

Abstract
Accelerated aging syndromes represent a valuable source of information about the molecular mechanisms involved in normal aging. Here, we describe a progeroid syndrome that partially phenocopies Hutchinson-Gilford progeria syndrome (HGPS) but also exhibits distinctive features, including the absence of cardiovascular deficiencies characteristic of HGPS, the lack of mutations in LMNA and ZMPSTE24, and a relatively long lifespan of affected individuals. Exome sequencing and molecular analysis in two unrelated families allowed us to identify a homozygous mutation in BANF1 (c.34G>A [p.Ala12Thr]), encoding barrier-to-autointegration factor 1 (BAF), as the molecular abnormality responsible for this Mendelian disorder. Functional analysis showed that fibroblasts from both patients have a dramatic reduction in BAF protein levels, indicating that the p.Ala12Thr mutation impairs protein stability. Furthermore, progeroid fibroblasts display profound abnormalities in the nuclear lamina, including blebs and abnormal distribution of emerin, an interaction partner of BAF. These nuclear abnormalities are rescued by ectopic expression of wild-type BANF1, providing evidence for the causal role of this mutation. These data demonstrate the utility of exome sequencing for identifying the cause of rare Mendelian disorders and underscore the importance of nuclear envelope alterations in human aging.
Main Text
Aging is a very complex process that affects most biological functions of the organism, but its molecular basis remain largely unknown.1 Over the last few years, our knowledge of the molecular mechanisms underlying human aging has gained new insights from studies on premature aging syndromes that cause the early development of multiple phenotypes normally associated with advanced age.2,3 Most of these human progeroid syndromes are caused by defects in DNA repair systems, but recent studies have shown that alterations in nuclear envelope formation and dynamics are involved in the development of accelerated aging syndromes.4 Thus, patients with Hutchinson-Gilford progeria syndrome (HGPS [MIM 176670]) carry mutations in LMNA (MIM 150330), which encodes two major components of the nuclear envelope, the lamins A and C.5,6 In addition, other progeroid syndromes, such as restrictive dermopathy (RD [MIM 275210]) and mandibuloacral dysplasia (MADB [MIM 608612]), are caused by mutations in ZMPSTE24 (also known as FACE1 [MIM 606480]),7,8 which encodes a metalloprotease involved in prelamin A maturation.9 However, there may still be other classes of genetic mutations responsible for the development of accelerated aging in patients who lack mutations in all previously described genes associated with these devastating diseases.
The recent availability of high-throughput sequencing technologies has now made it feasible to address personal genome projects that could uncover the precise genetic causes of human diseases.10,11 In addition to whole-genome sequencing, exome sequencing has been successfully used to identify mutations responsible for genetic disorders of unknown cause.12–15 In this work, we have used this approach to identify the disease-causing mutations in patients who were originally diagnosed with a progeroid syndrome that phenocopies features of HGPS and mandibuloacral dysplasia but whose mutational analysis of candidate genes did not reveal any change in LMNA or ZMPSTE24.
To gain insights into the molecular mechanisms implicated in putative accelerated aging, we studied patients with progeroid syndromes without mutations in known candidate genes. Affected individuals are members of two unrelated Spanish families, from distant regions of the country (family A is from Gran Canaria; family B is from Castilla). DNA samples were isolated from peripheral-blood leukocytes via standard techniques. The experiments were conducted in accordance with the guidelines of the Comité Científico de la Fundación Centro Médico de Asturias, and written informed consent was obtained from each individual providing biological samples. The pedigrees for both families are shown in Figure 1. The first patient studied (II-1, family A; Figure 1) was the second child born to consanguineous third cousins (coefficient of inbreeding, F = 1/64). Both parents and his four siblings were healthy, and there was no significant clinical family history. The patient exhibited normal development until 2 years of age. From that age, he experienced failure to thrive and his skin became dry and atrophic with small light-brown spots over the thorax, scalp, and limbs. He also developed a generalized lipoatrophy, severe osteoporosis, and marked osteolysis. The atrophic facial subcutaneous fat pad and the marked osteolysis of the maxilla and mandible result in a typical pseudosenile facial appearance with micrognatia, prominent subcutaneous venous patterning, a convex nasal ridge, and proptosis (Figure 1A and Figure S1 available online). His cognitive development was completely normal. Despite an initial diagnosis of HGPS or MAD, some of the clinical features suggested that this patient could have a distinct progeroid syndrome. The age of the patient (31 years, which is very unusual given that most HGPS patients die in their teenage years), his height (145 cm, underestimated as a result of his scoliosis), the presence of eyebrows and eyelashes, the persistence of scalp hair until the age of 12 (which never disappeared completely), the very severe osteolysis (of the mandible, clavicles, ribs, distal phalanges, and radius), and the absence of coronary dysfunction, atherosclerosis, or metabolic anomalies finally led to the diagnosis of atypical progeria. A second patient (II-1, family B; Figure 1), a 24-year-old man showing a phenotype almost identical to the index case (Figure 1A and Figure S2), was also analyzed. Despite thorough cardiovascular examination (echocardiogram, stress test, cardiac computed tomography, Doppler study of supra-aortic trunks), neither of the patients showed signs of ischemia or atherosclerosis, both cardinal features of HGPS.16 Moreover, the lack of mutations in LMNA and ZMPSTE24 would also be consistent with the hypothesis that these patients have a different progeroid syndrome.
Figure 1.
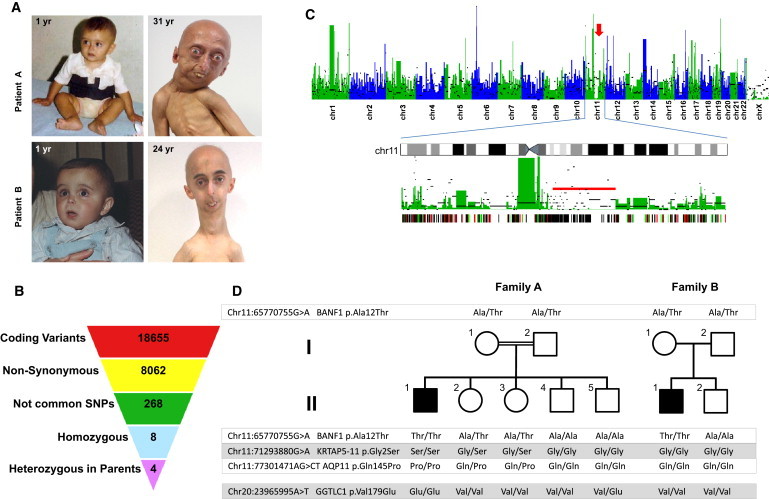
Identification of a Mutation in BANF1 by Exome Sequencing in a Patient with Atypical Progeria
(A) The appearance of both patients included in the study at 1 year of age (left), evidence of progeroid features in patient A at 31 years of age (top), and evidence of such features in patient B at 24 years (bottom). Clinical characteristics include facial abnormalities due to severe bone changes, small chin, convex nasal ridge, and prominent eyes. The presence of eyebrows and eyelashes are characteristics of atypical progeria.
(B) Scheme showing the filtering procedure used for identifying candidate genes in study of this progeroid syndrome, assuming a recessive inheritance model. Coding variants were filtered by retaining only those causing amino acid substitution. Common polymorphisms present in either dbSNP131 or in ten unrelated individual genomes were excluded. Homozygous variants that were present in heterozygosity in both parents were finally selected.
(C) Manhattan plot showing the density of heterozygous variants obtained from exome sequencing data in 50 Kb nonoverlapping windows of coding-sequence. The density of homozygous variants per window is indicated by black bars. An arrow indicates the presence of a homozygosity track on chromosome 11. Below is a detailed view of chromosome 11, showing a long stretch of homozygosity (red bar), and a plot showing mutations present in the patient and the mother (red lines), in the patient and the father (green lines), and in the patient and both parents (black lines).
(D) Pedigrees and results from sequencing of the four candidate gene variants in both families.
To evaluate this possibility at the molecular level, we first performed exon enrichment, followed by massively parallel sequencing on DNA samples from the proband (II-1, family A; Figure 1) and both parents. Three micrograms of genomic DNA was fragmented and hybridized with the use a SureSelect Human All Exon Kit (Agilent, Palo Alto, CA) together with the Paired-End Sample Preparation Kit from Illumina in accordance with the manufacturers' protocols. The captured DNA fragments were sequenced with the Genome Analyzer IIx (Illumina, San Diego, CA), with the use of two lanes per sample and 52 cycles, resulting in more than 60 million paired reads per sample. Reads were aligned to the reference genome (GRCh37) with the Burrows-Wheeler Aligner (BWA 0.5.7),17 and SAMtools 0.1.718 was used for removal of PCR duplicates and initial SNP calling. All single-nucleotide variants were required to have a minimum SNP quality of 40, supported by reads in both orientations, and to be no fewer than three bases from an indel. Common variants present in either dbSNP131 or in ten personal genomes of Spanish origin were filtered. Homozygous variants were identified with the use of custom scripts and verified by visual inspection. More than 98% of the coding exome was covered by at least one read in the three individuals, and more than 90% was covered by at least ten reads. Although these extremely rare diseases can be caused by dominant de novo mutations, we first assumed an autosomal-recessive mode of inheritance because of the consanguinity of healthy parents. We first searched for homozygous variants and applied a model of identity by descent (IBD). Out of the 18,655 coding variants found in the proband, 8062 produced nonsynonymous changes, from which 96% were present either in dbSNP131 or in several unrelated individuals from whom genomic data were available at our group (Figure 1B). Only four of the uncommon remaining variants were homozygous in the proband and heterozygous in the parents. Interestingly, three of the four candidate genes (BANF1 [MIM 603811], KRTAP5-11, and AQP11 [MIM 609914]) were located in a long contiguous stretch of homozygosity on chromosome 11q13 (Figure 1C), whereas the fourth one (GGTLC1 [MIM 612338]) was located on chromosome 20p11. SNP array analysis revealed that the first region contained two copies in the proband, precluding the occurrence of a large deletion in this patient. In addition, all nucleotide variants detected in this region were also present in both the father and the mother, indicating that this homozygosity track was not due to uniparental disomy but was independently inherited from the parents.
The presence of three candidate genes in a small locus of less than 13 Mb would likely result in linkage disequilibrium between the three variants, thus making difficult the identification of the individual alteration responsible for this disease. Therefore, we performed PCR amplification and capillary sequencing of the four variants mentioned above in the studied family and in the second family, with a similar progeroid syndrome and without mutations in either LMNA or ZMPSTE24. We found that the affected individual from the second family (II-1, family B; Figure 1) had the same homozygous mutation (chr11:65770755G>A) in BANF1 (NM_001143985.1, c.34G>A [p.Ala12Thr]) that was originally identified in the patient from family A, although sequencing of the coding regions of AQP11, KRTAP5-11, and GGTLC1 did not reveal additional mutations (Figure 1D and Figure S3). Even though both families are of different geographical origins and they did not report any known relationship with each other, both patients shared a common homozygous haplotype comprising four SNPs in BANF1 (rs14157, G/G; rs1786171, C/C; rs1786172, G/G; rs56984820, −/−), which suggests the occurrence of a founder mutation. All nonaffected members from both families were either heterozygous or did not have the BANF1 mutation, and sequencing of more than 400 chromosomes from individuals of the Spanish population failed to detect this variant, confirming that it is not a common polymorphism. Together, these data strongly support the causal role of this mutation in BANF1 as the genetic alteration responsible for this progeroid syndrome.
BANF1 encodes a protein of 89 amino acids called barrier-to-autointegration factor 1 (BAF), which forms dimers and is implicated in nuclear envelope assembly.19 The affected residue (Ala12) has been highly conserved through evolution from fish to humans (Figure 2A), suggesting an important role in the structure or function of this small protein. BAF interacts with DNA as well as with different proteins,19–21 including lamin A, which is mutated in HGPS, reinforcing the role of BAF in the segmental progeroid syndrome exhibited by the patients described herein. A three-dimensional model of mutant BAF shows that the mutated residue is located on the surface of the protein22,23 (Figure 2B). Nevertheless, the p.Ala12Thr mutation is not predicted to affect BAF dimerization or its binding to either DNA or emerin, raising the possibility that this amino acid substitution could impair the interaction with other proteins, its subcellular localization, or its stability. For examination of the effect of the BAF p.Ala12Thr mutation at the protein level, a skin biopsy was obtained from both patients and from a parent (I-1, family A) and used to establish cell cultures of primary dermal fibroblasts. Control cells (AG10803 human skin fibroblasts) were obtained from the Coriell Cell Repository. We analyzed BAF expression in primary fibroblasts from both patients (homozygous for the mutation), from a heterozygous carrier (I-1, family A), and from control fibroblasts, using immunoblotting with mouse monoclonal anti-BAF (ab88464, Abcam), which recognizes two bands corresponding to phosphorylated (slower migrating) and nonphosphorylated (faster migrating) BAF.24 Fibroblasts homozygous for the p.Ala12Thr mutation had very low levels of BAF protein as compared to control fibroblasts, whereas heterozygous fibroblasts had intermediate levels (Figure 2C). Quantitative RT-PCR experiments using a specific TaqMan expression assay for BANF1 (Applied Biosystems, Foster City, CA) revealed that BANF1 mRNA levels were not significantly downregulated in the mutant cells, indicating that this mutation affects the stability of the protein rather than having an effect at the mRNA level (Figure 2D).
Figure 2.
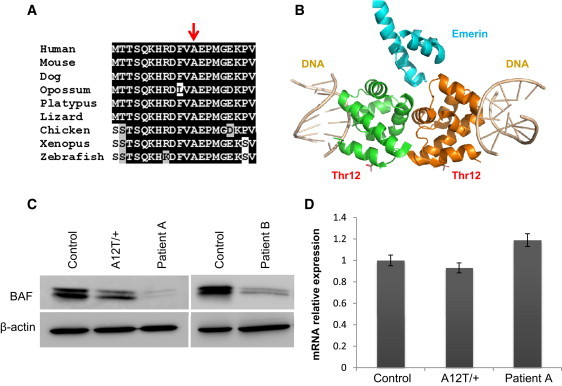
Alteration of BAF Structure in Cells Homozygous for the p.Ala12Thr Mutation
(A) Sequence alignment of the first 20 residues of human BAF to its orthologs from the indicated species, showing the evolutionary conservation of the Ala12 residue (indicated with an arrow).
(B) Three-dimensional model of a BAF dimer (green and orange in the figure) in complex with emerin (blue) and DNA (gray). To generate the model, the structure of the BAF dimer-emerin complex (PDB 2ODG) was aligned with the structure of a BAF-DNA complex (PDB 2BZF) with the use of PyMOL. The position of the Thr residue in mutant BAF is indicated.
(C) Immunoblot analysis of BAF in control human fibroblasts along with fibroblasts from patients homozygous for the p.Ala12Thr mutation and from the mother (heterozygous carrier) of the family A patient. The immunoblots shown are representative of three independent experiments.
(D) BANF1 mRNA relative levels in fibroblasts homozygous or heterozygous for the p.Ala12Thr mutation, determined by qRT-PCR. Values correspond to the mean of triplicates. Error bars represent standard deviation of the mean.
A morphological analysis of the nuclei from mutant fibroblasts revealed profound nuclear abnormalities, including blebs and aberrations previously described in other laminopathies5–8,25,26 (Figure 3). Immunofluorescence analysis using antibodies against lamin A/C and emerin, interaction partners of BAF and structural constituents of the nuclear lamina,27,28 revealed significant differences in the subcellular distribution of emerin between control cells and fibroblasts from these patients (Figure 3), whereas alterations in lamin A/C distribution were not evident under the same conditions (Figure S4). Emerin was specifically located in the nuclear lamina of normal fibroblasts, whereas in fibroblasts homozygous for the p.Ala12Thr mutation, emerin lost its nuclear distribution and was found predominantly in the cytoplasm, with some minor staining in the nuclear lamina.
Figure 3.
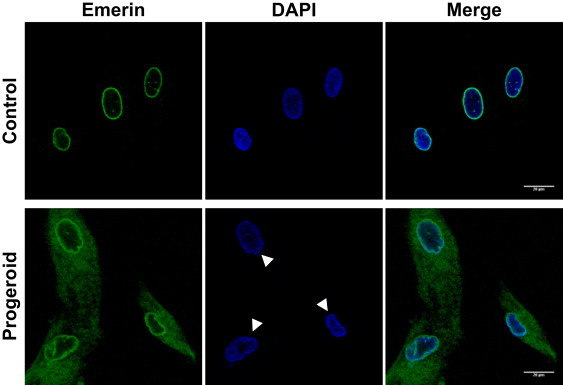
Nuclear Envelope Alterations in Fibroblasts Homozygous for the p.Ala12Thr Mutation in BAF
Emerin distribution was analyzed by immunofluorescence with the use of an anti-emerin rabbit polyclonal antibody (ab14208, Abcam) and by confocal microscopy (Leica SP2) in primary fibroblasts from the progeroid patient II-1 (family A) and in control fibroblasts. Nuclei with structural abnormalities can be observed in progeroid fibroblasts.
To further confirm the causal role of the BAF p.Ala12Thr mutation in this process, we generated an EGFP-BAF protein by cloning the coding sequence of human BAF in the polylinker region of pEGFP-C1 (Clontech, Palo Alto, CA). We transiently transfected mutant fibroblasts with this expression vector encoding an EGFP-BAF fusion protein, and confocal microscopy analysis revealed that ectopic expression of EGFP-BAF in these progeroid fibroblasts rescued the nuclear abnormalities. Thus, EGFP-BAF-positive cells recovered a normal nuclear morphology, in contrast to the aberrations observed in control cells transfected with EGFP (Figure 4). These findings are consistent with the reported relevance of BAF in nuclear lamina dynamics.29,30 In fact, experimental reduction of BAF by RNAi in human cells results in the formation of nuclear envelope alterations and abnormal localization of emerin,29 similar to the findings observed in these progeroid patients. Together, these results show that the p.Ala12Thr mutation in BAF identified in patients with this progeroid syndrome causes an abnormal distribution of components of the nuclear lamina, which are likely responsible for the nuclear abnormalities observed in this laminopathy. The phenotypic expression of laminopathies is highly variable, and although our two patients have features that are also exhibited in HGPS and mandibuloacral dysplasia, their clinical findings differ in several aspects. Neither of our patients has signs of atherosclerosis or cardiac ischemia (fatal heart attacks and strokes at a mean age of 13 years are a common feature of HGPS). Additionally, our patients do not have insulin resistance, diabetes mellitus, or hypertriglyceridemia, all of which are usual features of mandibuloacral dysplasia. Finally, it is remarkable that despite the phenotypic overlapping between patients with HGPS or mandibuloacral dysplasia and those with the segmental progeria described herein, the observed differences are of utmost importance to patients and their families. Thus, it seems that those patients with the progeroid syndrome caused by BANF1 mutation are not at increased risk of acute myocardial infarcts, cerebrovascular accidents, or diabetes mellitus. In contrast, they suffer profound skeletal abnormalities that affect their quality of life (both patients experience pain, dysfunction, and disability) and occasionally may result in life-threatening complications, such as pulmonary hypertension secondary to severe scoliosis. Therefore, palliation of osseous manifestations is a priority in our patients, given their relatively long lifespan.
Figure 4.
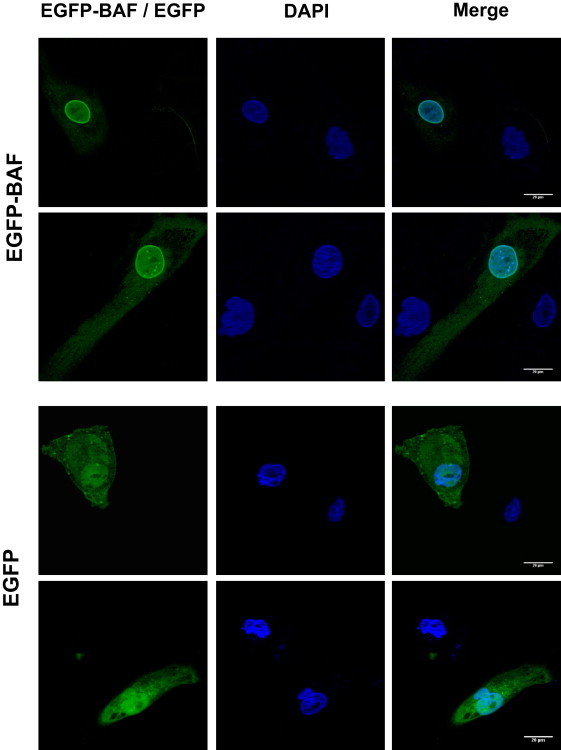
Rescue of the Nuclear Abnormalities by BANF1 Ectopic Expression
Fibroblasts from patient II-1 (family A) were transiently transfected with an expression vector encoding EGFP-BAF or EGFP alone as control and were analyzed by confocal microscopy. Untransfected progeroid cells present in the same preparation show frequent nuclear abnormalities (arrowheads). These structural alterations are rescued by BANF1 ectopic expression. Two independent representative fields are shown.
In summary, the finding of mutations in BANF1 associated with a progeroid syndrome may open therapeutic approaches for patients with this condition, as has been the case for children with HGPS (ClinicalTrials.gov; NCT00731016, NCT00425607, NCT00916747).31–33 Furthermore, the fact that BAF is functionally connected to the nuclear envelope, together with the previous finding that prelamin A isoforms are accumulated during normal and pathological aging,5,6,34–36 underscores the importance of the nuclear lamina for human aging and may provide new mechanistic insights about this complex, multifactorial, and universal process.
Acknowledgments
We thank the atypical progeria patients and their families for participating in this study, and especially N.M.O. and G.R.P. for their courage and enthusiasm. We also thank Y. Español for help with confocal microscopy; A. Ramsay and G. Velasco for helpful comments; S. Álvarez, M. Fernández, and R. Álvarez for excellent technical assistance; and the staff of the Centro Médico de Asturias for their kind assistance. This work has been supported by grants from Ministerio de Ciencia e Innovación-Spain, PCTI-FICYT Asturias, Fundación Centro Médico de Asturias, Fundación María Cristina Masaveu Peterson, and the European Union (FP7 MicroEnviMet). C.L-O. is an investigator in the Botin Foundation. The Instituto Universitario de Oncología is supported by Obra Social Cajastur and Acción Transversal del Cáncer-RTICC.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClinicalTrials.gov, http://clinicaltrials.gov
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Vijg J., Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454:1065–1071. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin G.M., Oshima J. Lessons from human progeroid syndromes. Nature. 2000;408:263–266. doi: 10.1038/35041705. [DOI] [PubMed] [Google Scholar]
- 3.Burtner C.R., Kennedy B.K. Progeria syndromes and ageing: what is the connection? Nat. Rev. Mol. Cell Biol. 2010;11:567–578. doi: 10.1038/nrm2944. [DOI] [PubMed] [Google Scholar]
- 4.Ramírez C.L., Cadiñanos J., Varela I., Freije J.M., López-Otín C. Human progeroid syndromes, aging and cancer: new genetic and epigenetic insights into old questions. Cell. Mol. Life Sci. 2007;64:155–170. doi: 10.1007/s00018-006-6349-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C.L., Munnich A., Le Merrer M., Lévy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 6.Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navarro C.L., Cadiñanos J., De Sandre-Giovannoli A., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W.J., Wagner A., Giuliano F. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005;14:1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal A.K., Fryns J.P., Auchus R.J., Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 9.Pendás A.M., Zhou Z., Cadiñanos J., Freije J.M., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodríguez F., Tryggvason K., López-Otín C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 10.Roach J.C., Glusman G., Smit A.F., Huff C.D., Hubley R., Shannon P.T., Rowen L., Pant K.P., Goodman N., Bamshad M. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lupski J.R., Reid J.G., Gonzaga-Jauregui C., Rio Deiros D., Chen D.C., Nazareth L., Bainbridge M., Dinh H., Jing C., Wheeler D.A. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker J., Semler O., Gilissen C., Li Y., Bolz H.J., Giunta C., Bergmann C., Rohrbach M., Koerber F., Zimmermann K. Exome Sequencing Identifies Truncating Mutations in Human SERPINF1 in Autosomal-Recessive Osteogenesis Imperfecta. Am. J. Hum. Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merideth M.A., Gordon L.B., Clauss S., Sachdev V., Smith A.C., Perry M.B., Brewer C.C., Zalewski C., Kim H.J., Solomon B. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margalit A., Brachner A., Gotzmann J., Foisner R., Gruenbaum Y. Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol. 2007;17:202–208. doi: 10.1016/j.tcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Montes de Oca R., Shoemaker C.J., Gucek M., Cole R.N., Wilson K.L. Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners. PLoS ONE. 2009;4:e7050. doi: 10.1371/journal.pone.0007050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segura-Totten M., Wilson K.L. BAF: roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 2004;14:261–266. doi: 10.1016/j.tcb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Cai M., Huang Y., Suh J.Y., Louis J.M., Ghirlando R., Craigie R., Clore G.M. Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J. Biol. Chem. 2007;282:14525–14535. doi: 10.1074/jbc.M700576200. [DOI] [PubMed] [Google Scholar]
- 23.Bradley C.M., Ronning D.R., Ghirlando R., Craigie R., Dyda F. Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 2005;12:935–936. doi: 10.1038/nsmb989. [DOI] [PubMed] [Google Scholar]
- 24.Nichols R.J., Wiebe M.S., Traktman P. The vaccinia-related kinases phosphorylate the N' terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol. Biol. Cell. 2006;17:2451–2464. doi: 10.1091/mbc.E05-12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Espada J., Varela I., Flores I., Ugalde A.P., Cadiñanos J., Pendás A.M., Stewart C.L., Tryggvason K., Blasco M.A., Freije J.M., López-Otín C. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol. 2008;181:27–35. doi: 10.1083/jcb.200801096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R., Collins F.S. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bengtsson L., Wilson K.L. Multiple and surprising new functions for emerin, a nuclear membrane protein. Curr. Opin. Cell Biol. 2004;16:73–79. doi: 10.1016/j.ceb.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Haraguchi T., Koujin T., Segura-Totten M., Lee K.K., Matsuoka Y., Yoneda Y., Wilson K.L., Hiraoka Y. BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 2001;114:4575–4585. doi: 10.1242/jcs.114.24.4575. [DOI] [PubMed] [Google Scholar]
- 29.Haraguchi T., Koujin T., Osakada H., Kojidani T., Mori C., Masuda H., Hiraoka Y. Nuclear localization of barrier-to-autointegration factor is correlated with progression of S phase in human cells. J. Cell Sci. 2007;120:1967–1977. doi: 10.1242/jcs.03461. [DOI] [PubMed] [Google Scholar]
- 30.Haraguchi T., Kojidani T., Koujin T., Shimi T., Osakada H., Mori C., Yamamoto A., Hiraoka Y. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J. Cell Sci. 2008;121:2540–2554. doi: 10.1242/jcs.033597. [DOI] [PubMed] [Google Scholar]
- 31.Capell B.C., Olive M., Erdos M.R., Cao K., Faddah D.A., Tavarez U.L., Conneely K.N., Qu X., San H., Ganesh S.K. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl. Acad. Sci. USA. 2008;105:15902–15907. doi: 10.1073/pnas.0807840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varela I., Pereira S., Ugalde A.P., Navarro C.L., Suárez M.F., Cau P., Cadiñanos J., Osorio F.G., Foray N., Cobo J. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008;14:767–772. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- 33.Worman H.J., Fong L.G., Muchir A., Young S.G. Laminopathies and the long strange trip from basic cell biology to therapy. J. Clin. Invest. 2009;119:1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dechat T., Pfleghaar K., Sengupta K., Shimi T., Shumaker D.K., Solimando L., Goldman R.D. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scaffidi P., Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. [Google Scholar]
- 36.Varela I., Cadiñanos J., Pendás A.M., Gutiérrez-Fernández A., Folgueras A.R., Sánchez L.M., Zhou Z., Rodríguez F.J., Stewart C.L., Vega J.A. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.