

Abstract
Renal diseases in mitochondrial cytopathies are a group of rare diseases that are characterized by frequent multisystemic involvement and extreme variability of phenotype. Most frequently patients present a tubular defect that is consistent with complete De Toni-Debré-Fanconi syndrome in most severe forms. More rarely, patients present with chronic tubulointerstitial nephritis, cystic renal diseases, or primary glomerular involvement. In recent years, two clearly defined entities, namely 3243 A > G tRNALEU mutations and coenzyme Q10 biosynthesis defects, have been described. The latter group is particularly important because it represents the only treatable renal mitochondrial defect. In this paper, the physiopathologic bases of mitochondrial cytopathies, the diagnostic approaches, and main characteristics of related renal diseases are summarized.
1. The Mitochondrial Respiratory Chain
Mitochondria exert multiple roles in cells; in addition to ATP synthesis through oxidative phosphorylations (OXPHOS), they are at the crossroad of numerous metabolic pathways, contribute to heat production, and control cell-cycle/apoptosis and regulation of several anaplerotic reactions [1, 2]. OXPHOS occur within the respiratory chain (RC) that is composed of four protein complexes (complexes I–IV). These proteins transfer electrons and protons across the inner mitochondrial membrane generating the electrochemical gradient for ATP synthesis, which is performed by the ATP synthase (complex V) (reviewed in [1–4]). Within the mitochondrial RC, ubiquinone or coenzyme Q10 (CoQ10) plays a crucial role in accepting and shuttling electrons (Figure 1) [5]. CoQ10 is a ubiquitous lipophylic vitamin-like substance that is found in high concentrations in tissues with elevated energy turnover (heart, brain, liver, and kidney). In humans, CoQ10 is comprised of a quinone group and a tail of 10 isoprenyl units (Figure 1). It is endogenously synthesized through a multienzyme mitochondrial complex [6], that is encoded by at least 16 genes (PDSS and COQ genes) (Figure 1). CoQ10 is also a cofactor for several dehydrogenases, a modulator of the mitochondrial permeability transition pore (MPT) that acts as a gating channel for apoptosis, a cofactor for pyrimidine biosynthesis, and an important antioxidant [7]. Approximately 0.2% of oxygen molecules are not reduced into water during OXPHOS and form reactive oxygen species (ROS) that can be converted by the Fenton reaction into highly reactive hydroxyl radicals (•OH), causing oxidative damage of mitochondrial DNA (mtDNA), peroxidation of lipids and proteins, and activation of the MPT [1, 2] (Figure 1). Accumulation of free radicals plays a crucial role in the physiopathology of many mitochondrial diseases [8]. In normal conditions, ROS are scavenged by superoxide dismutase, catalase, glutathione peroxidase, and thioredoxin peroxidase (Figure 1); CoQ10, vitamin C, vitamin E, and other small molecules also contribute to the mitochondrial defense arsenal against oxidation [1, 5].
Figure 1.
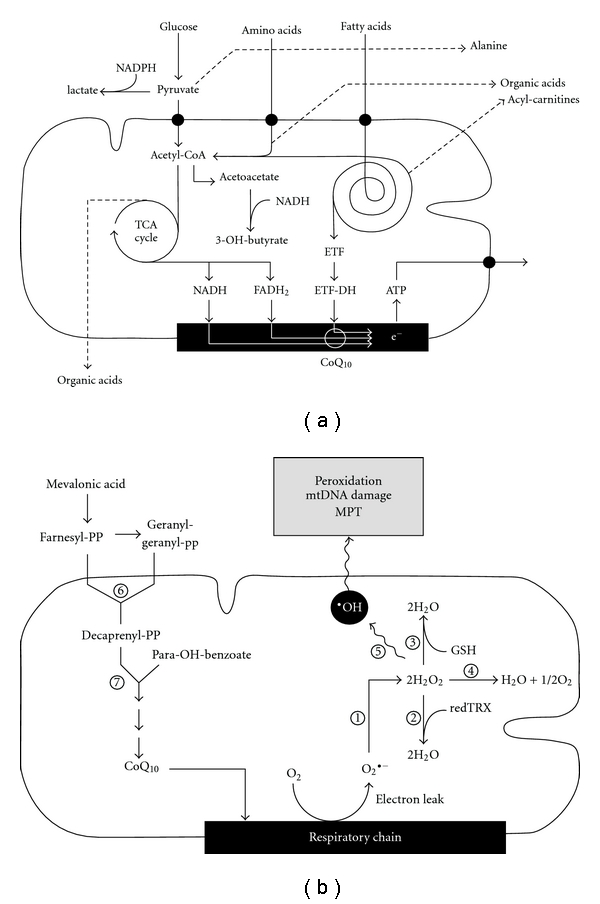
Schematic representation of different metabolic processes involved in cellular energy metabolism. (a) shows the interrelationships between the oxidative pathways of glucose, aminoacid, and fatty acid, which lead to ATP production in the mitochondrial RC enzymes (black box). White lines illustrate the electron flow through CoQ10 in the RC. Dashed lines indicate substrates that generate abnormal metabolites in mitochondrial cytopathies (i.e., lactate, alanine, organic acids, and acyl-carnitines). (b) shows the enzymatic cascade for the biosynthesis of CoQ10 from mevalonic acid (left) and the principal mitochondrial enzymes with antioxidant function (right): (1) superoxide dismutase, (2) thioreductase, (3) glutathione peroxidase, (4) catalase, (5) Fenton reaction, (6) prenyl diphosphate synthetase subunit 1 and 2 (PDSS1 and PDSS2), and (7) CoQ biosynthesis enzymes (COQ2–COQ8).
2. Genetics of Mitochondrial Diseases
mtDNA is composed of a 16,569 bp circular string that encodes for 37 genes, including all 22 tRNAs, 2 rRNA subunits, and 13 structural proteins of the RC; the remaining 75 proteins that compose the RC and other structural or functional mitochondrial proteins (more than 1000) are encoded by nuclear genes [3, 9]. In particular and relevant to several mitochondrial disorders, the biogenesis of the RC requires a number of assembly factors that are encoded by nuclear genes; although these proteins are not structural components of mature RC complexes, they control correct folding and maturation of protein subunits, and the delivery and insertion of prosthetic groups into the holoenzymes of the RC [10]. Mitochondria are transferred from mothers to their progeny in the oocyte; therefore, genetic diseases involving mitochondrial genes follow a maternal inheritance. The first mutations in mitochondrial genes have been reported in the late 1980s [10–13]. Since then, a large number of mutations in mtDNA have been reported and are collected in the MITOMAP human mitochondrial genome database (http://www.mitomap.org/). Their prevalence in the general population may be as high as 1-2 : 10000 live births [14]; mutations can be maternally inherited or sporadic. In addition, mtDNA disorders can follow a Mendelian pattern of inheritance if they are secondary to mutations of nuclear genes that control mtDNA copy number or integrity [15]. As opposed to nuclear genes, the genetic information encoded by mtDNA is present in hundreds of copies per cell; mutations may affect all mtDNA copies (homoplasmy) or only a portion of the mtDNA endowment of cells (heteroplasmy). Symptoms of patients with heteroplasmic mutations depend on the relative proportion between mutated and wild-type mtDNA copies [8]. Cell dysfunction generally occurs when the proportion of mutated mtDNA exceeds a given threshold; tissues with high metabolic rates such as brain, skeletal muscles, heart, and renal tubules are particularly exposed [3]. The degree of heteroplasmy is also variable from oocyte to oocyte, causing significant differences in disease expression among siblings [15].
Mutations in nuclear DNA may affect genes involved in mtDNA maintenance and replication, mitochondrial protein synthesis, protein subunits of individual complexes (they are common for complex I, but rare for other RC complexes), and assembly factors [1, 3]. Despite Mendelian inheritance, high variability in the clinical expression also characterizes nuclear mutations. Phenotypes associated with individual genes, however, tend to be more homogeneous; SURF1 mutations, for example, cause Leigh syndrome, SCO2 mutations are always associated with cardiomyopathy, complex I deficiencies (regardless of the gene involved) tend to present with isolated encephalopathy, and tubulopathy is a common feature of BCS1L mutations. Differences among patients with mutations in the same gene can be ascribed to the severity of individual mutations, degree of residual activities, modulating genes, or to the redundancy of the system [8].
3. Clinical Symptoms of Mitochondrial Cytopathies
Nearly all organs can be affected in mitochondrial cytopathies, resulting in very heterogeneous clinical presentations. Skeletal muscles are very frequently affected (myopathy, hypotonia, and exercise intolerance). Exercise intolerance is a common complaint that is often mislabeled as “psychogenic,” “chronic fatigue syndrome,” or “rheumatic fibromyalgia” [15]. Central nervous symptoms develop over time in most patients; virtually all types of neurological symptoms have been described in these disorders, including apnea, hypotonia, lethargy, psychomotor regression, ataxia, stroke-like episodes, hemiparesis, spasticity, seizures, dementia, leukodystrophy, myoclonus, cortical blindness, migraine, polyneuropathy (sensory and/or motor), and neurogenic bladder. Sensorineural deafness and cardiac diseases (myocardiopathy, arrhythmias, and heart block) are also commonly observed but may remain subclinical and should always be excluded. Endocrine complications include diabetes mellitus, hypoparathyroidism, hypothyroidism, hyporeninemic hypoaldosteronism, and growth hormone deficiency. Gastrointestinal symptoms may be related to liver dysfunction, to intestinal dysmotility (vomiting, diarrhea, and pseudoobstruction), or malabsorption. Hematological signs include sideroblastic anemia, neutropenia, and thrombocytopenia. Many patients have ocular problems, including progressive external ophthalmoplegia, ophthalmoparesis, pigmentary retinal degeneration, ptosis, cataract, optic atrophy, and blindness. Finally, various skin and hair lesions have also been described [16].
From the renal stand point, patients may present with signs of tubulopathy, proteinuria, nephrotic syndrome, tubulointerstitial nephritis, cystic kidney disease, myoglobinuria, or renal failure (see below).
Although the above list of symptoms highlights the extreme variability of phenotypes, a major characteristic of these diseases is the progressive involvement of different organs over time.
To date, more than 40 clinical syndromes have been described, based on the association of different symptoms [1, 3].
4. Diagnostic Approaches to Mitochondrial Cytopathies
When suspecting a mitochondrial defect, the first step is generally to measure serum lactate, which is frequently elevated. In oligosymptomatic renal diseases, serum lactate may be normal, but urine lactate is generally elevated. Brain lactate can also be directly measured in the cerebrospinal fluid or estimated by brain MR spectroscopy. If lactate levels are normal, further genetic studies are not usually recommended. The diagnostic workup requires a combination of different approaches, including biochemistry and enzymology analyses, molecular genetics, pathology (histology, histochemistry, and electron microscopy), and neuroradiology studies.
Measurement of urine organic acids by gas-chromatography/mass spectrometry (GC-MS) represents a helpful tool for diagnosing mitochondrial cytopathies (Figure 2). Impaired RC activity causes the accumulation of reduced NADH/NADPH promoting the conversion of acetoacetate into 3OH-butyrate in the mitochondrion and the conversion of pyruvate into lactate in the cytosol (Figure 1). These compounds are often observed in excess in urines, in association with intermediary products of the Krebs cycle (e.g., 2-ketoglutarate, fumarate, malate, or succinate). In some cases, specific profiles of urinary organic acid in combination with abnormal patterns of blood acylcarnitines allows the diagnosis of specific defects, such as ethylmalonic encephalopathy (ETHE) or SUCLA2-, SUCLG1-, and TMEM70-related diseases [17–20]. In addition, low cytosolic ATP impairs the activity of the γ-glutamyl cycle, which generates glutathione using the energy provided by the hydrolysis of ATP. We have observed in several cases of renal mitochondrial cytopathies increased urinary excretion 5-oxoproline, the upstream metabolite of the γ-glutamyl cycle; this finding is not specific of mitochondrial dysfunctions but is highly evocative of a mitochondrial defect if found in conjunction with high urinary excretion of 3OH-butyrate and lactate. Further indications may be obtained by quantitative analysis of plasma aminoacids, which typically shows high alanine (Figure 1) and/or low citrulline levels [21]. These tests require specialized laboratories, but represent first-line analyses allowing to investigate mitochondrial cytopathies with minimal invasiveness.
Figure 2.
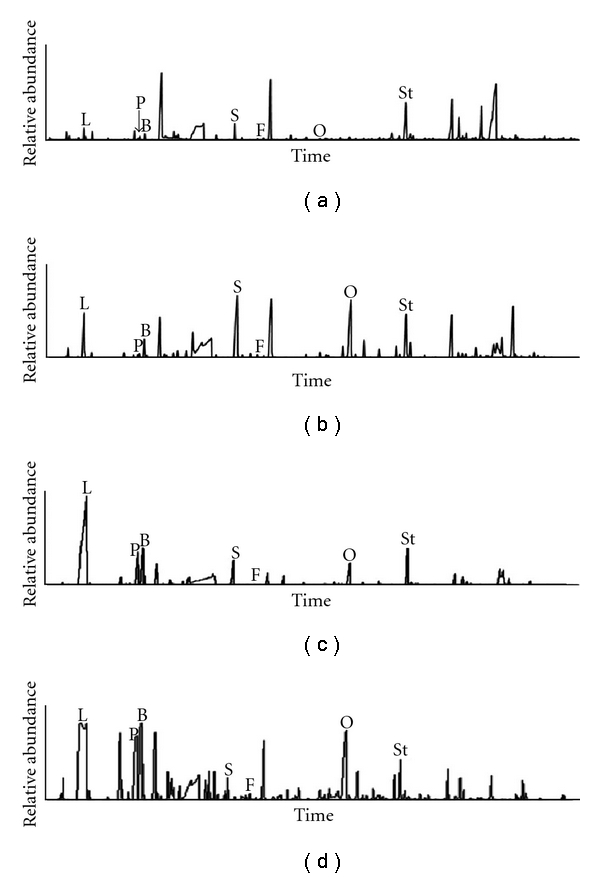
Urine organic acids chromatograms in mitochondrial cytopathies. (a) Control patient. The other panels show the urine chromatograms of their patients with a mitochondrial cytopathy presenting with a generalized tubular defect (b), with De Toni-Debré-Fanconi syndrome (c), or with congenital nephritic syndrome (d). Note the marked increase in lactate (L) excretion in all 3 patients, which was associated with increased urinary excretion of 3-OH-butyrate (B) and 5-oxoproline (O). Other metabolites such as pyruvate (P), succinate (S), and fumarate (F) may also be found in excess.
Further investigations usually require to obtain tissue samples; the general rule is to perform tests on samples collected from the most affected organs. However, in some cases, this approach may be unreasonably aggressive, and studies can be performed on cultured fibroblasts. Measurement of the RC complexes in the kidney, for example, may require an open surgical biopsy to obtain enough material. Similarly, CoQ10 determination has been traditionally performed on skeletal muscle [22], which is an invasive procedure in infants. Fortunately, the metabolic defects observed in skeletal muscles, heart, liver, or kidneys are generally present in fibroblasts, which can be easily expanded and shipped to specialized laboratories for diagnosis. In addition and contrary to other biopsy specimens, treatment of metabolic defects (CoQ10 biosynthesis defects, e.g.) can be started before obtaining a skin biopsy, because oral supplementations do not influence the results of analyses performed on cultured fibroblasts [23, 24].
Several methods have been developed to assess the RC activity in tissues (reviewed in [25]). Polarographic studies are usually used to measure oxygen consumption in the presence of oxidative substrates such as pyruvate, glutamate, malate, or succinate. Spectrophotometric studies allow to measure the activity of each RC complex; in addition, they allow to test the combined activity of [complex II + complex III] that depends on CoQ10. When the combined activity of [complex II + complex III] is markedly lower than the activity of each complex tested separately, results strongly suggest a ubiquinone biosynthesis defect [26].
In tissues sections, the activity of mitochondrial enzymes, such as cytochrome c oxidase (COX) and succinate dehydrogenase (SDH), can be easily assessed with histochemistry techniques [3, 27–29]. These assays are routinely performed on frozen muscle biopsy specimens but can also be applied to other tissues, such as the renal cortex. Because COX is encoded in part by mtDNA, and SDH is entirely encoded by nuclear genes, these studies can demonstrate heteroplasmy by showing cells with high SDH activity secondary to compensatory mitochondrial proliferation and low COX activity [30]; in other cases, they may show a more diffuse decrease in the activity of both enzymes. Electron microscopy, when available, generally demonstrates abnormal mitochondria, proliferation of mitochondria, or mitochondria depletion (Figure 3). Depletion of mitochondria is particularly apparent in proximal tubular cells, which are very rich in these organelles; mitochondrial proliferation in podocytes of patients with steroid-resistant nephritic syndrome (SRNS) is very evocative of a CoQ10 defect.
Figure 3.
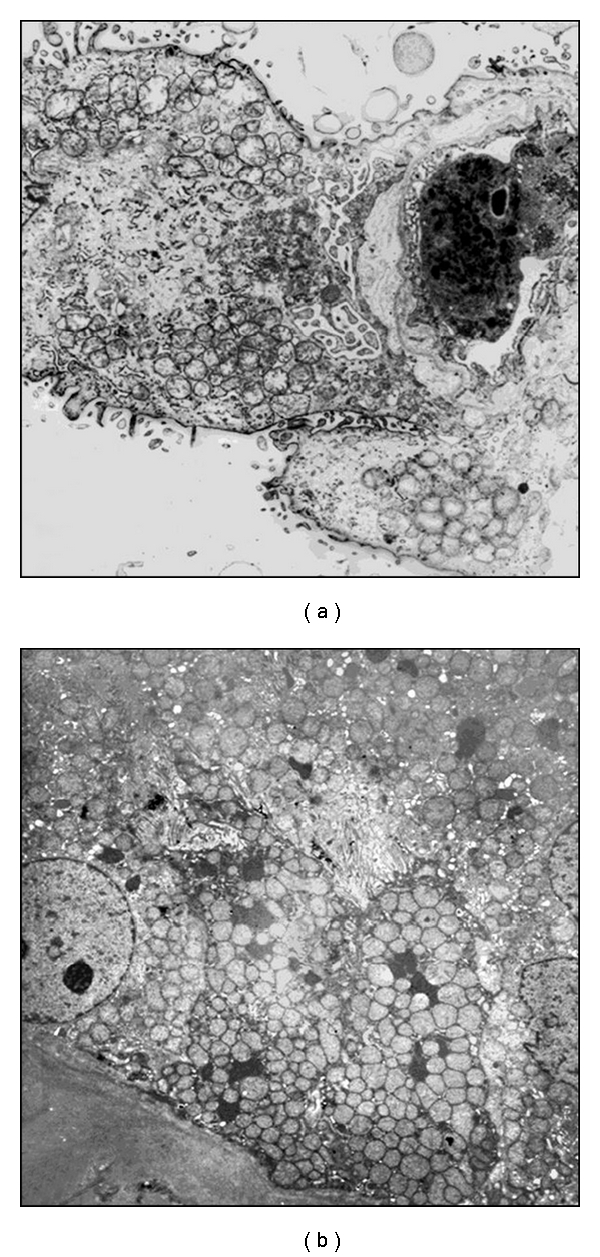
Electron microscopy studies. Two examples of intense proliferation of abnormal mitochondria (swollen mitochondria with simplified or absent mitochondrial cristae) in a podocyte of a patient with a CoQ10 biosynthesis defect (a) and in the proximal tubular cells of a child with severe De Toni-Debré-Fanconi syndrome (b).
5. Renal Mitochondrial Diseases
Kidney involvement is more frequently reported in children than in adults [31]. Several renal diseases have been reported over the past 2 decades, including tubular disorders, chronic tubulointerstitial nephritis, cystic renal disease, and glomerular diseases [31]. In addition, two distinct entities that have primarily glomerular involvement have been identified; these include mtDNA mutations in the tRNALEU gene and CoQ10 biosynthesis defects.
This paper is restricted to mitochondrial cytopathies that affect primarily OXPHOS. However, inherited disorders of mitochondrial fatty acid oxidation can also present with renal involvement. In a large series of 107 patients, Saudubray et al. have reported a tubulopathy and transient renal failure in more than 25% of cases [32]. Since massive rhabdomyolysis represents a frequent event during episodes of acute metabolic decompensation, acute renal failure is generally secondary to myoglobinuria in these patients. Transient renal tubular acidosis has been observed in carnitine palmitoyltransferase type 1 deficiency in combination with Reye-like syndrome [33]. Deficiency of carnitine palmitoyltransferase II causes a neonatal onset lethal multiorgan disease with cystic kidney dysplasia associated with dysmorphic features, central nervous system malformations, liver failure, and cardiomyopathy [34, 35]. Similar findings can be observed in glutaric aciduria type II (or multiple acyl-CoA dehydrogenase deficiency), an autosomal recessive defect of mitochondrial energy metabolism [36]. In both conditions, cystic kidneys can be detected prenatally or at birth through routine ultrasonography examination, showing hyperechoic and enlarged kidneys. These abnormalities are also observed in Zellweger syndrome and other disorders of peroxisomal β-oxidation suggesting that, regardless of the subcellular localization of the biochemical defect, abnormalities of fatty acid oxidation can lead to abnormal organogenesis.
5.1. Tubular Defects
Proximal tubular cells are very rich in mitochondria. Not surprisingly, the most frequent renal tubular finding is a proximal tubular defect, which has been reported in more than 60 patients; of these, 39 have been summarized by Niaudet and Rotig in 1997 [31]; 21 additional patients could be identified in the literature [37–42], including a large Spanish cohort reported by Martín-Hernández et al. in 2005 [43]. In approximately one-third of patients, the tubulopathy corresponded to overt De Toni-Debré-Fanconi syndrome. The remaining patients had more restricted and generally less overt tubular losses and presented with proximal renal tubular acidosis, glycosuria, hyperphosphaturia, and/or aminoaciduria. Nearly all patients had extrarenal symptoms, although cases of isolated tubulopathy have been reported [38, 39], indicating that serum and urine lactate should be investigated in all patients presenting with idiopathic De Toni-Debré-Fanconi syndrome [43]. From a biochemical stand point, the most frequent findings are complex III and/or complex IV defects, followed by complex I defects. From a genetic stand point, all types of mutations have been reported, but large mtDNA deletions are particularly frequent. Tubular involvement is a relatively frequent feature in severe, neonatal-onset RC defects with autosomal recessive inheritance and multisystem involvement; among genes associated with this phenotype are COX10, BCS1L, RRM2B, MRPS22, and SARS2 [44–48]. Symptoms were present in the neonatal period in one-third of patients and in 80% of cases by 2 years of age [31]. Renal biopsies, when available, showed chronic tubulointerstitial changes with damaged proximal tubular epithelia; electron microscopy often showed proliferation of abnormal mitochondria (Figure 3) [31, 37]. Few patients with a Bartter-like phenotype have also been reported [30, 49]. Finally, severe hypomagnesemia is often mentioned in the descriptions of patients with mitochondrial tubulopathies [50].
Of notice, abnormal renal tubular findings remain subclinical (or are overlooked because of the prominence of neurological symptoms) in nearly 2/3 of patients with tubulopathy [43].
When approaching patients with a mitochondrial tubulopathy, clinicians should keep in mind that mitochondrial damage can also be secondary to other causes, including metabolic diseases (tyrosinemia type I, e.g., [51]), drugs (ifosfamide, e.g., [52]), or toxic agents, in particular heavy metals (cadmium, e.g., [53]). A De Toni-Debré-Fanconi syndrome secondary to antimitochondrial antibodies in two patients with primary biliary cirrhosis has also been described [54].
5.2. Chronic Tubulointerstitial Nephritis and Cystic Diseases
Rare cases presenting with chronic renal failure secondary to tubulointerstitial nephritis, without evidence of a primary tubular defect, have been reported; they all had extrarenal symptoms [55–57]. Cystic renal changes have also been rarely described [58–60].
5.3. Sporadic Cases of Glomerular Involvement
Sclerotic glomerular lesions are often described in renal mitochondrial diseases and are probably secondary to tubular and tubulointerstitial lesions. At least nine patients presenting with primary glomerular lesions have been described in the literature [31, 58, 61–63]. They were generally diagnosed with proteinuria and/or hematuria. Some presented with congenital nephrotic syndrome. All patients had or developed over time neurological symptoms (encephalomyopathy); most patients progressed to chronic renal failure if they survived their extrarenal symptoms. The renal pathology, when available, was consistent with focal segmental glomerular sclerosis (FSGS); by EM, depletion or proliferation of abnormal mitochondria in glomerular cells has been described.
5.4. Renal Disease in tRNALEU Gene Mutations
The 3243 A > G point mutation in the leucine tRNA gene is the most prevalent mtDNA defect. This mutation was initially described in children with MELAS syndrome (myopathy, encephalomyopathy, lactic acidosis, and stroke-like episodes) [2, 3]; however, investigations of mothers that carried the same mutation showed that the clinical spectrum can be more restricted to diabetes mellitus, deafness, and/or neuromuscular symptoms [2, 64]; nearly 1% of the diabetic population carries this mutation [65]. In 1997, a large systematic screening of diabetic patients with a history of maternally inherited diabetes and/or sensorineural hearing loss showed a disproportional number of patients with end-stage renal failure secondary to a proteinuric renal disease in this subset of patients. Since then, at least 27 cases (and their relatives) have been described [64–72]. Patients with deafness and proteinuria can also be misleadingly diagnosed with Alport syndrome; a total of 90 Alport patients have been screened in two studies for a MELAS mutation, allowing to identify 2 misdiagnosed cases [65, 69].
Overall, 2/3 of reported patients are females; diabetes and/or deafness is generally present in the proband's mother and other family members [65, 67–69]. The age at diagnosis ranges from 14 to 50 years. The prevalent renal pathology finding is consistent with FSGS. Four cases of chronic tubulointerstitial nephritis and one case presenting with cystic kidney disease have also been described [64, 72]. A peculiar vasculopathy with hyalinosis of small arteries and myocyte necrosis has been noticed in 2 reports [66, 67].
All patients had high-urinary protein excretion; nephrotic syndrome developed in approximately one-third of cases. Proteinuria generally began in the second or third decade of life, with the youngest patient diagnosed at the age 5 [64]. Most patients were hypertensive; two female patients developed pre-eclampsia [66]. Chronic or end-stage renal failure developed within 10 years in approximately 50% of cases.
Nearly 80% of patients had sensorineural deafness or diabetes mellitus at diagnosis; some, in particular younger patients, developed these symptoms during followup [64–72]. Other reported findings include neuromuscular symptoms, retinal dystrophy, and cardiomyopathy. Serum lactate are generally normal.
5.5. CoQ10 Biosynthesis Defects
Primary CoQ10 deficiencies deserve a special place among mitochondrial renal defects because they represent the only treatable mitochondrial disorder. They were first reported in 1989 in association with myopathy and encephalopathy [22], and later with cerebellar ataxia [73].
The link between CoQ10 and renal disease was established in 2000 when three siblings were diagnosed with progressive encephalopathy and SRNS; neurological symptoms improved significantly after treatment with oral ubidecarenone [74]. Two additional siblings that developed SRNS at 1 year of age were reported in 2005 [75]. The first child developed progressive encephalomyopathy and stroke-like episodes at 18 months but improved after oral CoQ10 therapy, while the younger sister, who was diagnosed following her brother disease, was treated immediately after developing proteinuria and never developed neurological symptoms [76]. Mutations in the COQ2 gene were identified in these two siblings as the first report of a genetic defect associated with primary CoQ10 deficiency [77]. COQ2 encodes for parahydroxybenzoate polyprenyl transferase, the enzyme that joins the polyprenoid tail to the quinone group of CoQ10 [78] (Figure 1). COQ2 mutations have been found in four additional patients, all presenting with congenital or early-onset SRNS [26, 79].
Mutations in two other genes involved in the biosynthesis of CoQ10 have been identified in patients with similar clinical features, namely, in the PDSS2 gene (1 patient) [80] and in the COQ6 gene (11 patients from 5 different kindreds) [81]. Symptoms always began within the first years of life; SRNS was the presenting symptom in most cases, and unless treated, renal disease progressed to end-stage renal failure within few years. Other clinical features included deafness and encephalomyopathy in COQ6 patients; severe forms with neonatal onset may also present with liver failure and severe lactic acidosis [26, 79].
Other genes required for CoQ10 biosynthesis can present with different phenotypes: COQ9 mutations, for example (1 patient), cause a severe multisystem disorder with a renal tubulopathy, but no apparent glomerular involvement [82]; patients with mutations in the COQ8 or PDSS1 have no apparent renal disease [78, 83, 84].
The renal pathology varies from focal segmental glomerulosclerosis to collapsing glomerulopathy [26, 85]; electron microscopy generally shows numerous dysmorphic mitochondria in the cytoplasm of podocytes [26, 85].
Our understanding of CoQ10 has largely benefitted from the availability of the kd mouse model that recapitulates the renal phenotype of many CoQ10-deficient patients. These animals were described in the early 1970s [86], but their genetic defect was identified only in 2008, when it was shown that they harbor a homozygous mutation in the PDSS2 gene [87]. Kidneys are normal at birth and develop progressive interstitial nephritis associated with focal segmental glomerulosclerosis or collapsing glomerulopathy; most animals progress to end-stage renal disease by 4–8 months of age and die of renal failure [88]. Glomerular podocytes play a central role in this animal model, while tubular dilatations and interstitial nephritis represent a downstream consequence of the glomerular disease, as demonstrated by conditional knock-out experiments of the PDSS2 gene in podocytes or tubular cells [87]. Furthermore, it has been shown that dietary supplementation with CoQ10 prevents proteinuria and renal changes in mutant mice [89].
To date, the pathogenesis of CoQ10 deficiency remains unclear. In particular, the prevalence of lesions in glomerular cells, which are less energy dependant than tubular cells, raises the possibility that cell damage may not be entirely related to the role of CoQ10 in bioenergetics. Silencing the COQ6 gene in cultured podocytes for example, results in increased apoptosis [81]. In addition, growth of CoQ10-deficient fibroblasts can be corrected by uridine, suggesting that impairment of nucleotide metabolism (CoQ10 is required for the biosynthesis of pyrimidines) may also play a role in the pathogenesis of these disorders [24]. The important role of CoQ10 as an antioxidant may also be responsible for glomerular damage; an inverse relationship between the severity of CoQ10 deficiency and ROS production has been demonstrated in patient's fibroblasts [90, 91]; this hypothesis, however, is not substantiated by in vitro data showing that quinone analogues such as idebenone, which are good antioxidants but cannot rescue the mitochondrial respiratory defect, are probably not effective in the treatment of these diseases [74, 92]. CoQ10-deficient cells also display increased autophagy [93]. Finally, a number of studies in kd mice indicate that environmental factors are important in the development and progression of renal disease. For example, it has been shown that calorie restriction dramatically increases survival of these animals, while protein restriction has no effect [94]; other studies have shown that placing mice in a germ-free environment slows disease progression, underscoring the complexity of factors that are involved in the pathophysiology of CoQ10 renal defects [95].
Regardless of the mechanisms underlying CoQ10 defects, one of the most important aspects is the clinical response to oral supplementations. Initial reports failed to show benefits on renal lesions because patients had already advanced kidney disease [74, 75]. Conversely, when treatment was initiated immediately after the onset of renal symptoms in one girl, prompt reduction of proteinuria was observed; this patient has normal renal function nearly 8 years after starting treatment [76]. A similar response has also been documented in 2 patients with COQ6 mutations [81]. Empirically, CoQ10 doses of 30–50 mg/Kg/day have been used, but appropriate pharmacokinetic studies are lacking [76, 81], whereas PDSS2 mutant mice were treated with doses of 200 mg/Kg/day [89]. Different pharmaceutical formulations are commercially available; aqueous or oleous suspensions should probably be preferred to tablet preparations that contain crystalline forms of CoQ10 [96].
In summary, current evidences indicate that ubiquinone treatment should be started very early to prevent the development of irreversible lesions, especially in brain and kidneys [76]. A suspicion of a CoQ10 deficiency should always arise in patients with early-onset SRNS, especially when podocyte mitochondrial abnormalities are observed by electron microscopy, in the presence of lactic acidosis or when neurologic or muscular symptoms are present.
References
- 1.Finsterer J. Mitochondriopathies. European Journal of Neurology. 2004;11(3):163–186. doi: 10.1046/j.1351-5101.2003.00728.x. [DOI] [PubMed] [Google Scholar]
- 2.Di Donato S. Multisystem manifestations of mitochondrial disorders. Journal of Neurology. 2009;256(5):693–710. doi: 10.1007/s00415-009-5028-3. [DOI] [PubMed] [Google Scholar]
- 3.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. New England Journal of Medicine. 2003;348(26):2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 4.Fosslien E. Mitochondrial medicine—molecular pathology of defective oxidative phosphorylation. Annals of Clinical and Laboratory Science. 2001;31(1):25–67. [PubMed] [Google Scholar]
- 5.Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Developmental Disabilities Research Reviews. 2010;16(2):183–188. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casarin A, Jimenez-Ortega JC, Trevisson E, et al. Functional characterization of human COQ4, a gene required for Coenzyme Q10 biosynthesis. Biochemical and Biophysical Research Communications. 2008;372(1):35–39. doi: 10.1016/j.bbrc.2008.04.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artuch R, Salviati L, Jackson S, Hirano M, Navas P. Coenzyme Q10 deficiencies in neuromuscular diseases. Advances in Experimental Medicine and Biology. 2009;652:117–128. doi: 10.1007/978-90-481-2813-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeviani M, Carelli V. Mitochondrial disorders. Current Opinion in Neurology. 2007;20(5):564–571. doi: 10.1097/WCO.0b013e3282ef58cd. [DOI] [PubMed] [Google Scholar]
- 9.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annual Review of Neuroscience. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 10.Sacconi S, Trevisson E, Pistollato F, et al. hCOX18 and hCOX19: two human genes involved in cytochrome c oxidase assembly. Biochemical and Biophysical Research Communications. 2005;337(3):832–839. doi: 10.1016/j.bbrc.2005.09.127. [DOI] [PubMed] [Google Scholar]
- 11.Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242(4884):1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 12.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331(6158):717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 13.Moraes CT, Shanske S, Tritschler HJ, et al. mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. American Journal of Human Genetics. 1991;48(3):492–501. [PMC free article] [PubMed] [Google Scholar]
- 14.McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease—its impact, etiology, and pathology. Current Topics in Developmental Biology. 2007;77:113–155. doi: 10.1016/S0070-2153(06)77005-3. [DOI] [PubMed] [Google Scholar]
- 15.Bertini E. Mitochondrial encephalomyopathies and related syndromes: brief review. In: Cappa M, Maghnie M, Loche S, Bottazzo GF, editors. Endocrine Involvement in Developmental Syndromes. Vol. 14. Basel, Switzerland: Karger; 2009. pp. 38–52. [DOI] [PubMed] [Google Scholar]
- 16.Bodemer C, Rötig A, Rustin P, et al. Hair and skin disorders as signs of mitochondrial disease. Pediatrics. 1999;103(2):428–433. doi: 10.1542/peds.103.2.428. [DOI] [PubMed] [Google Scholar]
- 17.Tiranti V, D'Adamo P, Briem E, et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. American Journal of Human Genetics. 2004;74(2):239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrozzo R, Dionisi-Vici C, Steuerwald U, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130(3):862–874. doi: 10.1093/brain/awl389. [DOI] [PubMed] [Google Scholar]
- 19.Valayannopoulos V, Haudry C, Serre V, et al. New SUCLG1 patients expanding the phenotypic spectrum of this rare cause of mild methylmalonic aciduria. Mitochondrion. 2010;10(4):335–341. doi: 10.1016/j.mito.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Cízková A, Stránecký V, Mayr JA, et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nature Genetics. 2008;40(11):1288–1290. doi: 10.1038/ng.246. [DOI] [PubMed] [Google Scholar]
- 21.Rabier D, Diry C, Rotig A, et al. Persistent hypocitrullinaemia as a marker for mtDNA NARP T 8993 G mutation? Journal of Inherited Metabolic Disease. 1998;21(3):216–219. doi: 10.1023/a:1005391300203. [DOI] [PubMed] [Google Scholar]
- 22.Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(7):2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montero R, Sánchez-Alcázar JA, Briones P, et al. Analysis of Coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clinical Biochemistry. 2008;41(9):697–700. doi: 10.1016/j.clinbiochem.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 24.López-Martín JM, Salviati L, Trevisson E, et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Human Molecular Genetics. 2007;16(9):1091–1097. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods in Cell Biology. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- 26.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. Journal of the American Society of Nephrology. 2007;18(10):2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 27.Mita S, Schmidt B, Schon EA, DiMauro S, Bonilla E. Detection of “deleted” mitochondrial genomes in cytochromeoxidase-deficient muscle fibers of a patient with Kearns—Sayre syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(23):9509–9513. doi: 10.1073/pnas.86.23.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santorelli FM, Sciacco M, Tanji K, et al. Multiple mitochondrial DNA deletions in sporadic inclusion body myositis: a study of 56 patients. Annals of Neurology. 1996;39(6):789–795. doi: 10.1002/ana.410390615. [DOI] [PubMed] [Google Scholar]
- 29.Szabolcs MJ, Seigle R, Shanske S, Bonilla E, DiMauro S, D'Agati V. Mitochondrial DNA deletion: a cause of chronic tubulointerstitial nephropathy. Kidney International. 1994;45(5):1388–1396. doi: 10.1038/ki.1994.181. [DOI] [PubMed] [Google Scholar]
- 30.Emma F, Pizzini C, Tessa A, et al. "Bartter-like" phenotype in Kearns—Sayre syndrome. Pediatric Nephrology. 2006;21(3):355–360. doi: 10.1007/s00467-005-2092-5. [DOI] [PubMed] [Google Scholar]
- 31.Niaudet P, Rotig A. The kidney in mitochondrial cytopathies. Kidney International. 1997;51(4):1000–1007. doi: 10.1038/ki.1997.140. [DOI] [PubMed] [Google Scholar]
- 32.Saudubray JM, Martin D, de Lonlay P, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. Journal of Inherited Metabolic Disease. 1999;22(4):488–502. doi: 10.1023/a:1005556207210. [DOI] [PubMed] [Google Scholar]
- 33.Falik-Borenstein ZC, Jordan SC, Saudubray JM, et al. Brief report: renal tubular acidosis in carnitine palmitoyltransferase type 1 deficiency. New England Journal of Medicine. 1992;327(1):24–27. doi: 10.1056/NEJM199207023270105. [DOI] [PubMed] [Google Scholar]
- 34.Sharma R, Perszyk AA, Marangi D, Monteiro C, Raja S. Lethal neonatal carnitine palmitoyltransferase II deficiency: an unusual presentation of a rare disorder. American Journal of Perinatology. 2003;20(1):25–32. doi: 10.1055/s-2003-37952. [DOI] [PubMed] [Google Scholar]
- 35.North KN, Hoppel CL, de Girolami U, Kozakewich HP, Korson MS. Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. Journal of Pediatrics. 1995;127(3):414–420. doi: 10.1016/s0022-3476(95)70073-0. [DOI] [PubMed] [Google Scholar]
- 36.Whitfield J, Hurst D, Bennett MJ, Sherwood WG, Hogg R, Gonsoulin W. Fetal polycystic kidney disease associated with glutaric aciduria type II: an inborn error of energy metabolism. American Journal of Perinatology. 1996;13(3):131–134. doi: 10.1055/s-2007-994309. [DOI] [PubMed] [Google Scholar]
- 37.Au KM, Lau SC, Mak YF, et al. Mitochondrial DNA deletion in a girl with Fanconi's syndrome. Pediatric Nephrology. 2007;22(1):136–140. doi: 10.1007/s00467-006-0288-y. [DOI] [PubMed] [Google Scholar]
- 38.Kuwertz-Bröking E, Koch HG, Marquardt T, et al. Renal Fanconi syndrome: first sign of partial respiratory chain complex IV deficiency. Pediatric Nephrology. 2000;14(6):495–498. doi: 10.1007/s004670050802. [DOI] [PubMed] [Google Scholar]
- 39.Mochizuiki H, Joh K, Kawame H, et al. Mitochondrial encephalomyopathies preceded by de-Toni-Debré-Fanconi syndrome or focal segmental glomerulosclerosis. Clinical Nephrology. 1996;46(5):347–352. [PubMed] [Google Scholar]
- 40.Gilbert RD, Emms M. Pearson's syndrome presenting with Fanconi syndrome. Ultrastructural Pathology. 1996;20(5):473–475. doi: 10.3109/01913129609016351. [DOI] [PubMed] [Google Scholar]
- 41.Matsutani H, Mizusawa Y, Shimoda M, et al. Partial deficiency of cytochrome c oxidase with isolated proximal renal tubular acidosis and hypercalciuria. Child Nephrology and Urology. 1992;12(4):221–224. [PubMed] [Google Scholar]
- 42.Eviatar L, Shanske S, Gauthier B, et al. Kearns-Sayre syndrome presenting as renal tubular acidosis. Neurology. 1990;40(11):1761–1763. doi: 10.1212/wnl.40.11.1761. [DOI] [PubMed] [Google Scholar]
- 43.Martín-Hernández E, García-Silva MT, Vara J, et al. Renal pathology in children with mitochondrial diseases. Pediatric Nephrology. 2005;20(9):1299–1305. doi: 10.1007/s00467-005-1948-z. [DOI] [PubMed] [Google Scholar]
- 44.Antonicka H, Leary SC, Guercin GH, et al. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Human Molecular Genetics. 2003;12(20):2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 45.Visapää I, Fellman V, Vesa J, et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. American Journal of Human Genetics. 2002;71(4):863–876. doi: 10.1086/342773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acham-Roschitz B, Plecko B, Lindbichler F, et al. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Molecular Genetics and Metabolism. 2009;98(3):300–304. doi: 10.1016/j.ymgme.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 47.Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. Journal of Medical Genetics. 2007;44(12):784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial Seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. American Journal of Human Genetics. 2011;88(2):193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goto Y, Itami N, Kajii N, Tochimaru H, Endo M, Horai S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns—Sayre syndrome. Journal of Pediatrics. 1990;116(6):904–910. doi: 10.1016/s0022-3476(05)80648-1. [DOI] [PubMed] [Google Scholar]
- 50.Katsanos KH, Elisaf M, Bairaktari E, Tsianos EV. Severe hypomagnesemia and hypoparathyroidism in Kearns-Sayre syndrome. American Journal of Nephrology. 2001;21(2):150–153. doi: 10.1159/000046239. [DOI] [PubMed] [Google Scholar]
- 51.Endo F, Sun MS. Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells. Journal of Inherited Metabolic Disease. 2002;25(3):227–234. doi: 10.1023/a:1015646400182. [DOI] [PubMed] [Google Scholar]
- 52.Nissim I, Horyn O, Daikhin Y, et al. Ifosfamide-induced nephrotoxicity: mechanism and prevention. Cancer Research. 2006;66(15):7824–7831. doi: 10.1158/0008-5472.CAN-06-1043. [DOI] [PubMed] [Google Scholar]
- 53.Thévenod F. Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiology. 2003;93(4):87–93. doi: 10.1159/000070241. [DOI] [PubMed] [Google Scholar]
- 54.Lino M, Binaut R, Noël LH, et al. Tubulointerstitial nephritis and fanconi syndrome in primary biliary cirrhosis. American Journal of Kidney Diseases. 2005;46(3):e41–e46. doi: 10.1053/j.ajkd.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 55.Ueda Y, Ando A, Nagata T, et al. A boy with mitochondrial disease: asymptomatic proteinuria without neuromyopathy. Pediatric Nephrology. 2004;19(1):107–110. doi: 10.1007/s00467-003-1318-7. [DOI] [PubMed] [Google Scholar]
- 56.Tzen CY, Tsai JD, Wu TY, et al. Tubulointerstitial nephritis associated with a novel mitochondrial point mutation. Kidney International. 2001;59(3):846–854. doi: 10.1046/j.1523-1755.2001.059003846.x. [DOI] [PubMed] [Google Scholar]
- 57.Rötig A, Goutières F, Niaudet P, et al. Deletion of mitochondrial DNA in a patient with chronic tubulointerstitial nephritis. Journal of Pediatrics. 1995;126(4):597–601. doi: 10.1016/s0022-3476(95)70359-4. [DOI] [PubMed] [Google Scholar]
- 58.Hameed R, Raafat F, Ramani P, Gray G, Roper HP, Milford DV. Mitochondrial cytopathy presenting with focal segmental glomerulosclerosis, hypoparathyroidism, sensorineural deafness, and progressive neurological disease. Postgraduate Medical Journal. 2001;77(910):523–526. doi: 10.1136/pmj.77.910.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gürgey A, Özalp I, Rötig A, et al. A case of Pearson syndrome associated with multiple renal cysts. Pediatric Nephrology. 1996;10(5):637–638. doi: 10.1007/s004670050178. [DOI] [PubMed] [Google Scholar]
- 60.Müller Höcker J, Walther JU, Bise K, Pongratz D, Hübner G. Mitochondrial myopathy with loosely coupled oxidative phosphorylation in a case of Zellweger syndrome. A cytochemical-ultrastructural study. Virchows Archive B: Cell Pathology Including Molecular Pathology. 1984;45(2):125–138. doi: 10.1007/BF02889859. [DOI] [PubMed] [Google Scholar]
- 61.Güçer S, Talim B, Aşan E, et al. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy: report of two cases with special emphasis on podocytes. Pediatric and Developmental Pathology. 2005;8(6):710–717. doi: 10.1007/s10024-005-0058-z. [DOI] [PubMed] [Google Scholar]
- 62.Unal S, Kalkanoğlu HS, Kocaefe C, et al. Four-month-old infant with focal segmental glomerulosclerosis and mitochondrial DNA deletion. Journal of Child Neurology. 2005;20(1):83–84. doi: 10.1177/08830738050200011304. [DOI] [PubMed] [Google Scholar]
- 63.Goldenberg A, Ngoc LH, Thouret MC, et al. Respiratory chain deficiency presenting as congenital nephrotic syndrome. Pediatric Nephrology. 2005;20(4):465–469. doi: 10.1007/s00467-004-1725-4. [DOI] [PubMed] [Google Scholar]
- 64.Guéry B, Choukroun G, Noël LH, et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. Journal of the American Society of Nephrology. 2003;14(8):2099–2108. doi: 10.1097/01.asn.0000080180.51098.02. [DOI] [PubMed] [Google Scholar]
- 65.Jansen JJ, Maassen JA, van der Woude FJ, et al. Mutation in mitochondrial tRNA(Leu(UUR)) gene associated with progressive kidney disease. Journal of the American Society of Nephrology. 1997;8(7):1118–1124. doi: 10.1681/ASN.V871118. [DOI] [PubMed] [Google Scholar]
- 66.Moulonguet Doleris L, Hill GS, Chedin P, et al. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy. Kidney International. 2000;58(5):1851–1858. doi: 10.1111/j.1523-1755.2000.00356.x. [DOI] [PubMed] [Google Scholar]
- 67.Löwik MM, Hol FA, Steenbergen EJ, Wetzels JF, van den Heuvel LP. Mitochondrial tRNALeu(UUR) mutation in a patient with steroid-resistant nephrotic syndrome and focal segmental glomerulosclerosis. Nephrology Dialysis Transplantation. 2005;20(2):336–341. doi: 10.1093/ndt/gfh546. [DOI] [PubMed] [Google Scholar]
- 68.Hotta O, Inoue CN, Miyabayashi S, Furuta T, Takeuchi A, Taguma Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney International. 2001;59(4):1236–1243. doi: 10.1046/j.1523-1755.2001.0590041236.x. [DOI] [PubMed] [Google Scholar]
- 69.Cheong HI, Chae JH, Kim JS, et al. Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatric Nephrology. 1999;13(6):477–480. doi: 10.1007/s004670050641. [DOI] [PubMed] [Google Scholar]
- 70.Kurogouchi F, Oguchi T, Mawatari E, et al. A case of mitochondrial cytopathy with a typical point mutation for MELAS, presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation. American Journal of Nephrology. 1998;18(6):551–556. doi: 10.1159/000013406. [DOI] [PubMed] [Google Scholar]
- 71.Nakamura S, Yoshinari M, Doi Y, et al. Renal complications in patients with diabetes mellitus associated with an A to G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Diabetes Research and Clinical Practice. 1999;44(3):183–189. doi: 10.1016/s0168-8227(99)00051-0. [DOI] [PubMed] [Google Scholar]
- 72.Hirano M, Konishi K, Arata N, et al. Renal complications in a patient with A-to-G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Internal Medicine. 2002;41(2):113–118. doi: 10.2169/internalmedicine.41.113. [DOI] [PubMed] [Google Scholar]
- 73.Musumeci O, Naini A, Slonim AE, et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56(7):849–855. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- 74.Rötig A, Appelkvist EL, Geromel V, et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356(9227):391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 75.Salviati L, Sacconi S, Murer L, et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology. 2005;65(4):606–608. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- 76.Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. New England Journal of Medicine. 2008;358(26):2849–2850. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 77.Quinzii C, Naini A, Salviati L, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. American Journal of Human Genetics. 2006;78(2):345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forsgren M, Attersand A, Lake S, et al. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochemical Journal. 2004;382(2):519–526. doi: 10.1042/BJ20040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mollet J, Giurgea I, Schlemmer D, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. Journal of Clinical Investigation. 2007;117(3):765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.López LC, Schuelke M, Quinzii CM, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphote synthase subunit 2 (PDSS2) mutations. American Journal of Human Genetics. 2006;79(6):1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heeringa SF, Chernin G, Chaki M, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. Journal of Clinical Investigation. 2011;121(5):2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duncan AJ, Bitner-Glindzicz M, Meunier B, et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. American Journal of Human Genetics. 2009;84(5):558–566. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lagier-Tourenne C, Tazir M, López LC, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. American Journal of Human Genetics. 2008;82(3):661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mollet J, Delahodde A, Serre V, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. American Journal of Human Genetics. 2008;82(3):623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barisoni L, Madaio MP, Eraso M, Gasser DL, Nelson PJ. The kd/kd mouse is a model of collapsing glomerulopathy. Journal of the American Society of Nephrology. 2005;16(10):2847–2851. doi: 10.1681/ASN.2005050494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lyon MF, Hulse EV. An inherited kidney disease of mice resembling human nephronophthisis. Journal of Medical Genetics. 1971;8(1):41–48. doi: 10.1136/jmg.8.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peng M, Falk MJ, Haase VH, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genetics. 2008;4(4) doi: 10.1371/journal.pgen.1000061. Article ID e1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peng M, Jarett L, Meade R, et al. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney International. 2004;66(1):20–28. doi: 10.1111/j.1523-1755.2004.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saiki R, Lunceford AL, Shi Y, et al. Coenzyme Q10 supplementation rescues renal disease in Pdss2 kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. American Journal of Physiology Renal Physiology. 2008;295(5):F1535–F1544. doi: 10.1152/ajprenal.90445.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Quinzii CM, López LC, Von-Moltke J, et al. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB Journal. 2008;22(6):1874–1885. doi: 10.1096/fj.07-100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Quinzii CM, López LC, Gilkerson RW, et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB Journal. 2010;24(10):3733–3743. doi: 10.1096/fj.09-152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.López LC, Quinzii CM, Area E, et al. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011897. Article ID e11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rodríguez-Hernández A, Cordero MD, Salviati L, et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5(1):19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- 94.Fernandes G, Yunis EJ, Miranda M, Smith J, Good RA. Nutritional inhibition of genetically determined renal disease and autoimmunity with prolongation of life in kdkd mice. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(6):2888–2892. doi: 10.1073/pnas.75.6.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hallman TM, Peng M, Meade R, Hancock WW, Madaio MP, Gasser DL. The mitochondrial and kidney disease phenotypes of kd/kd mice under germfree conditions. Journal of Autoimmunity. 2006;26(1):1–6. doi: 10.1016/j.jaut.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hatanaka J, Kimura Y, Lai-Fu Z, Onoue S, Yamada S. Physicochemical and pharmacokinetic characterization of water-soluble Coenzyme Q(10) formulations. International Journal of Pharmaceutics. 2008;363(1-2):112–117. doi: 10.1016/j.ijpharm.2008.07.019. [DOI] [PubMed] [Google Scholar]