

Abstract
Among closely related taxa, proteins involved in reproduction generally evolve more rapidly than other proteins. Here, we apply a functional and comparative genomics approach to compare functional divergence across a deep phylogenetic array of egg-laying and live-bearing vertebrate taxa. We aligned and annotated a set of 4,986 1 : 1 : 1 : 1 : 1 orthologs in Anolis carolinensis (green lizard), Danio rerio (zebrafish), Xenopus tropicalis (frog), Gallus gallus (chicken), and Mus musculus (mouse) according to function using ESTs from available reproductive (including testis and ovary) and non-reproductive tissues as well as Gene Ontology. For each species lineage, genes were further classified as tissue-specific (found in a single tissue) or tissue-expressed (found in multiple tissues). Within independent vertebrate lineages, we generally find that gonadal-specific genes evolve at a faster rate than gonadal-expressed genes and significantly faster than non-reproductive genes. Among the gonadal set, testis genes are generally more diverged than ovary genes. Surprisingly, an opposite but nonsignificant pattern is found among the subset of orthologs that remained functionally conserved across all five lineages. These contrasting evolutionary patterns found between functionally diverged and functionally conserved reproductive orthologs provide evidence for pervasive and potentially cryptic lineage-specific selective processes on ancestral reproductive systems in vertebrates.
1. Introduction
Over the past 550 million years, evolutionary processes have generated a diverse array of vertebrate species. Taxa that include fishes, birds, reptiles, and mammals evolved unique suites of adaptations allowing them to prosper in the most extreme sea, air, and land environments. Vertebrate diversity spans morphological innovations, developmental programming, cellular responses, as well as behaviors and life histories, and such differences become increasingly evident when taxa are compared across deep phylogenies. Studying the evolutionary patterns of functional change across this subphylum provides an opportunity to understand the evolutionary processes that have been important throughout vertebrate evolution. Yet, to date, clear common functional signatures that are in rapid flux across all vertebrate taxa have not been identified indicating the historical presence of a variety of niche- and lineage-dependent selective processes.
While functional evolutionary signals are not apparent across diverse phylogenetic lineages, when more closely related species such as sister species or multiple species within a single genus are compared, reproductive traits consistently reveal high diversity among species. This reproductive signature has been known for centuries, beginning with Linnaeus' binomial classification system [1]. Charles Darwin, in his 1871 treatise on sexual selection, also catalogued highly differentiated secondary sexual organs between closely related bird and mammal species [2]. Over a century later, William Eberhard described the diversity of morphological differences found in male secondary sexual traits, including vertebrate genitalia [3]. Both Darwin and Eberhard explain this higher male variance as the result of female mate choice or male-male competition on sexually selected traits within populations. The last three decades have amassed more vertebrate examples including cichlids [4], frogs [5], and primates [6] indicating that selection on reproductive traits may be a common underlying evolutionary process in vertebrates.
Studying rates of morphological character change demonstrates how certain functional classes evolve relative to others and provides a lens into evolutionary processes of the past. While this framework works well on closely related species, signatures diminish when applied to distantly related taxa due to the presence of lineage-specific rates of development, selective constraints, and genetic architectural differences [7]. In addition, there are many processes in which the selected phenotype may be hidden or cryptic to human observers. Such phenotypes often occur at the molecular level and include immune response [8], gametic interactions [9], and pheromonal exchange [10]. To systematically understand the relative roles of different functional classes in the evolutionary history of vertebrates, and hence the role of certain selective processes, it would be instructive to employ a common and unbiased framework on a representative sample of taxa.
With the availability of annotated genomic sequences across an ever-expanding number of taxa in addition to associated functional data (e.g., ESTs, GO) that can link genes to function, an operational framework is emerging that compares rates of functional change across varying degrees of phylogenetic relatedness [11, 12]. By applying this functional and comparative genomics approach, we now can use normalized information from sequences to infer how functional categories of genes have changed in the past. Combining the two domains of time and function can provide valuable information about the history of these lineages, in particular, how certain selective forces act upon certain reproductive processes such as gamete recognition, oogenesis, spermatogenesis, and adult behavior.
In this paper, we quantify the rates of change among reproductive and non-reproductive genes in five distantly related vertebrate lineages. We functionally categorize ~5,000 orthologs using available testis, ovary, and non-reproductive EST libraries in each species and find that individual vertebrate lineages generally follow a pattern of greater divergence in genes solely expressed in the gonads compared to genes expressed in non-reproductive tissue. In most cases, the testis appears to be driving gonadal divergence. However, an opposing pattern emerges when we compare evolutionary rates among the much smaller subset of tissue-expressed genes that have remained functionally conserved across vertebrates (dNtestis < dNovary < dNnon-reproductive). Using this framework, we are beginning to unmask a pattern of rapid and cryptic molecular evolution on lineage-specific reproductive features that are part of conserved developmental processes, thus, providing a common underlying genetic basis of functional evolutionary change in the vertebrate subphylum.
2. Materials and Methods
2.1. Orthology and Estimates of Divergence
Protein coding genes from A. carolinensis, G. gallus, D. rerio, M. musculus, and X. tropicalis were used in this analysis. Orthologs for each species pair were obtained from BioMart (http://uswest.ensembl.org/biomart/index.html). Orthologs were filtered so that only transitive sets of 1 : 1 : 1 : 1 : 1 orthologs remained, producing 4,986 sets of 5-species orthologs. We excluded all paralogous relationships (including 10,122 1 : 1 : 2 : 1 : 1 relationships, where “2” denotes paralogous sequences from the zebrafish lineage) in order to maintain a relatively ambiguous ortholog set. The protein coding CDS and amino acid sequence of each gene's longest transcript were also obtained from BioMart: in the case of transcript length ties, the transcript with the lower incremental Ensemble ID number was used. Multiple sequence alignments for each orthologous set of proteins were generated using MUSCLE (version 3.8; [13]) and then back-translated using corresponding CDS and a custom Perl script (available from CJG on request). All 1 : 1 : 1 : 1 : 1 alignments in addition to their associated functional assignments will be made available via lizardbase (http://www.lizardbase.org/) as an active link to current A. carolinensis annotations in lizardbase's genome browser, JBrowse, and lizardbase's Resources Page. All alignments will also be made available on the Resources page in lizardbase.
A protein distance matrix was calculated for each protein alignment using the Jones-Taylor-Thornton (JTT) model in the prodist program from the Phylip suite of phylogenetic programs (version 3.69; [14]). Consensus phylogenetic trees were generated using concatenated sequences from both CDS and its associated protein sequences (See in Supplementary Material available online at doi:10.4061/2011/274975 Supplementary Figure 1). For a given gene from each species, the mean of its four orthologous protein distances was used as one of two estimates of sequence divergence. A matrix of nonsynonymous substitutions per nonsynonymous site, dN, was also estimated for each codon alignment using Nei and Gojobori's method [15] using the SNAP Perl program [16], and its mean dN across four orthologs was used as an estimate of sequence divergence.
2.2. Functional Annotation Using EST Libraries and Gene Ontologies
ESTs from each of the five species were filtered as “normal adult” tissue from NCBI's dbEST (downloaded in October 2009) and assigned to species-specific tissue libraries (see Supplementary Table 1) based on either organ or tissue fields in the Genbank record. EST sequences were locally indexed and aligned to genes from the same species using a standalone version of blastn (version 2.22; [17]). EST-to-gene alignments of at least 100 nucleotides, 90% identity, and an E-value of e − 20 were used as alignment criteria. For each of the five species lineages, genes with at least three ESTs (i.e., hits) meeting the above alignment criteria were assigned to seven non mutually exclusive functional classes: (1) genes with hits in only the testis were classified as testis-specific; (2) genes with hits in the testis and another tissue(s) were classified as testis-expressed; (3) genes with hits in only the ovary were classified as ovary-specific; (4) genes with hits in the ovary and another tissue(s) were classified as ovary-expressed; (5) genes with hits in only the testis and/or ovary were classified as gonadal-specific; (6) genes with hits in the testis and/or ovary, in addition to non-reproductive tissue(s), were classified as gonadal-expressed; (7) genes with hits from an assortment of non-reproductive tissues (see Supplementary Table 1) that were neither testis nor ovary were classified as non-reproductive. Thus, for each of the five species, genes with sufficient EST coverage fell into at least one functional class (Table 1). The difference between the mean dN of each reproductive class and the mean dN of the non-reproductive class was tested using an unpaired two-sample two-sided Wilcoxon rank sum test ([18]; Figure 1).
Table 1.
Functional classification of reproductive (testis, ovary, gonadal) and non-reproductive orthologs in vertebrate species. Genes were assigned to at least one of seven functional categories (see Section 2.2 for explanation). Non-reproductive genes are found in neither the testis nor ovary EST libraries but are present in other tissues.
| Functional classification | A. carolinensis | D. rerio | X. tropicalis | G. gallus | M. musculus |
|---|---|---|---|---|---|
| Testis-specific | 55 | 49 | 129 | 12 | 43 |
| Testis-expressed | 613 | 2511 | 2825 | 1011 | 1659 |
| Ovary-specific | 87 | 21 | 13 | 16 | 0 |
| Ovary-expressed | 889 | 2422 | 1367 | 2033 | 350 |
| Gonadal-specific | 243 | 126 | 187 | 53 | 45 |
| Gonadal-expressed | 1109 | 3142 | 3109 | 2447 | 1848 |
| Non-reproductive | 243 | 537 | 613 | 940 | 2159 |
| Total annotated genes (out of 4986) | 1352 | 3679 | 3722 | 3387 | 4007 |
Figure 1.
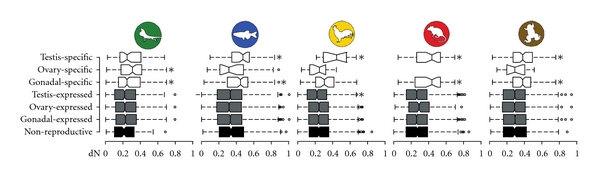
Protein divergence versus functional class across vertebrate lineages. Boxplots show the distribution of dN, nonsynonymous substitutions per nonsynonymous site in seven functional classes for each of the five species, A. carolinensis, D. rerio, G. gallus, M. musculus, and X. tropicalis. The three tissue-specific classes are found on the top (nonshaded), tissue-expressed classes are below in grey, and the non-reproductive functional class is indicated on the bottom, in black. Asterisks on the right-hand side of a boxplot signifies a highly significant (P < 0.001) difference in mean, as given by the Wilcoxon rank sum test, when compared to the non-reproductive class. No ovary-specific genes were identified in M. musculus.
We also compared evolutionary rates in functionally conserved genes, that is, those orthologs that do not change functional class across all five lineages, according to our EST annotations. Interestingly, we were not able to identify a single gonadal-specific gene, but were able to identify functionally conserved subsets of testis-expressed (n = 95), ovary-expressed (n = 16), and non-reproductive (n = 3) orthologs. Figures 2(a)–2(g) provides Venn diagrams for all species combinations in each functional class. Figure 3 compares dN across four (nonzero) functional classes.
Figure 2.
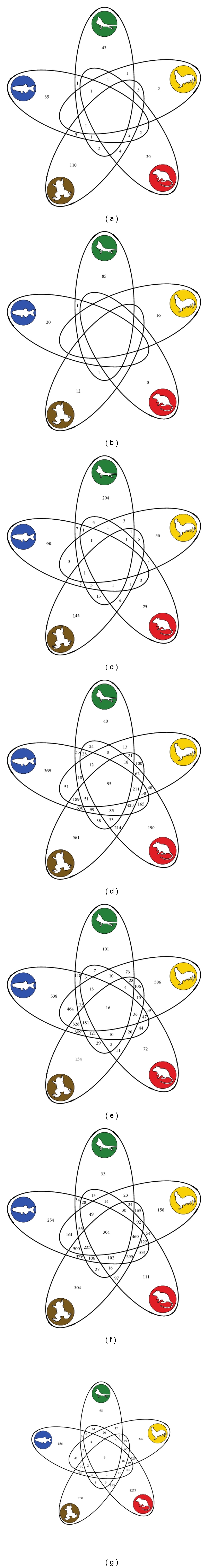
Venn diagrams of common functionally conserved genes across all five vertebrate species. For each of the seven functional classes, the number of genes found in all combination of species intersections and exclusions are listed. (a) testis-specific, (b) ovary-specific, (c) gonadal-specific, (d) testis-expressed, (e) ovary-expressed, (f) gonadal-expressed, (g) non-reproductive.
Figure 3.
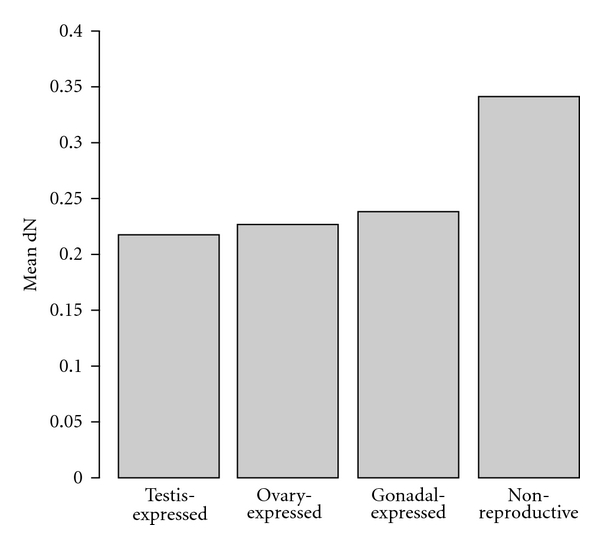
dN among functionally conserved classes across all five vertebrate species. Only four of the seven functional classes contained genes that were found in the same functional class across zebrafish, Anolis, Xenopus, chicken, and mouse. Functional classes were not significantly different from each other.
To complement the functional annotations generated by ESTs, we linked the 10% most diverged orthologs to the GO categories, Biological Process (BP) and Cellular Component (CC) in each species. GeneMerge [19] was used to test for statistically significant over-represented functional terms. A “word cloud” that relates the frequency of each GO term to its font size was generated for four of the five species (Figure 4, Supplementary Figure 2). X. tropicalis was excluded from this analysis due to its sparse GO term set.
Figure 4.

Word-size frequency distribution of Gene Ontology (GO) terms for the most diverged orthologs in A. carolinensis. Associated GO terms for the top 10% diverged ortholog subset are displayed according to size, based on the frequency of that term. GO terms from Biological Process (BP) and Cellular Component (CC) were used. Similar GO-based word-size frequencies based on 10% most diverged orthologs from M. musculus, G. gallus, and D. rerio are found in Supplementary Figure 2.
3. Results and Discussion
In this study, we chose five distantly related vertebrate species that fit the following criteria: (1) the presence of a well-assembled and freely available genome sequence, (2) the existence of well-curated gene models, (3) the availability of appreciable numbers of testis, ovary, and non-reproductive ESTs at dbEST, and (4) the condition that all five species, together, represent divergent clades thus presenting a deep vertebrate phylogeny with a diverse breadth of functional differences. After filtering out alignments that were of poor quality or had ambiguous orthologous relationships, a consensus tree-based off-concatenated CDS sequences from 4,986 orthologs was generated using the five vertebrate species. The tree's topology was well supported in 100% of 1000 bootstrap replicates (Supplementary Figure 1). A concatenated protein tree-demonstrated the same topology and support (not shown) and mirrored published vertebrate phylogenies (e.g., [20]). We note that these ~5,000 orthologs represent a relatively “well-behaved” and conserved gene set that do not possess paralogs in any of the five lineages. This study focuses on 1 : 1 : 1 : 1 : 1 orthologs and ignores complications arising from neo-/subfunctionalization caused by gene duplication events [21, 22], particularly those found in the zebrafish lineage after an ancient duplication event [23].
We used the extensive EST libraries publically available for each species in order to categorize genes into functional classes. Our objective was to generate a standardized sample of genes in each of the reproductive and non-reproductive functional classes, for each species. Historically, EST libraries were originally developed to assist in the genome annotation process (e.g., [24]). The quantity, quality, specificity, and tissue-diversity of EST libraries vary considerably across species (see Supplementary Table 1) and are largely a function of each research community's priorities and preferences for each of the five sequenced genomes. Since our principal objective is to compare reproductive versus. non-reproductive levels of molecular divergence in vertebrates, we sought to generate pooled gene samples derived from testis and ovary (i.e., reproductive) and non-reproductive tissue (any adult tissue that does not contain a sex-specific organ or tissue). In addition, genes from tissue-specific (or tissue-limited) classes were differentiated from “tissue-expressed” genes that are expressed more ubiquitously. This approach enables us to compare functional gene classes using relatively large sample sizes and ample statistical power.
A total of seven functional classes were assigned to genes in each of the five species (see Section 2). Table 1 summarizes the number of genes that are contained in each functional class for each species. It is important to note that the proportion of reproductive (e.g., testis, ovary) to non-reproductive genes in each species is not necessarily indicative of the total fraction of reproductive genes found in each genome but, again, reflects each community's specialized interests in generating certain libraries. In addition, overall EST library coverage can be different by an order of magnitude. For example, at last count, the mouse has nearly 5 million ESTs deposited in dbEST, while the green lizard has only 150,000 ESTs. The broader EST coverage in mouse may explain why our screen failed to identify any ovary-specific genes in this taxon. In contrast, since the anoles EST set includes only three non-reproductive tissues at a lower coverage than other species, this may also explain the relatively high number of ovary-specific genes in this species. With such large differences in EST coverage in each of the five species, it is important to understand the limits of these analyses.
Overall, our results provide evidence of a general pattern of rapid reproductive change over deep vertebrate lineages. Each of the five vertebrates demonstrate significantly higher protein divergence in gonadal genes compared to non-reproductive genes (Figure 1). Rapidly evolving testis genes appear to be driving much of the pattern of higher gonadal-specific gene divergence in these lineages: four of the five taxa—zebrafish, Xenopus, chicken, and mouse—all share significantly higher testis-specific divergence. Interestingly, these three taxa include two of the more basal taxa, Xenopus and zebrafish (Supplementary Figure 1), supporting that this pattern spans broad phylogenetic groups across the vertebrate subphylum. In green lizards, we observe a contrasting pattern of gonadal divergence as ovary-specific genes appear to be driving the significantly higher divergence of gonadal genes (but see caveat above). Thus, while we see a general pattern of significantly higher divergence among reproduction-specific genes across all vertebrate lineages, there may be large differences in the subset of reproductive genes that are diverging.
In Drosophila, we also see a similar pattern of rapidly evolving gonadal genes from EST libraries. Reproductive genes from the testis and ovary and non-reproductive genes from the brain have been used to characterize sexually dimorphic expression patterns [25–27] as well as to compare the evolution of reproductive genes relative to non-reproductive genes [28–30]. A recent study using 12 genomes in Drosophila and an extensive EST set from D. melanogaster also found that rates of evolution among testis-expressed genes are significantly higher than genes expressed in the ovary or head [12]. A number of studies in mammals have also demonstrated a similar pattern of higher divergence rates in male reproductive genes [11, 31–33].
This higher divergence of reproductive genes, and in particular, male-specific proteins, supports the hypothesis that sexual selection may be an important driver of evolutionary change and extends sexual selection theory to the level of molecules such as those found in gametogenesis and fertilization [34–36]. The strength of this molecular signature indicates the pervasive and cryptic nature of this process: much of this pattern would remain hidden without a comparative and systematic treatment of genome-wide sequence data. We also note that reproductive proteins, particularly those regulating sperm development, are of particular interest to researchers studying mechanisms of reproductive isolation because hybrid male sterility may be the product of the rapid evolution of male reproductive genes: spermatogenesis appears to be a selected target of hybrid male fertility breakdown [37–41]. In addition, there is mounting evidence that positive selection drives the evolution of genes controlling key transitions during both spermatogenesis and oogenesis [42, 43].
Other functional classes of testis-associated genes have also been found in Drosophila. Genes encoding proteins secreted by male accessory glands (Acps), the ejaculatory duct, and the ejaculatory bulb, as well as many components of D. melanogaster seminal fluid, are known to be rapidly evolving. These proteins are transferred from the male to the female along with sperm during mating and mediate a series of postmating events [44–46]. Furthermore, there is ample evidence of adaptive evolution at several loci that encode D. melanogaster seminal fluid proteins [47–52]. Whether a similar signal among secretory reproductive classes is found in vertebrate lineages is an intriguing question.
While a clear pattern of rapid testis-specific divergence emerges from our lineage-specific annotations, we then asked whether the same evolutionary pattern holds across genes that have maintained a similar function across all five vertebrate species. In other words, what are the relative rates of evolutionary change across functionally conserved classes? Surprisingly, the numbers of genes per class were drastically reduced to the point that only four classes—testis-expressed, ovary-expressed, gonadal-expressed, and non-reproductive genes—share genes in common across all five species (Figure 2). Furthermore, a decreasing but nonsignificant trend of evolutionary rates was found among these four functional classes: dNnon-reproductive > dNgonad-expressed > dNovary-expressed > dNtestis-expressed. Overall, this functionally conserved group describes a subset of the data with a contrasting evolutionary pattern, thereby demonstrating that testis-specific genes are affected by a variety of evolutionary forces. In a recent study, Dean et al. [33] performed a genomic and proteomic study on six tissue types from the male reproductive tract of mouse (excluding testis) and found that one tract, the seminal vesicle, had significantly higher rates of divergence while the other five tracts showed significantly lower rates of divergence when compared to other proteins. Our results demonstrate a similar high variance of evolutionary rates within the testis.
A. carolinensis was the outlier of the five vertebrate taxa with a significantly higher divergence among ovary-specific genes. Ovaries have also been shown to be sites of rapid divergence in D. melanogaster as part of a molecular coevolutionary process between sperm and egg. A number of rapidly evolving genes have been found expressed in the female reproductive tract and potentially secreted [53] or induced in the female reproductive tract by mating [54–56]. Further characterization of the green anole genome, in addition to other Lepidosauria genomes and genomic resources that will soon be available, will allow us to address whether female lizards are indeed driving sexual selective processes and whether this is a common lineage-specific process among squamate reptiles or simply an artifact of EST functional annotation.
While aligning orthologs to ESTs offers a powerful approach for functional annotation, it is important to procure a more granular understanding of process, function and localization. Therefore, we took the 10% most diverged orthologs and associated each species' corresponding gene to its Gene Ontology (GO), namely, Biological Process (BP) and Cellular Component (CC). A word cloud in which the font size is a function of the frequency of statistically over-represented functional phrases in diverged orthologs is shown for A. carolinensis in Figure 4. GO-associated word frequencies for fish, mouse, and chicken are found in Supplementary Figure 2. The density of each word cloud for each species reflects the amount of curation effort in Gene Ontology within these species' communities. As expected, we don't see much overlap between the GO and EST approaches to functional annotation. Reproductive function is a poorly annotated ontological class harboring a level of characterization that will not substantially improve until more geneticists and molecular biologists study reproductive loci in greater detail.
4. Concluding Remarks
Patterns parsed from extant genomes can inform us about the underlying evolutionary processes that have acted upon lineages in the past. As a functional class, reproductive-specific genes are more rapidly evolving than other functional gene classes, and it appears that testis genes are driving this pattern of divergence in the majority of vertebrate lineages. This work sets the stage for a more nuanced analysis of divergence leveraged against function across diverse taxa. With more genomes and ESTs generated, greater effort can be afforded to better estimate the probability that a gene is a member of a particular functional class, even when the number of ESTs and libraries are quite different between species. Newer data types such as RNAseq will certainly help solve the sampling bias problem with better coverage and more tissues sampled. Future studies that include paralogous sequences to evaluate birth/death processes and de novo gene functionalization models (including incorporating the large number of paralogs from zebrafish) in the context of functional class will also be useful in addressing the role of reproductive genes in vertebrate evolution.
It is remarkable that across very distant phylogenetic lineages, we detect the same evolutionary patterns found among closely related species: high lineage-specific reproductive diversity and, in particular, a high variance in male reproductive characters. These parallel patterns support the contention that sexual selection on both morphological and molecular characters may be an important, common, and pervasive feature of vertebrate evolution.
Supplementary Material
A catalog of the EST libraries used in this study are listed in Supplementary Table 1.
Supplementary Figure 1 displays the five-species vertebrate phylogeny based off concatenated CDS sequences.
The word-size frequency distribution of Gene Ontology (GO) terms for the most diverged orthologs in M. musculus, G. gallus, and D. rerio are found in Supplementary Figure 2.
Acknowledgments
The authors would like to thank Dr. Tonia Hsieh (Temple University) for guidance and support during all stages of this project. They also thank Dr. Ed Braun (University of Florida) for technical advice and the Beaty Biodiversity Research Centre at the University of British Columbia for use of their SciBorg cluster. C. J. Grassa dedicates his contributions to Thomas and Sarah Grassa.
Abbreviations
- GO:
Gene ontology
- EST:
Expressed sequence tag
- CDS:
Coding sequence
- BP:
Biological process
- CC:
Cellular component
- dN:
Nonsynonymous substitutions per nonsynonymous site.
References
- 1.Linnaeus C. Systema Naturae. Nieuwkoop, The Netherlands: 1735. reprinted in 1964 by B. de Graaf. [Google Scholar]
- 2.Darwin CR. The Descent of Man, and Selection in Relation to Sex. London, UK: John Murray; 1871. [Google Scholar]
- 3.Eberhard WG. Sexual Selection and Animal Genitalia. Cambridge, Mass, USA: Harvard University Press; 1985. [Google Scholar]
- 4.Salzburger W, Meyer A. The species flocks of East African cichlid fishes: recent advances in molecular phylogenetics and population genetics. Naturwissenschaften. 2004;91(6):277–290. doi: 10.1007/s00114-004-0528-6. [DOI] [PubMed] [Google Scholar]
- 5.Boul KE, Funk WC, Darst CR, Cannatella DC, Ryan MJ. Sexual selection drives speciation in an Amazonian frog. Proceedings of the Royal Society B. 2007;274(1608):399–406. doi: 10.1098/rspb.2006.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harcourt AH, Gardiner J. Sexual selection and genital anatomy of male primates. Proceedings of the Royal Society B. 1994;255(1342):47–53. doi: 10.1098/rspb.1994.0007. [DOI] [PubMed] [Google Scholar]
- 7.Roux J, Robinson-Rechavi M. Developmental constraints on vertebrate genome evolution. PLoS Genetics. 2008;4(12) doi: 10.1371/journal.pgen.1000311. Article ID e1000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korber B, LaBute M, Yusim K. Immunoinformatics comes of age. PLoS Computational Biology. 2006;2(6, article e71) doi: 10.1371/journal.pcbi.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swanson WJ, Vacquier VD. The rapid evolution of reproductive proteins. Nature Reviews Genetics. 2002;3(2):137–144. doi: 10.1038/nrg733. [DOI] [PubMed] [Google Scholar]
- 10.Shirangi TR, Dufour HD, Williams TM, Carroll SB. Rapid evolution of sex pheromone-producing enzyme expression in Drosophila. PLoS Biology. 2009;7(8) doi: 10.1371/journal.pbio.1000168. Article ID e1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo-Davis CI, Kondrashov FA, Hartl DL, Kulathinal RJ. The functional genomic distribution of protein divergence in two animal phyla: coevolution, genomic conflict, and constraint. Genome Research. 2004;14(5):802–811. doi: 10.1101/gr.2195604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haerty W, Jagadeeshan S, Kulathinal RJ, et al. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics. 2007;177(3):1321–1335. doi: 10.1534/genetics.107.078865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felsenstein J. PHYLIP (Phylogeny Inference Package) Version 3.6. Seattle, Wash, USA: Department of Genome Sciences, University of Washington; 2005. [Google Scholar]
- 15.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular Biology and Evolution. 1986;3(5):418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 16.Korber B. HIV signature and sequence variation analysis. In: Rodrigo AG, Learn GH, editors. Computational Analysis of HIV Molecular Sequences. chapter 4. Dodrecht, The Netherlands: Kluwer Academic; 2000. pp. 55–72. [Google Scholar]
- 17.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 18.Bauer DF. Constructing confidence sets using rank statistics. Journal of the American Statistical Association. 1972;67:687–690. [Google Scholar]
- 19.Castillo-Davis CI, Hartl DL. GeneMerge—post-genomic analysis, data mining, and hypothesis testing. Bioinformatics. 2003;19(7):891–892. doi: 10.1093/bioinformatics/btg114. [DOI] [PubMed] [Google Scholar]
- 20.Hedges SB. Molecular evidence for the origin of birds. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(7):2621–2624. doi: 10.1073/pnas.91.7.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154(1):459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 23.Taylor JS, Van de Peer Y, Braasch I, Meyer A. Comparative genomics provides evidence for an ancient genome duplication event in fish. Philosophical Transactions of the Royal Society B. 2001;356(1414):1661–1679. doi: 10.1098/rstb.2001.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Research. 2000;10(4):483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrews J, Bouffard GG, Cheadle C, Lü J, Becker KG, Oliver B. Gene discovery using computational and microarray analysis of transcription in the Drosophila melanogaster testis. Genome Research. 2000;10(12):2030–2043. doi: 10.1101/gr.10.12.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parisi M, Nuttall R, Edwards P, et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biology. 2004;5(6, article R40) doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh RS, Kulathinal RJ. Male sex drive and the masculinization of the genome. BioEssays. 2005;27(5):518–525. doi: 10.1002/bies.20212. [DOI] [PubMed] [Google Scholar]
- 28.Coulthart MB, Singh RS. High level of divergence of male-reproductive-tract proteins, between Drosophila melanogaster and its sibling species, D. simulans. Molecular Biology and Evolution. 1988;5(2):182–191. doi: 10.1093/oxfordjournals.molbev.a040484. [DOI] [PubMed] [Google Scholar]
- 29.Civetta A, Singh RS. High divergence of reproductive tract proteins and their association with postzygotic reproductive isolation in Drosophila melanogaster and Drosophila virilis group species. Journal of Molecular Evolution. 1995;41(6):1085–1095. doi: 10.1007/BF00173190. [DOI] [PubMed] [Google Scholar]
- 30.Jagadeeshan S, Singh RS. Rapidly evolving genes of Drosophila: differing levels of selective pressure in testis, ovary, and head tissues between sibling species. Molecular Biology and Evolution. 2005;22(9):1793–1801. doi: 10.1093/molbev/msi175. [DOI] [PubMed] [Google Scholar]
- 31.Good JM, Nachman MW. Rates of protein evolution are positively correlated with developmental timing of expression during mouse spermatogenesis. Molecular Biology and Evolution. 2005;22(4):1044–1052. doi: 10.1093/molbev/msi087. [DOI] [PubMed] [Google Scholar]
- 32.Turner LM, Chuong EB, Hoekstra HE. Comparative analysis of testis protein evolution in rodents. Genetics. 2008;179(4):2075–2089. doi: 10.1534/genetics.107.085902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dean MD, Clark NL, Findlay GD, et al. Proteomics and comparative genomic investigations reveal heterogeneity in evolutionary rate of male reproductive proteins in mice (Mus domesticus) Molecular Biology and Evolution. 2009;26(8):1733–1743. doi: 10.1093/molbev/msp094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Civetta A, Singh RS. Sex-related genes, directional sexual selection, and speciation. Molecular Biology and Evolution. 1998;15(7):901–909. doi: 10.1093/oxfordjournals.molbev.a025994. [DOI] [PubMed] [Google Scholar]
- 35.Singh RS, Kulathinal RJ. Sex gene pool evolution and speciation: a new paradigm. Genes and Genetic Systems. 2000;75(3):119–130. doi: 10.1266/ggs.75.119. [DOI] [PubMed] [Google Scholar]
- 36.Kulathinal RJ, Singh RS. The nature of genetic variation in sex and reproduction-related genes among sibling species of the Drosophila melanogaster complex. Genetica. 2004;120(1–3):245–252. doi: 10.1023/b:gene.0000017645.84748.dd. [DOI] [PubMed] [Google Scholar]
- 37.Kulathinal RJ, Singh RS. Cytological characterization of premeiotic versus postmeiotic defects producing hybrid male sterility among sibling species of the Drosophila melanogaster complex. Evolution. 1998;52(4):1067–1079. doi: 10.1111/j.1558-5646.1998.tb01834.x. [DOI] [PubMed] [Google Scholar]
- 38.Michalak P, Noor MAF. Genome-wide patterns of expression in Drosophila pure species and hybrid males. Molecular Biology and Evolution. 2003;20(7):1070–1076. doi: 10.1093/molbev/msg119. [DOI] [PubMed] [Google Scholar]
- 39.Haerty W, Singh RS. Gene regulation divergence is a major contributor to the evolution of Dobzhansky-Muller incompatibilities between species of Drosophila. Molecular Biology and Evolution. 2006;23(9):1707–1714. doi: 10.1093/molbev/msl033. [DOI] [PubMed] [Google Scholar]
- 40.Kulathinal RJ, Singh RS. The molecular basis of speciation: from patterns to processes, rules to mechanisms. Journal of Genetics. 2008;87(4):327–338. doi: 10.1007/s12041-008-0055-x. [DOI] [PubMed] [Google Scholar]
- 41.Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423(6941):715–719. doi: 10.1038/nature01679. [DOI] [PubMed] [Google Scholar]
- 42.Civetta A, Rajakumar SA, Brouwers B, Bacik JP. Rapid evolution and gene-specific patterns of selection for three genes of spermatogenesis in Drosophila. Molecular Biology and Evolution. 2006;23(3):655–662. doi: 10.1093/molbev/msj074. [DOI] [PubMed] [Google Scholar]
- 43.Bauer DuMont VL, Flores HA, Wright MH, Aquadro CF. Recurrent positive selection at Bgcn, a key determinant of germ line differentiation, does not appear to be driven by simple coevolution with its partner protein bam. Molecular Biology and Evolution. 2007;24(1):182–191. doi: 10.1093/molbev/msl141. [DOI] [PubMed] [Google Scholar]
- 44.Chapman T, Davies SJ. Functions and analysis of the seminal fluid proteins of male Drosophila melanogaster fruit flies. Peptides. 2004;25(9):1477–1490. doi: 10.1016/j.peptides.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 45.Wong A, Wolfner MF. Sexual behavior: a seminal peptide stimulates appetites. Current Biology. 2006;16(7):R256–R257. doi: 10.1016/j.cub.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Ram KR, Wolfner MF. Sustained post-mating response in Drosophila melanogaster requires multiple seminal fluid proteins. PLoS Genetics. 2007;3(12, article e23) doi: 10.1371/journal.pgen.0030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aguadé M, Miyashita N, Langley CH. Polymorphism and divergence in the Mst26A male accessory gland gene region in Drosophila. Genetics. 1992;132(3):755–770. doi: 10.1093/genetics/132.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsaur SC, Ting CT, Wu CI. Positive selection driving the evolution of a gene of male reproduction, Acp26Aa, of Drosophila: II. Divergence versus polymorphism. Molecular Biology and Evolution. 1998;15(8):1040–1046. doi: 10.1093/oxfordjournals.molbev.a026002. [DOI] [PubMed] [Google Scholar]
- 49.Aguadé M. Positive selection drives the evolution of the Acp29AB accessory gland protein in Drosophila. Genetics. 1999;152(2):543–551. doi: 10.1093/genetics/152.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Begun DJ, Whitley P, Todd BL, Waldrip-Dail HM, Clark AG. Molecular population genetics of male accessory gland proteins in Drosophila. Genetics. 2000;156(4):1879–1888. doi: 10.1093/genetics/156.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swanson WJ, Clark AG, Waldrip-Dail HM, Wolfner MF, Aquadro CF. Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(13):7375–7379. doi: 10.1073/pnas.131568198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mueller JL, Ravi Ram K, McGraw LA, et al. Cross-species comparison of Drosophila male accessory gland protein genes. Genetics. 2005;171(1):131–143. doi: 10.1534/genetics.105.043844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panhuis TM, Swanson WJ. Molecular evolution and population genetic analysis of candidate female reproductive genes in Drosophila. Genetics. 2006;173(4):2039–2047. doi: 10.1534/genetics.105.053611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lawniczak MKN, Begun DJ. A genome-wide analysis of courting and mating responses in Drosophila melanogaster females. Genome. 2004;47(5):900–910. doi: 10.1139/g04-050. [DOI] [PubMed] [Google Scholar]
- 55.Lawniczak MKN, Begun DJ. Molecular population genetics of female-expressed mating-induced serine proteases in Drosophila melanogaster. Molecular Biology and Evolution. 2007;24(9):1944–1951. doi: 10.1093/molbev/msm122. [DOI] [PubMed] [Google Scholar]
- 56.McGraw LA, Gibson G, Clark AG, Wolfner MF. Genes regulated by mating, sperm, or seminal proteins in mated female Drosophila melanogaster. Current Biology. 2004;14(16):1509–1514. doi: 10.1016/j.cub.2004.08.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A catalog of the EST libraries used in this study are listed in Supplementary Table 1.
Supplementary Figure 1 displays the five-species vertebrate phylogeny based off concatenated CDS sequences.
The word-size frequency distribution of Gene Ontology (GO) terms for the most diverged orthologs in M. musculus, G. gallus, and D. rerio are found in Supplementary Figure 2.