

Summary
An LC-MS method based on the use of high resolution Fourier transform ion cyclotron resonance mass spectrometry (FTIRCMS) for profiling oligonucleotides synthesis impurities is described.
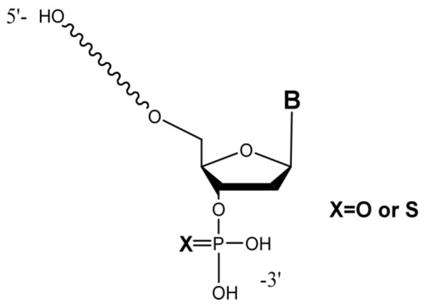
Oligonucleotide phosphorothioatediesters (phosphorothioate oligonucleotides), in which one of the non-bridging oxygen atoms at each phosphorus center is replaced by a sulfur atom, are now one of the most popular oligonucleotide modifications due to their ease of chemical synthesis and advantageous pharmacokinetic properties. Despite significant progress in the solid-phase oligomerization chemistry used in the manufacturing of these oligonucleotides, multiple classes of low-level impurities always accompany synthetic oligonucleotides. Liquid chromatography-mass spectrometry has emerged as a powerful technique for the identification of these synthesis impurities. However, impurity profiling, where the entire complement of low-level synthetic impurities is identified in a single analysis, is more challenging. Here we present an LC-MS method based the use of high resolution-mass spectrometry, specifically Fourier transform ion cyclotron resonance mass spectrometry (FTIRCMS or FTMS). The optimal LC-FTMS conditions, including the stationary phase and mobile phases for the separation and identification of phosphorothioate oligonucleotides, were found. The characteristics of FTMS enable charge state determination from single m/z values of low-level impurities. Charge state information then enables more accurate modeling of the detected isotopic distribution for identification of the chemical composition of the detected impurity. Using this approach, a number of phosphorothioate impurities can be detected by LC-FTMS including failure sequences carrying 3′-terminal phosphate monoester and 3′-terminal phosphorothioate monoester, incomplete backbone sulfurization and desulfurization products, high molecular weight impurities, and chloral, isobutyryl, and N3 (2-cyanoethyl) adducts of the full length product. When compared with low resolution LC-MS, ~60% more impurities can be identified when charge state and isotopic distribution information is available and used for impurity profiling.
Keywords: Synthetic oligonucleotides, phosphorothioatediesters, high resolution mass spectrometry, LC-FTMS, accurate mass measurement
INTRODUCTION
During the past decade there has been increased application of oligonucleotides (OGN) in the biotechnology market. Oligonucleotides are used as diagnostic tools and as a drug development platform allowing for the modulation of gene expression. Unmodified oligodeoxyribonucleotides and oligoribonucleotides typically have low specificity and therapeutic efficacy in vivo due to nuclease-mediated degradation. Those problems can be overcome by chemical alteration of their native structure via modifications of the phosphodiester backbone, ribose sugar and the heterocyclic bases [1].
Oligonucleotide phosphorothioatediesters (PS-OGN or phosphorothioate oligonucleotides), in which one of the non-bridging oxygen atoms at each phosphorus center is replaced by a sulfur atom, are now one of the most popular oligonucleotide modifications due to their ease of chemical synthesis and their advantageous pharmacokinetic properties. Various modifications of phosphorothioate oligonucleotides are commonly used as active pharmaceutical ingredients. The presence of sulfur in the oligomer increases its in vivo stability while retaining its charge and solubility. Continuous advancements of preclinical and clinical programs – approximately 70 oligonucleotide-based therapeutics are in clinical trials worldwide – have led to increased demand for large quantities of synthetic oligonucleotides [2].
Despite significant progress in solid-phase oligomerization chemistry, synthetic oligonucleotides still contain multiple classes of low-level impurities. These impurities, when present in oligonucleotide therapeutics, can influence not only their efficacy and toxicity but also lead to potential regulatory issues due to potential changes in the impurity profiles. Phosphorothioate oligonucleotides are more difficult to analyze than the corresponding phosphorodiester oligonucleotides because they carry diastereoisomers at each phosphorus center [3]. These diastereomers (2(n−1), where n is the length of the oligonucleotides) lead to increased sample complexity that cannot be fully resolved by existing analytical techniques [4, 5].
Impurity profiles can be used to better understand solid-phase oligomerization chemistry, yielding better control over manufacturing process. Due to the analytical challenges created by the physicochemical properties of the impurities arising from oligonucleotide synthesis, multiple techniques have been employed for the characterization of purified oligonucleotides including capillary gel electrophoresis-based methods (CGE-UV/fluorescence) [6], conventional liquid chromatography methods (HPLC-UV), and anion exchange (AEC) [7] and ion pairing (IP) chromatography.
While such techniques have been useful for characterizing purified oligonucleotides, mass spectrometry (MS) in combination with HPLC has become a powerful tool for the analysis of synthetic oligonucleotides and the impurities created during their synthesis [8–16]. LC-MS has been used to identify several classes of impurities in phosphorothioate oligonucleotide synthesis including failure sequences, polymerization products, side products resulting from inefficient sulfurization, depurination, deamination, n−1 terminal thio-monophosphates, adducts of acrylonitrile, and chloral adducts [15–20].
Prior LC-MS studies have addressed single impurity classes or have required enzymatic or acid hydrolysis to verify sequence information for the synthesis impurity. An alternative approach, based on accurate mass measurements and isotope pattern profiling, has been used for LC-MS screening of drug compounds and their metabolites [21, 22]. Because the analytical platform is similar and impurity profiling is conceptually related to metabolite screening, we sought to examine whether this approach could reveal equivalent or greater information on impurity profiles without requiring additional analysis of hydrolysis products.
Here we utilized ion pairing-reversed phase (IP-RP) HPLC with Fourier Transform Mass Spectrometry (FTMS) to identify the components of crude synthetic phosphorothioate oligonucleotides. LC-FTMS has been used previously for screening various mixtures [23–25], and its advantages of high resolving power and mass measurement accuracy are proposed to allow for impurity profiling of phosphorothioate oligonucleotide synthesis without the need for enzymatic hydrolysis or acid hydrolysis. In addition, we compare LC-MS generated impurity profiles for a phosphorothioate oligonucleotide using the high performance FTMS and a linear ion trap mass spectrometer.
EXPERIMENTAL
Materials
1,1,1,3,3,3-hexafluoroisopropanol (HFIP) was from Sigma (St. Louis, MO), water and methanol were from Burdick & Jackson (Muskegon, MI), and triethylamine (TEA) was from Fisher Scientific (Pittsburgh, PA). The unpurified crude 24-mer phosphorothioate oligonucleotide, 5′-TCG TCG TTT TGT CGT TTT GTC GTT-3′, was provided by Girindus America Inc. (Cincinnati, OH). The sample was evaporated to dryness and then re-suspended in autoclaved nanopure water for use. Polythymidilic acids (dT10, dT15 and dT20) used for instrument tuning and calibration were obtained from the University of Cincinnati DNA Core Facility.
Liquid Chromatography
A Hitachi HPLC system (Hitachi High-Technologies America, San Jose, CA) comprised of a L-7100 solvent pump, a L-7400 UV-Vis detector, and a D-7000 system controller was used for LC optimization. HPLC columns investigated for separation of oligonucleotide synthesis reaction products included a Clarity Oligo-RP 2.0 × 150 mm, 3 μm particle size (Phenomenex, Torrance, CA), an Xterra MS C18, 1.0 × 150 mm column, 3.5 μm particle size (Waters, Milford, MA), an Xbridge BEH C18 1.0 × 150 mm column, 3.5 μm particle size (Waters, Milford, MA), and an Xbridge OST C18 1.0 × 50 mm column, 2.5 μm particle size (Waters, Milford, MA). Mobile phase A consisted of 16 mM TEA/400 mM HFIP at pH 7.0 in water. Mobile phase B consisted of 16 mM TEA/400 mM HFIP in methanol. A variety of mobile phase gradients were investigated.
Liquid Chromatography Mass Spectrometry
High-resolution LC-MS was performed using a Hitachi HPLC system (Hitachi High-Technologies America, San Jose, CA) comprised of an L-7100 solvent pump, an L-7400 UV-Vis detector, and a D-7000 system controller connected in-line to a Thermo LTQ-FT™ (Thermo Scientific, Waltham, MA) mass spectrometer equipped with an ESI source. Low-resolution LC-MS was performed using a MicroAS autosampler, Surveyor MS Pump Plus HPLC system and Thermo LTQ XL™ (Thermo Scientific, Waltham, MA) mass spectrometer equipped with an ESI source.
For high-resolution LC-MS, the ESI source was set to 325 °C and 4.25 kV. Data from the FT-ICR cell, equipped with a 7.0 Tesla magnet, was acquired in scan event 1. The mass range was restricted to m/z 500 – 2000 to avoid interference from the HFIP dimer ion. Negative ion data was collected in full scan and profile data type with the resolution set to 100,000. For low-resolution LC-MS, the ESI source was set to 275 °C and 4.50 kV. For general data collection, negative ion data was collected in full scan with a mass range of m/z 500 – 2000 and profile data type. For sequence verification using collision-induced dissociation (CID), data-dependent acquisition of MS/MS spectra was used with a 2.0 isolation width and 35% normalization collision energy. Vendor supplied Xcalibur software was used for all data acquisition and processing.
RESULTS AND DISCUSSION
The phosphorothioate oligonucleotides characterized in this work were synthesized using typical elongation cycle consisting of four chemical reactions, performed on the solid support, separated by acetonitrile washing steps intended to remove excess reagents. The components of the elongation cycle include:
Acid-driven removal of the transient protecting group (DMTr) freeing the 5′-hydroxyl group of the support-bound oligonucleotide intermediate.
Elongation of the support-bound oligonucleotide intermediate via coupling of a protected nucleoside phosphoramidite in the presence of an acidic activator yielding phosphite triester intermediates.
Oxidative sulfurization of the formed trialkyl phosphite triester intermediate yielding phosphorothioate triester linkages.
Capping of the support bound unreacted 5′-hydroxyl groups of the support-bound oligonucleotide intermediate.
This elongation cycle is repeated until the desired oligonucleotide sequence is assembled. The final synthesis steps involved removal of the protecting groups from the phosphorothiotriester linkages, release from the solid support and removal of the protecting group from exocyclic amines and 5′-hydroxyl groups.
The solid-phase synthesis of oligonucleotides is known to result in formation of the failure sequences in addition to the desired full-length product (FLP) [26, 27]. It is anticipated that the synthesis of modified phosphorothioate oligonucleotides can result in additional impurities, due to inefficient sulfurization [15], which should be minimized during optimization of synthesis conditions in order to lessen potential regulatory concerns. To develop a broadly applicable LC-MS method for the identification and eventual quantification of low-level impurities in synthetic oligonucleotides, our first step was to establish LC-MS conditions and experimental parameters sufficient to identify most, if not all, of the anticipated synthesis impurities.
HPLC Considerations
Oligonucleotide separations using HPLC are typically implemented based on differences in the hydrophobicity or charge of the sample, and both of those parameters are related to the length of the oligonucleotides. Reversed phase HPLC has been the chromatographic method of choice for separations based on differences in hydrophobicity, and anion exchange HPLC has been the chromatographic method of choice for separations based on differences in charge. The criteria for developing and optimizing chromatographic methods include: resolution, detection sensitivity and compatibility with electrospray ionization (ESI) for coupled LC-MS methods.
Optimal LC-MS methods provide baseline separation of components via chromatography with minimal increase in mass spectral background, ion suppression, cation adducts to analytes, and damage to the instrument. Online LC-MS analysis with ESI provides an inherent advantage by concentrating analytes as they elute from the column. However, the continuous introduction of the column eluent leads an inherent disadvantage of the coupling of these systems as buffer salts and ion pairing agents sprayed into the instrument, especially at high amounts, increase the need for frequent cleaning, degrade instrument components, and decrease instrument performance.
Each chromatographic method has its own benefit for the purification and separation of oligonucleotides. Although not widely used for direct coupling with mass spectrometry, anion-exchange LC-MS is possible. The most effective AE-HPLC buffers typically describe the use of sodium or potassium salts (e.g., KCl), which are not compatible with ESI-MS of oligonucleotides. Thus, a compromise must be found between the separation step and the ionization method. The use of a volatile salt buffer, such as triethylammonium acetate (TEAA), does allow for the separation of oligonucleotides with direct on-line ESI-MS analysis. However, the signal-to-noise ratio for oligonucleotides is reduced with on-line AE-LC-MS due to an increase in the background arising from the mobile phase and so this approach was not pursued in this study.
Because ion pairing reversed phase HPLC (IP-RP-HPLC) has proven to be successful for the general separation of oligonucleotides and is available for coupling with ESI-MS, HPLC optimization focused on this approach. MS detection of phosphorothioate oligonucleotides is generally performed in negative-ion mode because of the presence of an acidic proton at each phosphorothioatediester bond along the backbone. However, various factors can affect signal intensity/ionization efficiency in the negative ion-mode including solution pH, mobile phase composition, presence of cations, salts, etc. It is important to keep pH of the sample near to 7 due to possible formation of additional sample impurities during method development. A low pH in the mobile phase can lead to depurination while a high pH in the mobile phase can induce deamination.
Mobile Phase
While TEAA buffers at pH~7 are amenable to IP-RP HPLC coupled with ESI-MS [10, 13], the use of HFIP as a mobile phase additive has been shown to improve electrospray ionization efficiency for the analysis of oligonucleotides [8]. For these studies, triethylamine was used as the ion pairing agent in combination with HFIP for improved ESI-MS results. A variety of mobile phase compositions were investigated to identify those that yielded the best separation of phosphorothioate FLP from any impurities. Higher TEA concentrations in the mobile phase improved separation efficiency, as expected, due to higher ion-pairing efficiency [28]. The maximum concentration of TEA that is soluble in 400 mM HFIP is 16 mM, and this concentration of TEA was found to be optimum for these analyses. This 400 mM HFIP/16 mM TEA buffer system in either water or in methanol with flow rate of 40 μL/min and gradient 15–75% B in 45 min (Table 1) resulted in satisfactory separation of most of the synthesis impurities. A mobile phase B composition of 200 mM HFIP/8 mM TEA in 50% MeOH/H2O was also investigated and found to yield improved separation at the expense of analysis time.
TABLE 1.
Mobile phase gradient used for LC-HRMS method optimization.
| Time (min) | A (%) | B (%) | Flow rate (μL/min) |
|---|---|---|---|
| 0 | 95 | 5 | 40 |
| 15 | 65 | 35 | 40 |
| 35 | 55 | 45 | 40 |
| 40 | 25 | 75 | 40 |
| 42 | 10 | 90 | 40 |
| 43 | 10 | 90 | 40 |
| 46 | 95 | 5 | 40 |
| 55 | 95 | 5 | 40 |
Column Selection
A stationary phase that can strongly retain ion-paired oligonucleotides, thus requiring a mobile phase with high organic content for analyte elution, is advantageous as the higher organic content can improve desolvation efficiency, which improves ionization and leads to higher mass spectrometry sensitivities. In this study, several stationary phases for IP-RP HPLC were evaluated for their ability to separate FLP phosphorothioate oligonucleotides from synthesis impurities. All columns evaluated were microbore HPLC columns with vendor-specific C18 stationary phases. The discriminating parameters for column choice included separation efficiency, column back-pressure at a 40 μL min−1 flow rate, and mass spectral background. In this study, the Xterra MS C18 and Xbridge BEH C18 from Waters were found to yield similar performance, with the Xbridge providing greater long-term stability and reproducibility.
The Xbridge BEH C18 1.0 × 150 mm column with 3.5 μm particle size was also compared to an Xbridge OST C18 1.0 × 50 mm column with 2.5 μm particle size. Although the smaller particle size will increase column efficiency due to shortening diffusion distances leading to improved mass transfer as evident in a Van Deemter plot, the back-pressure will increase especially for equal column lengths. Comparing the Xbridge BEH to the Xbridge OST, it was found that the BEH column resulted in higher separation efficiencies, most likely because this column contains a greater number of theoretical plates, N, than the OST as noted in Equation 1:
| (Equation 1) |
where L is the column length in cm and dp is the particle diameter in μm. The 15 cm long Xbridge BEH provides nearly a factor of 2 improvement in the number of theoretical plates than the 5 cm long Xbridge OST. Because separation efficiency, rather than speed of analysis, was the goal of this work, further investigations using UPLC were not investigated and it is noted that the Xbridge OST is not available in lengths greater than 5 cm.
Temperature Effects
Another strategy to reduce the detrimental effects of mass transfer on oligonucleotide separation is to perform the separation at an elevated column temperature. In this study, the effect of column temperature on separation was investigated using the previously identified optimal stationary phase and mobile phase conditions. At column temperatures between 25 °C and 45 °C, no significant improvement in separation efficiency was noted. At column temperatures of 50 °C and higher, the background signal in the mass spectrometer increased and suppression of ions from the sample mixture became more noticeable (data not shown). Based on these investigations of column temperature and separation efficiency, all subsequent studies were conducted at 27 °C.
Sample Loading
When attempting to identify low-level impurities from oligonucleotides syntheses, one challenge is to maximize detection of the impurities without overloading the column with the uninformative synthesis FLP. Chromatograms for FLP sample amounts less than 150 pmol injected on column yielded only a few impurity peaks using either UV or MS detection. The lowest amount of FLP sample that could be injected on column and still resulted in sufficient responses from the synthesis impurities was 200 pmol; however, at this amount injected on column, peak broadening in the region around FLP peak was noticeable. Attempts to use heart-cut fractions to reduce the FLP in the sample did not result in significant improvements in the chromatographic separation or MS detection of the synthesis impurities. Due to the extra analysis time required, further attempts to pre-fractionate the sample were not attempted.
Figure 1 contains the UV and MS chromatograms for a typical phosphorothioate oligonucleotide synthesis reaction mixture. The FLP in this example elutes around 37 min using the optimized mobile phase conditions, gradient program and stationary phase described above. As can be noted in this figure, a number of peaks in addition to the FLP are detected both before and after elution of the FLP. The lack of a strong ion signal in the total ion chromatogram from the mass spectrometry detection (Figure 1b) arises because dynamic exclusion was used to eliminate ions related to the FLP from the mass analyzer. For all of the synthesis mixtures investigated, the chromatograms in Figure 1 are representative of the complexity of sample impurities and challenges present when attempting to identify low-level components within a mixture containing primarily the FLP.
Figure 1.
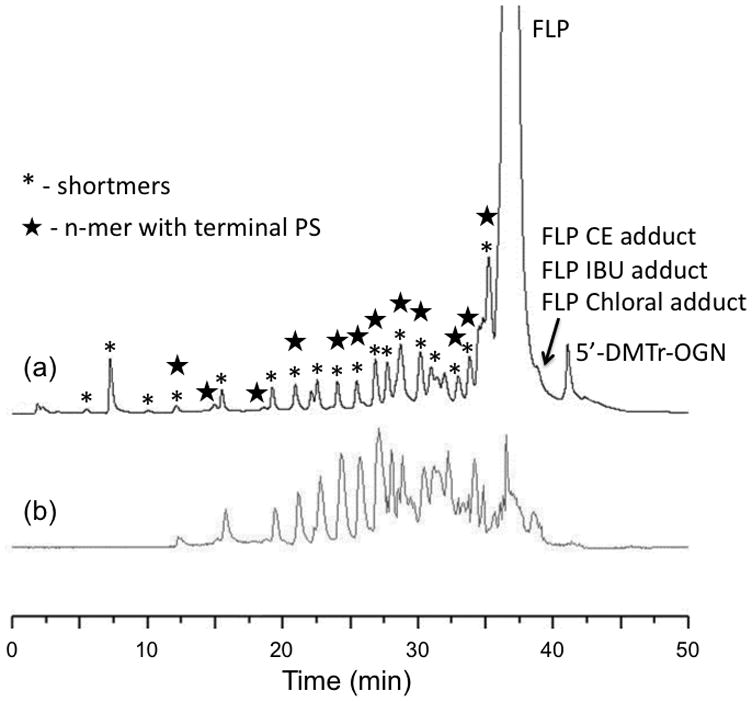
Separation of crude sample containing 24-mer phosphorothioate oligonucleotide and its impurities: (a) total ion chromatogram from LC-MS analysis and (b) UV trace at 260 nm. Mobile phase consisted of 400 mM HFIP/16.3 mM TEA in water (buffer A) or in methanol (buffer B).
Mass Spectrometry Optimization
As all of the expected synthesis impurities are related, in some way, to the FLP, in general the optimization of mass spectrometry parameters can be carried out using any standard oligonucleotides or the FLP itself. The only significant concern encountered in this work was the generation of nucleobase loss by nozzle-skimmer dissociation in the ESI source. Although depurination is also expected to arise as an impurity [15], the loss of purine bases (adenine or guanine) during the ESI-MS process will be sensitive to nozzle-skimmer conditions. For example, Figure 2 contains two representative mass spectra obtained from directly infusing a synthesis reaction mixture. In the top panel (Figure 2a), instrumental conditions are optimized to generate little to no nozzle-skimmer dissociation of the electrosprayed sample. In contrast, the bottom panel (Figure 2b) was obtained at instrumental conditions that, while reducing multiply-charged ions of the FLP, also promote nozzle-skimmer dissociation leading to nucleobase loss. In this example, guanine base loss (−151 Da) is detected at m/z 1885.25 (z = −4) from the FLP (m/z 1923.08, z = −4). To avoid ESI-generated artifacts in the identification of synthesis by-products, it is recommended that a range of MS instrumental parameters be investigated prior to initiating LC-MS characterization of the sample of interest.
Figure 2.
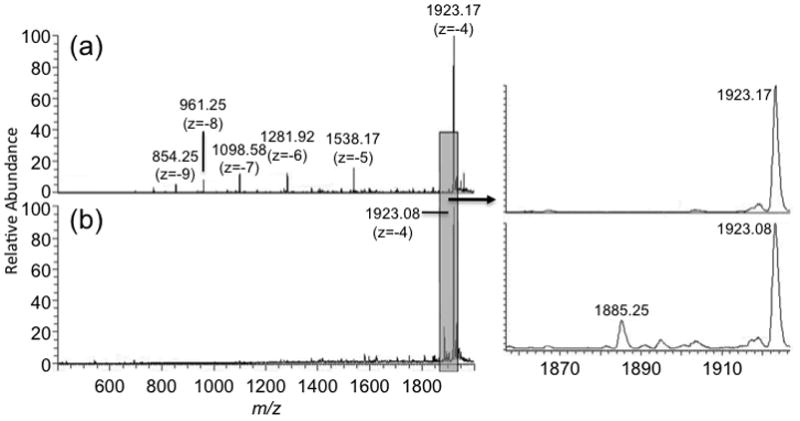
Comparison of mass spectra of crude sample obtained under different instrumental settings. A sample was injected by direct infusion with flow rate of 3 μL/min. (a) parameters obtained after tuning with dT20, which reduced nozzle-skimmer dissociation but generated a large charge state distribution; (b) parameters obtained after manually tuning instrument to reduce the charge state distribution, which leads to nozzle-skimmer dissociation (see text).
Low Resolution Mass Spectrometry
Initial studies using IP-RP-HPLC-MS focused on the suitability of a low-resolution mass spectrometer for identifying synthesis impurities. Although mass spectral resolving power is typical required for mixtures containing numerous components of similar molecular mass, for oligonucleotides synthesis samples, most of the expected impurities have significantly different masses and/or chromatographic retention time allowing use of quadrupole or quadrupole ion trap mass analyzers [15–17]. Thus, these initial LC-MS studies were conducted using a linear ion trap. Figure 3 contains representative data obtained during LC-MS analysis of a phosphorothioate oligonucleotides synthesis reaction mixture. Those impurities expected to be present that were identified when using the linear ion trap mass analyzer are listed in Table 2. As is evident by a review of the identified ions, only failure sequence products, ions resulting from depurination during synthesis, and ions containing a terminal phosphorothioate end group were readily identified.
Figure 3.
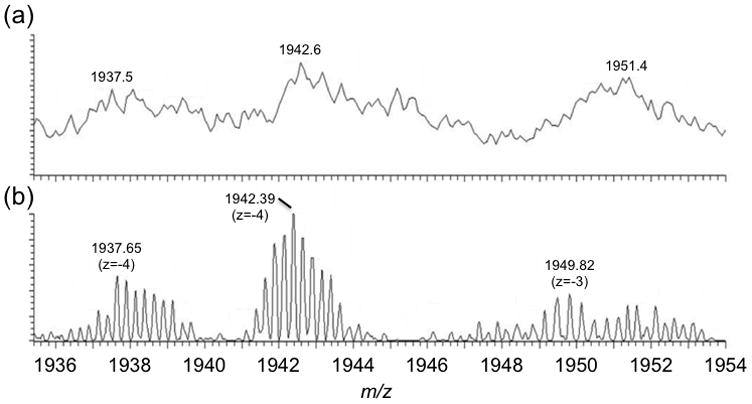
Comparison of (a) LC-MS spectrum and (b) LC-FTMS spectrum of low-level impurities. The use of high resolution FTMS enables isotopes to be resolved so that the charge state can be determined for the impurity.
TABLE 2.
Comparison of impurities identified via LC-low resolution MS and LC-high resolution MS.
| Low res (LTQ-XL) | High res (LTQ-FT) |
|---|---|
| Shortmers | Shortmers |
| n-mer with terminal PS | n-mer with terminal PS |
| depurination product | depurination product |
| n-mer with PO linkage | |
| DMTr 24-mer adduct | |
| Chloral 24-mer adduct | |
| CE adduct | |
| IBU adduct |
DMTr: 4,4-dimethoxytrityl; CE: 2-cyanoethyl; IBU: isobutyryl
Although high mass resolving power was not determined to be necessary for separation of multiple co-eluting ions, the drawback of a low resolving power mass analyzer is the inability to accurately assign ion charge state except in the few cases where singly or doubly charged ions were present. Because many FLP synthetic oligonucleotides are around the 20-mer range, generating singly or doubly charged ions from such large oligonucleotides is difficult. Moreover, low-level impurities of higher charge state are often indistinguishable from the background when using a low resolving power mass analyzer (Figure 3b). Thus, while a low resolution mass spectrometer can be used for the identification of particular synthesis impurities, a significant amount of information from the sample is missing without the ability to accurately determine the charge state of the eluting species, unless assumptions are made about the charge state of the m/z value analyzed [15–17].
High Resolution Mass Spectrometry
Because of the aforementioned difficulties, next we examined the use of a mass analyzer with greater resolving power, here an LTQ-FT mass spectrometer. High resolution mass spectrometry (HRMS) not only allows one to determine the charge state of the ions, which is necessary for calculating elemental compositions, but also enables modeling of the isotopic pattern based on chemical composition. These isotopic patterns provide additional information from the sample that can be used to identify low-level impurities [29–31].
Figure 4 provides an example of using isotope pattern modeling to more accurately identify an impurity within the sample. Figure 4a is the experimentally obtained data and Figure 4b is modeled data yielding the best fit to the experimental data. Based on the detected m/z, the charge state, and the isotope pattern, this impurity is identified as an (n−1) short-mer. Using this approach, many more synthesis impurities could be identified than were found using the low resolution linear ion trap mass analyzer (Table 2). In general, the mass accuracy for all identified impurities was typically < 3 ppm even for very low abundance ions.
Figure 4.
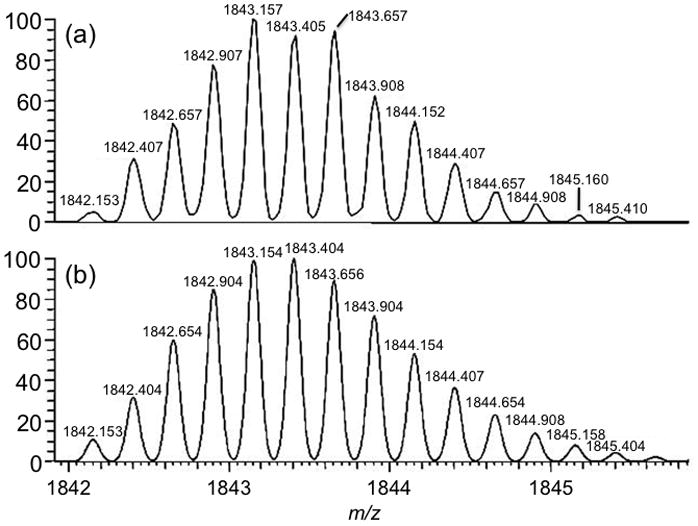
(a) Mass spectrum of impurity identified at m/z 1843 and (b) modeled data for (n−1) short-mer phosphorothioate oligonucleotide with chemical composition of C226H226N68O127P22S22. This chemical composition is consistent with the (n−1) short-mer sequence 5′-CG TCG TTT TGT CGT TTT GTC GTT-3′.
Identified Synthesis Impurities
Table 3 lists the phosphorothioate oligonucleotides synthesis impurities that can be identified by detecting a single charge state and using high resolution mass spectrometry to accurately identify the charge state combined with isotope distribution modeling to confirm the most likely elemental formula. The chemical structures of several of the impurities identified by this approach are shown in Figure 5, which are presented based on the most likely structure from the solid-phase synthesis approach used. Future work, possibly using MS/MS, is needed to confirm the exact identity of each impurity.
TABLE 3.
Impurities found for full-length product (FLP). While all short-mers were detected, only (n−1) included as representative in the table.
| Compound | m/z (observed) | m/z (theoretical) |
|---|---|---|
| n−1 short-mer | 1842.15354 | 1842.15287 |
| (n−1) PS | 1866.13788 | 1866.13872 |
| FLP depurination product | 1888.39372 | 1888.39506 |
| FLP with PO linkage | 1918.17083 | 1918.16439 |
| FLP CE adduct | 1935.66233 | 1935.66722 |
| FLP IBU adduct | 1939.93134 | 1939.92979 |
| FLP Chloral adduct | 1958.90085 | 1958.88796 |
| 5′-DMTr OGN | 1997.94883 | 1997.94321 |
DMTr: 4,4-dimethoxytrityl; CE: 2-cyanoethyl; IBU: isobutyryl.
Figure 5.
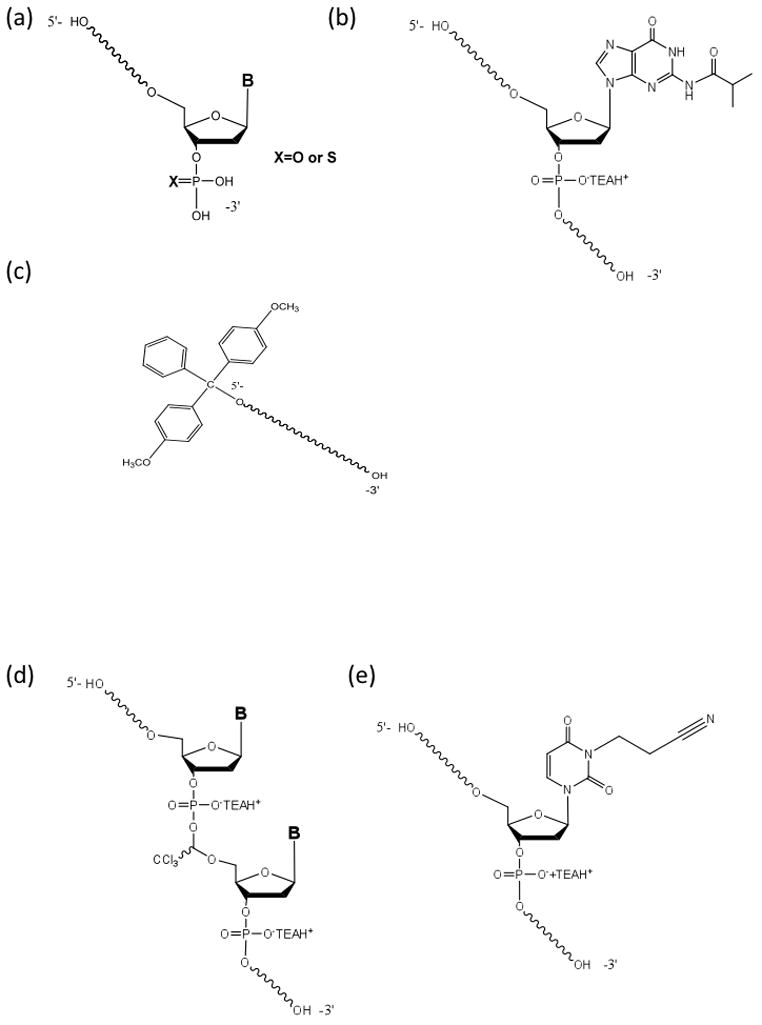
Structures of several of the classes of phosphorothioate oligonucleotides synthesis impurities identified by LC-HRMS profiling. (a) Failure sequences carrying 3′ terminal mono- and mono thiophosphates (n−1)PO or (n−1)PS. (b) Full length product isobutyryl adduct. (c) 5′-O 4,4-dimethoxytrityl-protected oligonucleotide or 5′-O 4,4-dimethoxytrityl-protected n−1 deletion sequences. (d) Full-length product chloral adduct. (e) Full-length product N3(2-cyanoethyl) adduct.
Internal (n−1) deletion sequences belong to a class of impurities typically linked to non-quantitative removal of the DMTr group or inefficient coupling followed by inefficient capping. These impurities can be mitigated by adjustments to the synthesis process parameters (contact time and the reagent composition).
Short-mers containing terminal monothiophosphate arise from the direct coupling of nucleoside phosphoramidites to the silanol groups of the solid support or reaction of the residual phosphitylating agent present in the phosphoramidite synthons. These impurities cannot be separated from the manufactured oligonucleotides during purification, thus, a reduction in these impurities is best addressed during manufacturing of the nucleoside phosphoramidite monomers or derivatization of the solid support.
N3-cyanoethyl adducts are related to an inefficient removal of the cyanoethyl protecting groups from the phosphorothiate triester linkages followed by generation of acrylonitrile and its subsequent addition to thymidine residues. These adducts can be minimized by optimization of the on-column removal of the cyanoethyl group.
Desulfurized FLP (containing one or multiple insertions of the phosphodiester linkages) can arise from two possible sources. First, despite the enhanced reactivity of some recently developed reagents, the sulfurization reaction is not quantitative at each elongation cycle. Second, sulfur loss can occur during treatment with concentrated aqueous ammonia at an elevated temperature during the deprotection step. This side reaction can be completely suppressed by the addition of 2-mercaptoethanol.
The isobutyryl protected FLP reveals that the deprotection conditions were less than optimal. These impurities can be reduced through an increase of the contact time or temperature.
The trichloroacetaldehyde modified FLP indicates the presence of trichloroacetaldehyde in the detritylating solution. Higher purity detritylation solution is necessary to eliminate this impurity.
5′-O-DMTr protected oligonucleotide products arise when the final detritylation step is inefficient. Elimination of these impurities requires optimization of the final detritylation step.
In addition, other ions were identified during LC-MS that arise from the analysis step rather than the oligonucleotides synthesis process itself. In particular, the major ions of this type arise from base depurination of the FLP (secondary peaks 135 u (Ade) or 151 u (Gua) less than the major peak), which are suspected to be generated by heating and/or in-source fragmentation occurring within the ESI source.
CONCLUSIONS
The combination of high resolution mass spectrometry with isotope distribution modeling enables impurity profiling of oligonucleotides synthesis reaction mixtures. In this work, we have identified the optimal IP-RP HPLC-ESI-MS conditions, including the stationary phase and mobile phases, for the separation and identification of phosphorothioate oligonucleotides and associated process-related impurities. Comparing high resolution mass spectrometry with low resolution mass spectrometry, we found that ~60% more impurities could be identified using mass spectral data obtained from the high resolution instrument because charge states can be indentified for low abundance impurities. This charge state information then enables the use of isotopic pattern modeling to confirm the chemical composition of any process-related impurities present in the mixture. In this work, we identified many of the expected synthesis impurities. The next step will be to develop this method further to obtain quantitative information related to the specific impurities detected. Such information can be used to improve and optimize oligonucleotides synthesis, as the presence of the particular families of impurities and their relative abundance in the synthesis mixture carries essential information about the performance of all steps involved in the elongation cycle.
Acknowledgments
Financial support for this work was provided by Girindus America, Inc., the National Institutes of Health (RR019900) and the University of Cincinnati.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson C, O’Keefe AD. Building oligonucleotide therapeutics using non-natural chemistries. Curr Opin Chem Biol. 2006;10:607–614. doi: 10.1016/j.cbpa.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Patil S, Rhodes D, Burgess D. DNA-based therapeutics and DNA delivery systems: a comprehensive review. AAPS J. 2005;7:E61–77. doi: 10.1208/aapsj070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu X. Recent Advances in the Stereocontrolled Synthesis of Antisense Phosphorothioates. Mini Rev Med Chem. 2006;6:319–330. doi: 10.2174/138955706776073439. [DOI] [PubMed] [Google Scholar]
- 4.Gilar M, Belenky A, Cohen AS. Polymer solutions as a pseudostationary phase for capillary electrochromatographic separation of DNA diastereomers. Electrophoresis. 2000;21:2999–3009. doi: 10.1002/1522-2683(20000801)21:14<2999::AID-ELPS2999>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal S, Tang JY, Brown DM. Analytical study of phosphorothioate analogues of oligodeoxynucleotides using high-performance liquid chromatography. J Chromatogr A. 1990;509:396–399. doi: 10.1016/s0021-9673(01)93098-5. [DOI] [PubMed] [Google Scholar]
- 6.Gilar M, Belenky A, Smisek DL, Bourque A, Cohen AS. Kinetics of phosphorothioate oligonucleotide metabolism in biological fluids. Nucl Acids Res. 1997;25:3615–3620. doi: 10.1093/nar/25.18.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourque AJ, Cohen AS. Quantitative analysis of phosphorothioate oligonucleotides in biological fluids using direct injection fast anion-exchange chromatography and capillary gel electrophoresis. J Chromatogr B. 1994;662:343–349. doi: 10.1016/0378-4347(94)00207-x. [DOI] [PubMed] [Google Scholar]
- 8.Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. Analysis of oligonucleotides by HPLC Electrospray Ionization Mass Spectrometry. Anal Chem. 1997;69:1320–1325. doi: 10.1021/ac960916h. [DOI] [PubMed] [Google Scholar]
- 9.Berger B, Holzl G, Oberacher H, Niederstatter H, Huber CG, Parson W. Single nucleotide polymorphism genotyping by on-line liquid chromatography-mass spectrometry in forensic science of the Y-chromosomal locus M9. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;782:89–97. doi: 10.1016/s1570-0232(02)00694-3. [DOI] [PubMed] [Google Scholar]
- 10.Holzl G, Oberacher H, Pitsch S, Stutz A, Huber CG. Analysis of biological and synthetic ribonucleic acids by liquid chromatography-mass spectrometry using monolithic capillary columns. Anal Chem. 2005;77:673–680. doi: 10.1021/ac0487395. [DOI] [PubMed] [Google Scholar]
- 11.Huber CG, Oberacher H. Analysis of nucleic acids by on-line liquid chromatography-mass spectrometry. Mass Spectrom Rev. 2001;20:310–343. doi: 10.1002/mas.10011. [DOI] [PubMed] [Google Scholar]
- 12.Oberacher H, Huber CG, Oefner PJ. Mutation scanning by ion-pair reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry (ICEMS) Hum Mutat. 2003;21:86–95. doi: 10.1002/humu.10155. [DOI] [PubMed] [Google Scholar]
- 13.Oberacher H, Niederstatter H, Parson W. Liquid chromatography-electrospray ionization mass spectrometry for simultaneous detection of mtDNA length and nucleotide polymorphisms. Int J Legal Med. 2007;121:57–67. doi: 10.1007/s00414-006-0117-7. [DOI] [PubMed] [Google Scholar]
- 14.Premstaller A, Oberacher H, Huber CG. High-performance liquid chromatography-electrospray ionization mass spectrometry of single- and double-stranded nucleic acids using monolithic capillary columns. Anal Chem. 2000;72:4386–4393. doi: 10.1021/ac000283d. [DOI] [PubMed] [Google Scholar]
- 15.Capaldi DC, Gaus HJ, Carty RL, Moore MN, Turney BJ, Decottignies SD, McArdle JV, Scozzari AN, Ravikumar VT, Krotz AH. Formation of 4:4′-dimethoxytrityl-C-phosphonate oligonucleotides. Bioorg Med Chem Lett. 2004;14:4683–4690. doi: 10.1016/j.bmcl.2004.06.088. [DOI] [PubMed] [Google Scholar]
- 16.Kurata C, Bradley K, Gaus H, Luu N, Cedillo I, Ravikumar VT, Van Sooy K, McArdle JV, Capaldi DC. Characterization of high molecular weight impurities in synthetic phosphorothioate oligonucleotides. Bioorg Med Chem Lett. 2006;16:607–614. doi: 10.1016/j.bmcl.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 17.Capaldi DC, Gaus H, Krotz AH, Arnold J, Carty RL, Moore MN, Scozzari AN, Lowery K, Cole DL, Ravikumar VT. Synthesis of High-Quality Antisense Drugs. Addition of Acrylonitrile to Phosphorothioate Oligonucleotides: Adduct Characterization and Avoidance. Org Process Res Dev. 2003;7:832–838. [Google Scholar]
- 18.Chen D, Yan Z, Cole DL, Srivatsa GS. Analysis of internal (n−1)mer deletion sequences in synthetic oligodeoxyribonucleotides by hybridization to an immobilized probe array. Nucleic Acids Res. 1999;27:389–395. doi: 10.1093/nar/27.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Z, Theruvathu JA, Farrel A, Burdzy A, Sowers LC. Characterization of synthetic oligonucleotides containing biologically important modified bases by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem. 2008;379:196–207. doi: 10.1016/j.ab.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rentel C, Wang X, Batt M, Kurata C, Oliver J, Gaus H, Krotz AH, McArdle JV, Capaldi DC. Formation of modified cytosine residues in the presence of depurinated DNA. J Org Chem. 2005;70:7841–7845. doi: 10.1021/jo050767f. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Yang Y. An algorithm for thorough background subtraction from high-resolution LC/MS data: application for detection of glutathione-trapped reactive metabolites. J Mass Spectrom. 2008;43:1181–1190. doi: 10.1002/jms.1390. [DOI] [PubMed] [Google Scholar]
- 22.Zhu P, Ding W, Tong W, Ghosal A, Alton K, Chowdhury W. A retention-time-shift-tolerant background subtraction and noise reduction algorithm (BgS-NoRA) for extraction of drug metabolites in liquid chromatography/mass spectrometry data from biological matrices. Rapid Commun Mass Spectrom. 2009;23:1563–1572. doi: 10.1002/rcm.4041. [DOI] [PubMed] [Google Scholar]
- 23.Marshall AG. Milestones in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry Technique Development. Int J Mass Spectrom. 2000;200:331–356. [Google Scholar]
- 24.Brown SC, Kruppa G, Dasseux J-L. Metabolomics applications of FT-ICR mass spectrometry. Mass Spectrom Rev. 2004;24:223–231. doi: 10.1002/mas.20011. [DOI] [PubMed] [Google Scholar]
- 25.Zhang LK, Rempel D, Pramanik BN, Gross ML. Accurate mass measurements by Fourier transform mass spectrometry. Mass Spectrom Rev. 2004;24:286–309. [Google Scholar]
- 26.Reese CB. Oligo- and poly-nucleotides: 50 years of chemical synthesis. Org Biomol Chem. 2005;3:3851–3868. doi: 10.1039/b510458k. [DOI] [PubMed] [Google Scholar]
- 27.Sinha ND, Michaud DP. Recent developments in the chemistry, analysis and control for the manufacture of therapeutic-grade synthetic oligonucleotides. Curr Opin Drug Discov Devel. 2007;10:807–818. [PubMed] [Google Scholar]
- 28.Gilar M, Fountain KJ, Budman Y, Neue UD, Yardley KR, Rainville PD, Russell Ii RJ, Gebler JC. Ion-pair reversed-phase high-performance liquid chromatography analysis of oligonucleotides: - Retention prediction. J Chromatogr A. 2002;958:167–182. doi: 10.1016/s0021-9673(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-de-Cossio J, Gonzalez LJ, Satomi Y, Betancourt L, Ramos Y, Huerta V, Amaro A, Besada V, Padron G, Minamino N, Takao T. Isotopica: a tool for the calculation and viewing of complex isotopic envelopes. Nucleic Acids Res. 2004;32:w674–w678. doi: 10.1093/nar/gkh423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu P, Tong W, Alton K, Chowdhury S. An Accurate-Mass-Based Spectral-Averaging Isotope-Pattern-Filtering Algorithm for Extraction of Drug Metabolites Possessing a Distinct Isotope Pattern from LC-MS Data. Anal Chem. 2009;81:5910–5917. doi: 10.1021/ac900626d. [DOI] [PubMed] [Google Scholar]
- 31.Koomen JM, Russell WK, Tichy SE, Russell DH. Accurate mass measurement of DNA oligonucleotide ions using high-resolution time-of-flight mass spectrometry. J Mass Spectrom. 2002;37:357–371. doi: 10.1002/jms.312. [DOI] [PubMed] [Google Scholar]