to verify that no PCR mutations were introduced during the cloning process. HEK293T cells were transiently transfected with the constructs by using TransIT-LT1 reagent (Mirus) and lysed with RIPA lysis buffer 48 hr after transfection. The DNAL1-Myc protein levels were studied by immunoblot with a Myc antibody (gift of Noah Isakov). Although the transfection conditions were identical, there was a marked reduction (7-fold) in the quantity of the mutated protein in comparison to the normal amount, as estimated with the GAPDH protein as a loading control (not shown). We tested the possibility that the observed low level of the mutant protein is caused by its instability. Forty-eight hours after transfection of HEK293T with the DNAL1-Myc plasmid, cycloheximide (CMX, Sigma-Aldrich), an inhibitor of protein translational elongation in eukaryotes, was added (to a final concentration of 10 mg/ml). The quantities of mutated and control DNAL1-Myc proteins, starting from approximately equal quantities, were compared by immunoblot analysis at 3 and 6 hr after the addition of CMX. A representative experiment, demonstrating faster reduction in the quantity of the mutant protein than in that of the normal protein, is shown in Figure 3A. To quantify this reduction, we performed three independent experiments. The intensity of the immunoblot signals was measured with the Image Lab software (Bio-Rad). The densitometric ratio of DNAL1-Myc to GAPDH at the time of addition of CMX was established as 100%, and the ratio of the DNAL1-Myc to GAPDH at 3 and 6 hr was compared to time 0. Indeed, the quantity of the mutated protein was reduced by 40% and 94% at 3 and 6 hr, respectively, whereas the normal protein was reduced only by 10% and 30%. The difference between the normal and mutated protein at the two time points was evaluated by a two-tailed Student's t test, under an assumption of unequal variance (p < 0.01 and p < 0.001 at 3 hr and 6 hr, respectively) (Figure 3B).
Figure 3.

Effect of the Mutation on the Stability of the DNAL1 Protein
(Top panel) A representative immunoblot analysis presenting DNAL1 protein at 0, 3, and 6 hr after addition of CMX. GAPDH is shown as a loading control. (Bottom panel) Quantification of the DNAL1-Myc stability relative to GAPDH for three experiments. The densitometric ratio of DNAL1-Myc to GAPDH at the time of addition of CMX was established as 100% and the ratio of the DNAL1 to GAPDH at 3 and 6 hr is compared to time 0. The data is presented as mean ± standard deviation. The difference between the normal and mutated protein at the two time points was evaluated by a two-tailed Student t test, assuming unequal variance. ∗∗p = 0.006, ∗∗∗p = 0.001. The following abbreviations are used: MUT, mutated DNAL1 protein; and WT, normal DNAL1 protein.
In humans, dynein is composed of two heavy chains (∼500 kD), two intermediate chains (70–125 kD), and several light chains (15–30 kD).30 The heavy chains form the globular heads and stems of the complex. In Chlamydomonas two molecules of LC1 are tightly associated with AAA nucleotide-binding domains of the γ heavy chain.28,31 The interaction of the LC1 is mediated through a central LRR section that folds as a cylindrical, right-handed spiral formed from six β-β-α motifs. This central cylinder is flanked by terminal helical subdomains. The C-terminal helical domain juts out from the cylinder and is proposed to interact with the dynein heavy chain. The position of the mutated aspargine is just at the last LRR consensus that stabilizes the turn between the β strand and the α helix. In humans, the ortholog of the γ heavy chain is encoded by two genes, DNAH5 and DNAH8,30 and a similar association of DNAL1 to DNAH5 was also demonstrated29. It was also shown that Chlamydomonas LC1 interacts directly with tubulin and tethers the γ heavy motor unit to the A tubule of the outer-doublet microtubules within the axoneme31,32 (schematically presented in Figure 4A). To verify whether the change in the mutated protein effects binding between DNAL1 to dynein heavy chain and to tubulin, we tested their direct interaction by immunoprecipitation.
Figure 4.

Interaction of the Mutated and Normal DNAL1 Protein with the Dynein Heavy Chain and with Tubulin
(A) Schematic representation of the interaction of DNAL1 with the dynein heavy chain (DNAH) and tubulin.31,32
(B) Immunoprecipitation of dynein heavy chain. (a) Immunoprecipitation of the dynein heavy chain with the mutated (MUT) and normal (WT) DNAL1-Myc by Myc antibody probed with an anti-dynein heavy chain antibody. (b) The beads were incubated solely with the lysate mixture and axoneme and probed with an anti-dynein heavy chain antibody, thus presenting the background dynein heavy chain signal in the immunoprecipitation. (c) Input quantity of DNAL1-Myc for the reaction. (d) Input quantity of dynein heavy chain extracted from rat tracheas. The percentage in brackets refers to the loading on the gel of the lysate mixture with axoneme relative to the quantity used for the immunoprecipitation. The antibody used for the Immunoblot analysis is denoted at the right side.
(C) Immunopercipitation of α-tubulin. (a) IP of α-tubulin with the mutated (MUT) and normal (WT) DNAL1-Myc by Myc antibody probed with an anti-α-tubulin antibody. (b) The beads were incubated solely with the lysate mixture and axoneme and probed with an anti-α-tubulin antibody, thus presenting the background of α-tubulin signal in the immunoprecipitation. (c) Input quantity of α-tubulin for the reaction. (d) Input quantity of DNAL1. The percentage in brackets refers to the loading on the gel of the lysate mixture with axoneme relative to the quantity used for the immunoprecipitation. The antibody used for the Western analysis is denoted at the right side.
The stability of the mutated DNAL1 after the incubation with the axonemal extracts and with the primary antibody at 4°C was comparable to the normal DNAL1 (not shown).
To obtain sufficient amounts of dynein heavy chain proteins and microtubules for immunoprecipitation, we prepared axonemal extracts from rat tracheas. Tracheas with larynx were removed from rats and placed in ice-cold saline. After excision of the larynx and removal of excess connective tissue and fat, the tracheas were transferred to fresh ice-cold saline. High-salt axonemal extracts were prepared as previously described.33 We produced mutant and normal DNAL1-tagged proteins by transfecting HEK293T cells with the respective DNAL1-Myc plasmids and incubated cell extracts containing equal quantities of the normal and mutated DNAL1-tagged proteins (72 μg and 500 μg of the normal and mutated protein extracts, respectively) for 24 hr at 4°C with equal amounts of rat axonemal extracts (230 μg for each extract). Myc antibodies were used for immunoprecipitation of the tagged DNAL1, and the quantity of dynein heavy chain and tubulin in the immunoprecipitate was tested by dynein heavy chain antibody (Abcam, catalog number ab6305) and α-tubulin antibody (B-5-1-2, Abcam, catalog number ab11304), respectively. Both dynein heavy chain and tubulin were readily coimmunoprecipitated with the normal DNAL1-tagged protein, whereas the mutated protein showed about 80% reduction in the immunoprecipitated quantities of both the dynein heavy chain and α-tubulin (Figures 4B and 4C). The stability of the mutated DNAL1 after the incubation with the axonemal extracts and with the primary antibody at 4°C was comparable to that of the normal DNAL1. From these results, it can be concluded that the physical interaction between the mutated DNAL1 protein and both dynein heavy chain and tubulin is damaged.
Most of the genetically characterized PCD variants exhibit mutations in genes that encode dynein arm components that are responsible for ciliary-beat generation.14–21 Using a total genome scan, we now provide evidence that a DNAL1 point mutation, inherited in a recessive manner, causes PCD in two Bedouin families. The patients had the classical presentation of PCD, characterized by complete situs inversus, early neonatal respiratory distress, and chronic sino-pulmonary infection leading to severe morbidity. PCD was confirmed by the laboratory results of very low nNO; a significant decrease in cilia movement, determined by light microscopy; and an absence of ODAs, determined by transmission electron microscopy. The identification of a mutation in DNAL1 demonstrates that the Chlamydomonas LC1 human ortholog, like other components of the ODA, is important for ciliary function in the airways. The presence of situs inversus in the patients indicates that DNAL1 is also necessary for the function of the cilia that produce the nodal flow essential for the determination of left-right asymmetry.10 The function of the orthologs of Chlamydomonas LC1 was intensively studied in model systems. RNAi silencing of expression of the LC1 ortholog of Trypanosoma brucei, TbLC1, resulted in the complete loss of the dominant tip-to-base beat that is a hallmark of trypanosome flagellar motility and the concomitant emergence of a sustained reverse beat that derived cell movement in reverse. Ultrastructure analysis revealed that the outer-arm dyneins are disrupted in TbLC1 mutants. Therefore, LC1 was shown to be required for stable dynein assembly and forward motility in T. brucei.34 The cloning and study of the LC1 ortholog of Phytophthora nicotianae (PnDLC1) demonstrated that RNAi-mediated silencing of PnDLC1 expression yielded transformants that released nonflagellate, nonmotile zoospores from their sporangia.35 The ciliated epithelium of the planarian Schmidtea mediterranea was used for dissecting the role of ODA motors in the coordination of the oscillations of neighboring cilia and the formation of metachronal synchrony of motile cilia.36 Metachronal synchrony is important for effective clearance of mucus and transport of other periciliary fluids over the surface of the epithelium. It is thought to be achieved by coupling between neighboring cilia by a system that provides mechanical feedback. Manipulation of the S. mediterranea by an RNAi vector so that there was an absence of the ODAs resulted in a significant decline in beat frequency and an inability of cilia to coordinate their oscillations and form metachronal waves. Specifically, similar manipulation to eliminate LC1 yields a similar phenotype even though, in contrast to other models, in this case the outer arms still assemble in the axoneme. The lack of metachrony was not due to a decrease in ciliary-beat frequency or to temporal variability in the beat cycle of impaired cilia. Therefore, it was suggested that LC1 acts in a mechanosensory feedback mechanism controlling ODA activity, and thus the ciliary beat, based on external conformational cues. This enables the coordination of ciliary beating and ultimately the formation of metachronal waves.36 These data are consistent with observations of a Chlamydomonas mutant (oda-2t), which lacks the outer arm heavy chain motor domain and LC1 and exhibits pronounced motility defects.37 Interestingly, in Chlamydomonas reinhardtii, a series of mutations affecting the amino acids that are crucial for the structure of the LC1α9 helix, which protrudes into the HC AAA+ domains and is important for the interaction between LC1 and heavy chain, demonstrated a dominant-negative adverse effect. The swimming velocity was reduced, and the flagella beat constantly out of phase and stalled near the power/recovery stroke switch point.32 These findings again support the suggestion that LC1 regulates the activity outer-arm dyneins through a conformational switch. Although the DNAL1 mutation replacing Asn150 with Ser causes instability of the mutated protein, it does not lead to its complete absence. Similarly, it does not completely abolish the binding to either dynein heavy chain or to tubulin. Thus, it is still possible that the Asn150Ser mutation leads to a partial loss of protein function and has a semidominant mode of transmission of the disease phenotype, that is, a typical PCD phenotype in homozygous individuals, whereas heterozygous carriers could have a much less severe phenotype. Although, we were not able to obtain nasal epithelial cells from a heterozygote, and thus cannot exclude a defect in the structure or movement of the cilia, there is no support for a dominant effect on the basis of clinical data; None of the parents or the hetrozygous siblings suffered from any of the symptoms that affected the of PCD patients (e.g., reactive airway disease, chronic nasal discharge, chronic serous otitis media, situs inversus). Moreover, there were no reports of any infertility problems, the families having numerous children.
To date, the structure of LC1 of C. reinhardtii and its interactions with the γ heavy chain have been resolved.28,31,38 The human (DNAL1) and murine (Dnal1) orthologs of the Chlamydomonas LC1 gene were identified and in silico protein analysis showed complete conservation of the LC1/γ heavy chain-binding motif in DNAL1 and actual binding of DNAL1 to DNAH5,29 one of the two human orthologs of the Chlamydomonas γ heavy chain.30 LC1 binds to the γ heavy chain motor domain and becomes crosslinked to a relatively small region between the P1 ATP hydrolytic site and the P2 or P3 nucleotide-binding site.31,38 LC1 is a member of the LRR subclass defined by SDS22+ (which contains 22-residue repeats).This LRR motif structure consists of a β strand and a α helix interconnected via a tight turn. The turn is stabilized by a hydrogen-bonding network set up by the invariant Asn residue. The conserved hydrophobic and Asn side chains within the LRR consensus (LxxLxLxxNxL) pack to form the hydrophobic core. A single hydrophobic patch formed by the six central LRRs is exposed on the molecular surface of LC1 and is presumed to mediate interaction with the γ heavy chain. The C-terminal helical domain juts out from the cylinder, is adjacent to the hydrophobic surface within the repeat region that interacts with the dynein heavy chain, and is suggested to insert itself into the dynein ATP hydrolytic site.38 Attachment of LC1 to this region represents the only known example of an accessory polypeptide directly associated with the catalytic motor domain of the dynein heavy chain. The Asn at position 150 in DNAL1 aligns with the critical Asn that is in the last LRR consensus of LC1 and that stabilizes the turn between the β strand and the α helix by the side chain that forms hydrogen bonds in the loop structure. Additionally, this Asn is immediately adjacent to the C-terminal helical domain that inserts itself into the ATP hydrolytic site. Thus, the prediction that the mutation of this Asn to Ser will eliminate the fold agrees with the finding of the drastic reduction in the binding of the mutated DNAL1 to dynein relative to the normal protein because the replacement of the Asn's with Ser probably changes the pattern of hydrogen bonds in the loop.
The structural domain mediating the binding between LC1 of C. reinhardtii and tubulin is not yet defined. The missense mutations of amino acids crucial for the structure of the LC1 α9 helix, which is important for the interaction of LC1 and γ heavy chain, do not affect the binding to tubulin.32 The drastic reduction in binding that we observed with the p.Asn150Ser mutation might indicate the importance of this residue, and possibly the whole last LRR domain, to the interaction of DNAL1 with tubulin. It is possible that the loss of tethering of the dynein heavy chain to tubulin contributes to the recessive mode of inheritance of the PCD in our families, in contrast to the dominant negative effect observed for the missense mutations of LC1 of C. reinhardtii.32
Our study also demonstrates the importance of the Asn at position 150 for protein stability, in agreement with the proposals of the importance of the proper folding of the LRR domain for protein stability.39 Similarly, mutations that replace the critical Asn in LRR with Ser were demonstrated to cause several genetic diseases,40 such as congenital stationary night blindness type 1 (CSNB1)/X-linked congenital stationary night blindness (XLCSNB), which is caused by p.Asn312Ser in nyctalopin. Another example was observed in the ninth LRR domain of keratocan, causing autosomal recessive cornea plana (CNA2) in which the forward convex structure is flattened, leading to a decrease in refraction. PCD was also demonstrated to be caused by a mutation (p.Leu175Arg) disrupting a SDS22-like subfamily LRR structure of LRRC50.18 This LRR might participate in the structural link between the inner- and outer-row dyneins during their preassembly.18,19
In summary, although the importance of the human (DNAL1) ortholog of the Chlamydomonas LC1 gene was already appreciated, and mutations in this gene had been searched for in 86 PCD patients,29 we present evidence that a mutation of a critical amino acid in DNAL1 causes PCD. This mutation could help to elucidate the interaction between the DNAL1 to dynein heavy chain and to tubulin. Our finding adds DNAL1 as a gene mutated in PCD patients and an important component of our understanding of the types of mutations that cause PCD.
Acknowledgments
We are grateful to the patients and their families for their participation in this study. We are grateful to Noah Isakov, Ben Gurion University, for the generous gift of Myc antibodies. We thank Yona Lichtenfeld and Einat Nativ-Roth for the transmission electron microscope analyses. This work was supported by a grant from the Israel Science Foundation (to R.P.). V.C.S. is an investigator of the Howard Hughes Medical Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ciliaproteome, http://v3.ciliaproteome.org/cgi-bin/index.php
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Swiss Model tool, http://swissmodel.expasy.org/
References
- 1.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 2.Basu B., Brueckner M. Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr. Top. Dev. Biol. 2008;85:151–174. doi: 10.1016/S0070-2153(08)00806-5. [DOI] [PubMed] [Google Scholar]
- 3.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M. Primary ciliary dyskinesia: Current state of the art. Arch. Dis. Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rott H.D. Kartagener's syndrome and the syndrome of immotile cilia. Hum. Genet. 1979;46:249–261. doi: 10.1007/BF00273308. [DOI] [PubMed] [Google Scholar]
- 5.Jeganathan D., Chodhari R., Meeks M., Faeroe O., Smyth D., Nielsen K., Amirav I., Luder A.S., Bisgaard H., Gardiner R.M. Loci for primary ciliary dyskinesia map to chromosome 16p12.1-12.2 and 15q13.1-15.1 in Faroe Islands and Israeli Druze genetic isolates. J. Med. Genet. 2004;41:233–240. doi: 10.1136/jmg.2003.014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Callaghan C. Innate pulmonary immunity: Cilia. Pediatr. Pulmonol. Suppl. 2004;26:72–73. doi: 10.1002/ppul.70057. [DOI] [PubMed] [Google Scholar]
- 7.Kosaki K., Ikeda K., Miyakoshi K., Ueno M., Kosaki R., Takahashi D., Tanaka M., Torikata C., Yoshimura Y., Takahashi T. Absent inner dynein arms in a fetus with familial hydrocephalus-situs abnormality. Am. J. Med. Genet. A. 2004;129A:308–311. doi: 10.1002/ajmg.a.30177. [DOI] [PubMed] [Google Scholar]
- 8.Ibañez-Tallon I., Pagenstecher A., Fliegauf M., Olbrich H., Kispert A., Ketelsen U.P., North A., Heintz N., Omran H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 2004;13:2133–2141. doi: 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- 9.Storm van's Gravesande K., Omran H. Primary ciliary dyskinesia: Clinical presentation, diagnosis and genetics. Ann. Med. 2005;37:439–449. doi: 10.1080/07853890510011985. [DOI] [PubMed] [Google Scholar]
- 10.Baker K., Beales P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet.C Semin Med Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 11.Meeks M., Bush A. Primary ciliary dyskinesia (PCD) Pediatr. Pulmonol. 2000;29:307–316. doi: 10.1002/(sici)1099-0496(200004)29:4<307::aid-ppul11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 12.MacCormick J., Robb I., Kovesi T., Carpenter B. Optimal biopsy techniques in the diagnosis of primary ciliary dyskinesia. J. Otolaryngol. 2002;31:13–17. doi: 10.2310/7070.2002.19153. [DOI] [PubMed] [Google Scholar]
- 13.Wodehouse T., Kharitonov S.A., Mackay I.S., Barnes P.J., Wilson R., Cole P.J. Nasal nitric oxide measurements for the screening of primary ciliary dyskinesia. Eur. Respir. J. 2003;21:43–47. doi: 10.1183/09031936.03.00305503. [DOI] [PubMed] [Google Scholar]
- 14.Hornef N., Olbrich H., Horvath J., Zariwala M.A., Fliegauf M., Loges N.T., Wildhaber J., Noone P.G., Kennedy M., Antonarakis S.E. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 2006;174:120–126. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zariwala M.A., Leigh M.W., Ceppa F., Kennedy M.P., Noone P.G., Carson J.L., Hazucha M.J., Lori A., Horvath J., Olbrich H. Mutations of DNAI1 in primary ciliary dyskinesia: Evidence of founder effect in a common mutation. Am. J. Respir. Crit. Care Med. 2006;174:858–866. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Failly M., Saitta A., Muñoz A., Falconnet E., Rossier C., Santamaria F., de Santi M.M., Lazor R., DeLozier-Blanchet C.D., Bartoloni L., Blouin J.L. DNAI1 mutations explain only 2% of primary ciliary dykinesia. Respiration. 2008;76:198–204. doi: 10.1159/000128567. [DOI] [PubMed] [Google Scholar]
- 17.Barbato A., Frischer T., Kuehni C.E., Snijders D., Azevedo I., Baktai G., Bartoloni L., Eber E., Escribano A., Haarman E. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 2009;34:1264–1276. doi: 10.1183/09031936.00176608. [DOI] [PubMed] [Google Scholar]
- 18.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O'Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Rooijen E., Giles R.H., Voest E.E., van Rooijen C., Schulte-Merker S., van Eeden F.J. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J. Am. Soc. Nephrol. 2008;19:1128–1138. doi: 10.1681/ASN.2007080917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parvari R., Hershkovitz E., Kanis A., Gorodischer R., Shalitin S., Sheffield V.C., Carmi R. Homozygosity and linkage-disequilibrium mapping of the syndrome of congenital hypoparathyroidism, growth and mental retardation, and dysmorphism to a 1-cM interval on chromosome 1q42-43. Am. J. Hum. Genet. 1998;63:163–169. doi: 10.1086/301915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amir A.D., Bartal O., Morad E., Nagar T., Sheynin J., Parvari R., Chalifa-Caspi V. KinSNP software for homozygosity mapping of disease genes using SNP microarrays. Hum. Genomics. 2010;4:394–401. doi: 10.1186/1479-7364-4-6-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy-Litan V., Hershkovitz E., Avizov L., Leventhal N., Bercovich D., Chalifa-Caspi V., Manor E., Buriakovsky S., Hadad Y., Goding J., Parvari R. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am. J. Hum. Genet. 2010;86:273–278. doi: 10.1016/j.ajhg.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu H., Maciejewski M.W., Marintchev A., Benashski S.E., Mullen G.P., King S.M. Solution structure of a dynein motor domain associated light chain. Nat. Struct. Biol. 2000;7:575–579. doi: 10.1038/76804. [DOI] [PubMed] [Google Scholar]
- 29.Horváth J., Fliegauf M., Olbrich H., Kispert A., King S.M., Mitchison H., Zariwala M.A., Knowles M.R., Sudbrak R., Fekete G. Identification and analysis of axonemal dynein light chain 1 in primary ciliary dyskinesia patients. Am. J. Respir. Cell Mol. Biol. 2005;33:41–47. doi: 10.1165/rcmb.2004-0335OC. [DOI] [PubMed] [Google Scholar]
- 30.Pazour G.J., Agrin N., Walker B.L., Witman G.B. Identification of predicted human outer dynein arm genes: Candidates for primary ciliary dyskinesia genes. J. Med. Genet. 2006;43:62–73. doi: 10.1136/jmg.2005.033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benashski S.E., Patel-King R.S., King S.M. Light chain 1 from the Chlamydomonas outer dynein arm is a leucine-rich repeat protein associated with the motor domain of the gamma heavy chain. Biochemistry. 1999;38:7253–7264. doi: 10.1021/bi990466y. [DOI] [PubMed] [Google Scholar]
- 32.Patel-King R.S., King S.M. An outer arm dynein light chain acts in a conformational switch for flagellar motility. J. Cell Biol. 2009;186:283–295. doi: 10.1083/jcb.200905083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hastie A.T., Dicker D.T., Hingley S.T., Kueppers F., Higgins M.L., Weinbaum G. Isolation of cilia from porcine tracheal epithelium and extraction of dynein arms. Cell Motil. Cytoskeleton. 1986;6:25–34. doi: 10.1002/cm.970060105. [DOI] [PubMed] [Google Scholar]
- 34.Baron D.M., Kabututu Z.P., Hill K.L. Stuck in reverse: Loss of LC1 in Trypanosoma brucei disrupts outer dynein arms and leads to reverse flagellar beat and backward movement. J. Cell Sci. 2007;120:1513–1520. doi: 10.1242/jcs.004846. [DOI] [PubMed] [Google Scholar]
- 35.Narayan R.D., Blackman L.M., Shan W., Hardham A.R. Phytophthora nicotianae transformants lacking dynein light chain 1 produce non-flagellate zoospores. Fungal Genet. Biol. 2010;47:663–671. doi: 10.1016/j.fgb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Rompolas P., Patel-King R.S., King S.M. An outer arm Dynein conformational switch is required for metachronal synchrony of motile cilia in planaria. Mol. Biol. Cell. 2010;21:3669–3679. doi: 10.1091/mbc.E10-04-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z., Takazaki H., Nakazawa Y., Sakato M., Yagi T., Yasunaga T., King S.M., Kamiya R. Partially functional outer-arm dynein in a novel Chlamydomonas mutant expressing a truncated γ heavy chain. Eukaryot. Cell. 2008;7:1136–1145. doi: 10.1128/EC.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H., Blackledge M., Maciejewski M.W., Mullen G.P., King S.M. Relaxation-based structure refinement and backbone molecular dynamics of the dynein motor domain-associated light chain. Biochemistry. 2003;42:57–71. doi: 10.1021/bi026762j. [DOI] [PubMed] [Google Scholar]
- 39.Bella J., Hindle K.L., McEwan P.A., Lovell S.C. The leucine-rich repeat structure. Cell. Mol. Life Sci. 2008;65:2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsushima N., Tachi N., Kuroki Y., Enkhbayar P., Osaki M., Kamiya M., Kretsinger R.H. Structural analysis of leucine-rich-repeat variants in proteins associated with human diseases. Cell. Mol. Life Sci. 2005;62:2771–2791. doi: 10.1007/s00018-005-5187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.