

Abstract
Mitochondrial dysfunction is a common cause of peripheral neuropathies. While the role of neuron and axonal mitochondria in peripheral nerve disease is well appreciated, whether Schwann cell (SC) mitochondrial deficits contribute to peripheral neuropathies is unclear. Here, we examine how SC mitochondrial dysfunction affects axonal survival and contributes to the decline of peripheral nerve function by generating mice with SC-specific mitochondrial deficits. These mice (Tfam-SCKOs) were produced through the tissue-specific deletion of the mitochondrial transcription factor A gene (Tfam), which is essential for mitochondrial DNA (mtDNA) transcription and maintenance. Tfam-SCKOs were viable, but as they aged, they developed a progressive peripheral neuropathy characterized by nerve conduction abnormalities as well as extensive muscle denervation. Morphological examination of Tfam-SCKO nerves revealed early preferential loss of small unmyelinated fibers followed by prominent demyelination and degeneration of larger-caliber axons. Tfam-SCKOs displayed sensory and motor deficits consistent with this pathology. Remarkably, the severe mtDNA depletion and respiratory chain abnormalities in Tfam-SCKO mice did not affect SC proliferation or survival. Mitochondrial function in SCs is therefore essential for maintenance of axonal survival and normal peripheral nerve function, suggesting that SC mitochondrial dysfunction contributes to human peripheral neuropathies.
Introduction
Peripheral neuropathies are a heterogeneous group of disorders characterized by peripheral nerve abnormalities. With an incidence of up to 8% in elderly populations, peripheral neuropathies are a common cause of substantial morbidity and constitute a significant economic and societal burden (Hughes, 2002). The etiology for these diseases is varied, ranging from vascular and metabolic irregularities to genetic mutations. Despite this apparent diversity, mitochondrial derangements have emerged as a common cause of many types of peripheral neuropathy. A number of mutations that affect mitochondrial function are now thought to be responsible for several forms of inherited neuropathies (Niemann et al., 2006). Similarly, diabetic neuropathy, the most prevalent form of peripheral nerve disease, is also associated with abnormalities in mitochondrial morphology and metabolism (Fernyhough et al., 2003). With the hope of developing new therapies, therefore, much effort is being devoted to understanding how mitochondrial deficiencies influence nerve function.
In recent years, significant progress has been made in elucidating the role of axonal mitochondria in peripheral neuropathies (Baloh, 2008). Peripheral nerve axons, however, do not function in isolation. Schwann cells (SCs), the main glial cell type in the PNS, are intimately associated with all peripheral nerve axons and are essential for their long-term preservation and survival (Chen et al., 2003; Reddy et al., 2003; Meyer zu Horste et al., 2007). Mitochondrial dysfunction in SCs is thus likely to participate in the disease progression in peripheral neuropathies. In fact, pathological mitochondria in neuropathic nerves are often localized to SCs (Schroder, 1993; Kalichman et al., 1998). The contribution of SC mitochondrial dysfunction to the pathology observed in peripheral nerve diseases, however, is poorly understood and remains largely unexplored.
With the goal of understanding whether SC mitochondrial abnormalities affect axonal survival and contribute to the decline of patients suffering from peripheral nerve diseases, we generated mice with impaired mitochondrial function exclusively in SCs. These Tfam-SCKO mice were generated through the tissue-specific deletion of the mitochondrial transcription factor A gene (Tfam), which is required for mitochondrial DNA (mtDNA) transcription and replication (Larsson et al., 1998). Here, we show that mitochondrial function in SCs is essential for maintenance of axonal survival and normal peripheral nerve function. Importantly, SC mitochondrial dysfunction alone recapitulated a number of critical features of human neuropathies. Remarkably, the severe mtDNA depletion and respiratory chain abnormalities in Tfam-SCKO mice did not affect SC survival. SC mitochondrial dysfunction, therefore, is a likely contributor to the impairment of patients suffering from peripheral neuropathies.
Materials and Methods
Matings of transgenic animals.
All animal experiments were performed in compliance with institutional animal protocols. TfamloxP/loxP mice in a pure C57BL/6 background (Larsson et al., 1998) were crossed to P0-Cre mice also in a pure C57BL/6 background (Feltri et al., 1999). Compound heterozygotes (P0-Cre+/−, Tfam+/loxP) were then backcrossed to homozygous TfamloxP/loxP to generate the Tfam-SCKO mice (P0-Cre+/−, TfamloxP/loxP) and their control (ctrl) littermates (P0-Cre−/−, TfamloxP/loxP). TfamloxP/loxP mice were also crossed to homozygosity to Rosa26-YFP reporter mice. TfamloxP/loxP/Rosa26-YFP+/+ were then crossed to P0-Cre mice using an analogous breeding scheme. TfamloxP/loxP genotyping was performed as previously described (Larsson et al., 1998).
Immunohistochemistry and cell quantification.
The following primary and secondary antibodies were used: rabbit anti-GFP (1:1000; Invitrogen), chicken anti-GFP (1:1000; Aves), rabbit anti-βIII tubulin (1:1000; Covance), rabbit anti-phosphohistone 3 (1:200; Millipore), rabbit anti-Kv1.1 (1:100; Abcam), rabbit anti-Nav1.6 (1:100; Millipore), mouse anti-Caspr (1:1000; gift from Dr. Elior Peles, Weizmann Institute of Science, Rehovot, Israel), anti-rabbit Cy3 (1:500; Jackson ImmunoResearch Laboratories), streptavidin-conjugated Cy3 (1:500; Jackson ImmunoResearch Laboratories), anti-rabbit Alexa 488 (1:500; Jackson ImmunoResearch Laboratories), and anti-chicken Alexa 488 (1:500; Jackson ImmunoResearch Laboratories).
For immunohistochemical analysis of sciatic nerves, nerves were dissected, immersion fixed in 4% paraformaldehyde for 2 h, rinsed with PBS, and cryoprotected in 30% sucrose. Sciatic nerves were then embedded in Tissue-Tek OCT compound (Sakura Finetek) and sectioned at 6 μm. For analysis of DRGs, mice were transcardially perfused with 4% paraformaldehyde. The entire spinal cord was then dissected, postfixed in PFA overnight, rinsed in PBS, and cryoprotected in 30% sucrose. After cryoprotection, DRGs were dissected, embedded in Tissue-Tek OCT compound, and sectioned at 6 μm. For analysis of footpads, footpad skin was removed from the plantar surface of the hindpaw, immersion fixed in Zamboni's fixative for 2 h, rinsed with PBS, and cryoprotected in 30% sucrose. Skin was sectioned at 30 μm in a plane perpendicular to the skin surface. For analysis of teased nerve fibers, mouse sciatic nerves were dissected and incubated in 4% paraformaldehyde in PBS for 30 min at room temperature. The nerves were washed three times in PBS for 5 min and desheathed, and bundles of nerve were dissected with fine needles in PBS on Fisherbrand Superfrost Plus microscope slides. Slides were air dried for at least 2 h at room temperature and stored at −20°C until staining.
All frozen sections were immunostained by postfixing in ice-cold acetone at −20°C for 10 min and blocking in 10% horse serum (DRG and footpads) or 5% fish skin gelatin (sciatic nerve and teased fibers) in PBS–0.2% Triton for 1 h at room temperature. Sections were then incubated with primary antibody diluted in blocking buffer overnight at 4°C. Secondary antibody incubation was performed at room temperature for 1 h also in blocking buffer. TUNEL staining was performed as previously described (Grinspan et al., 1996). After all stainings, sections were mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories) for microscopic visualization. Images were captured using an upright microscope equipped for epifluorescence microscopy (Nikon 80i; CoolSnapES camera) and were processed using MetaMorph, ImageJ, and Gimp software using global adjustments in brightness and contrast.
All cell number quantifications were performed by counting the cells of interest in four randomly selected regions in longitudinal nerve sections from each animal assessed (at least three mice per genotype at each age) at a 20× magnification. Epidermal innervation density was quantified by tracing the dermal–epidermal border and determining the number of fibers that crossed this borders as previously described (Golden et al., 2010).
Western blotting.
Sciatic nerves were isolated, desheathed in PBS, and immediately frozen in liquid nitrogen. Lysates were prepared by homogenizing the tissue by sonication in a buffer containing 150 mm sodium chloride, 50 mm HEPES, 1% NP-40, 1 mm EDTA, 1 mm sodium fluoride, 1 mm sodium orthovanadate, and Complete protease inhibitor mixture (Roche Applied Science). The lysates were clarified by centrifugation at 14,000 rpm for 10 min and quantified using the MicroBCAProtein Assay kit (Pierce). For Western blotting, the proteins were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). Membranes were blocked in 5% milk in 0.5% TBS-Tween and incubated overnight with the appropriate primary antibody. Following incubation with secondary antibodies conjugated to HRP (GE Healthcare), membranes were developed with SuperSignal West Dura substrate (Pierce). The primary antibodies used were rabbit anti-Tfam (1:4000; Dr. Nils-Goran Larsson, Max Planck Institute for Biology of Ageing, Cologne, Germany), mouse anti-Porin 31HL (ab2) (1:200; Calbiochem), and mouse anti-tubulin (1:1000; Sigma-Aldrich). The secondary antibodies used were anti-mouse and anti-rabbit HRP (1:5000; Jackson ImmunoResearch Laboratories).
RNA and DNA preparation and quantitative real-time PCR.
Sciatic nerves were harvested from Tfam-SCKO mice at different ages and immediately frozen in liquid nitrogen. For RNA isolation, tissue was homogenized in Trizol reagent (Invitrogen) and total RNA prepared according to the manufacturer's protocol. For DNA isolation, tissue was digested and DNA isolated using DNeasy Blood and Tissue kit (QIAGEN) according to the manufacturer's protocol. DNA and RNA concentration were quantified using an ND-1000 spectrophotometer (Nanodrop Technologies).
For RNA, qRT-PCR cDNA was reverse transcribed from total RNA using MMLV reverse transcriptase (Invitrogen). qRT-PCR was performed using a SYBR Green-based detection system on a 7700 Sequence Detector instrument (Applied Biosystems) as described previously (Nagarajan et al., 2001). Glyceraldehyde-3-phosphate dehydrogenase expression was used to normalize samples and obtain relative expression values that were used to calculate fold changes. mtDNA content was also quantified by qRT-PCR using a SYBR Green-based detection system on a 7700 Sequence Detector instrument (Applied Biosystems) in a similar way as described previously (Nagarajan et al., 2001). Instead of cDNA, however, 15 ng of total DNA was used per reaction. Primers that recognized a region unique to the mitochondrial genome were used to determine the mtDNA content relative to a serial dilutions standard curve. mtDNA content values were normalized to nuclear DNA content as determined by a set of primers directed to the genomic locus of Smrt1. DNA and RNA qRT-PCR primer sequences were as follows (5′–3′): Tfam: forward (F), CAGGAGGCAAAGGATGATTC; reverse (R), ATGTCTCCGGATCGTTTCAC; GAPDH: F, TGCCCCCATGTTTGTGATG; R, TGTGGTCATGAGCCCTTCC; mt-ND2: F, CGCCCCATTCCACTTCTGATTACC; R, TTAAGTCCTCCTCATGCCCCTATG; mt-Cox1: F, GAACCCTCTATCTACTATTCGG; R, CAAGTCAGTTTCCAAAGCCT; SDHB: F, TGTAGAGAAGGCATCTGTGG; R, CGTAGAAGTTACTCAAATCAGGG; mtDNA: F, AAGTCGTAACAAGGTAAGCA; R, ATATTTGTGTAGGGCTAGGG; Nuc.DNA: F, GGGTATATTTTTGATACCTTCAATGAGTTA; R, TCTGAAACAGTAGGTAGAGACCAAAGC.
Mitochondrial isolation, respiratory enzyme activity measurements, and respirometry.
For mitochondrial isolation, four sciatic nerves were dissected, desheathed, mechanically dissociated, digested with collagenase for 20 min, and pooled together in 2 ml of homogenization buffer (HM) containing 0.22 m mannitol, 70 mm sucrose, 10 mm Tris-HCl, 0.5 mm EDTA, 1 mm EGTA, and 0.5% delipidated BSA. Nerves were homogenized for ∼2 min (12 strokes) using a Teflon-glass homogenizer turning at 200 rpm. The homogenate was then centrifuged at 800 × g for 10 min. The clarified supernatant was centrifuged at 8000 × g for 10 min, and the pellet was resuspended in 1 ml of HM buffer without BSA and spun once more at 7500 × g. The resulting pellet was resuspended in 250 μl of HM buffer without BSA. The protein concentration of the resuspended mitochondrial fractions was determined using a MicroBCAProtein Assay kit (Pierce). Five micrograms of protein from these mitochondrial preps were used to measure complex II and complex IV activity spectrophotometrically as previously described (Birch-Machin and Turnbull, 2001).
For high-resolution respirometry, two sciatic nerves were dissected, briefly digested with collagenase for 10 min, and then desheathed and fluffed using a pair of fine forceps. At this point, nerve respiration was measured in a 2 ml chamber using an OROBOROS Oxygraph 2k (Oroboros) as previously described (Mancuso et al., 2010).
Surgical procedure.
Mice were anesthetized by intraperitoneal injection of 2-2-2 tribromoethanol at a dose of 500 mg/kg. The sciatic nerve was exposed at the hip and a reproducible crush injury was created by using no. 5 jeweler's forceps for 30 s. The site of injury was marked with a single 10-0 nylon epineural suture. Nerve lesions were produced on the right side and the contralateral nerve was left intact and served as the uninjured control. At the appropriate time, nerves were harvested and processed for histology.
Nerve light and electron microscopy and morphometry.
Sciatic nerves from ctrl and Tfam-SCKO mice at different ages were dissected and placed in 3% glutaraldehyde overnight. After washing with phosphate buffer, nerves were postfixed in 1% osmium tetroxide in phosphate buffer overnight at 4°C. Specimens were then dehydrated in graded alcohols and embedded in 100% epoxy (Araldite 502). One-micrometer-thick plastic-embedded sections were prepared and stained with toluidine blue for light microscopy. For electron microscopy, thin sections were prepared, stained with uranyl acetate and lead citrate, and photographed with a JEOL 1200 electron microscope. All nerves underwent qualitative assessment of neural architecture followed by detailed histomorphometric analysis performed as previously described (Hunter et al., 2007).
Nerve electrophysiology and electromyography.
Electrophysiology and electromyography were performed on mice at 2, 4, and 8 months of age using a Viking Quest electromyography machine (Nicolet) as previously described (Baloh et al., 2009). Briefly, mice were anesthetized with Avertin and placed on a heating pad. For nerve electrophysiology, subcutaneous platinum subdermal EEG electrodes (0.4 mm diameter, 12 mm length) (Viasys) were used. Stimulating electrodes were placed just above the left ankle and the left sciatic notch for nerve stimulation. Recording electrodes were placed in the footpad. Evoked compound motor action potentials were obtained using supramaximal stimulation, and distance between the two sites of stimulating electrodes was used to calculate conduction velocity. For electromyography (EMG) recordings, a 27-gauge, Teflon-coated, monopolar needle electrode with a 70 × 500 μm recording surface (PRO-37SAF; Electrode Store) was used. A 29 gauge reference needle electrode (GRD-SAF; Electrode Store) was inserted subcutaneously in close approximation to the recording electrode. A subdermal ground electrode was placed on the back. The recording electrode was inserted into the tibialis anterior or gastrocnemius/soleus muscles, and spontaneous electrical activity was recorded for 90 s.
Muscle histology.
Gastrocnemius muscles were dissected fresh and immediately frozen in isopentane cooled in liquid nitrogen. Cryostat sections of gastrocnemius muscle were cut at 10 μm onto slides and stained with hematoxylin and eosin.
Nerve cytochrome oxidase staining.
Sciatic nerves were dissected fresh, placed in Tissue-Tek OCT compound, and immediately frozen in isopentane cooled in liquid nitrogen. Tissue was then sectioned at 6 μm. Sections were placed in an incubating solution containing sucrose (100 mg/ml), DAB (3,3′-diaminobenzidine tetrahydrochloride 2) (0.6 mg/ml), sodium phosphate buffer (final concentration, 0.05 m), catalase 3 (2.6 μg/ml), and cytochrome c (1.6 mg/ml) for 60 min at room temperature. After incubation, sections were washed three times with deionized water, dehydrated in a series of ascending alcohols and cleared with xylene, and mounted using Permount.
Behavioral testing.
Behavioral testing was performed using male and female Tfam-SCKO mice and littermate controls at 2 and 4 months of age. The experimenter was blind to the genotypes of the mice during data acquisition. Noxious heat sensitivity (Hargreaves test) was determined as previously described (Golden et al., 2010). Briefly, the thermal threshold was determined by measuring the withdrawal latency to a radiant heat source applied to the plantar surface of the hindpaw in five separate trials for each hindpaw with a 15 min interval between trials. The withdrawal threshold was determined by averaging the withdrawal latency obtained in each of the trials. Mechanical sensitivity was determined using the von Frey test. Beginning with the smallest filament and continuing from smallest to largest, calibrated von Frey filaments were pressed to the plantar surface of the hindpaw until the filament just bent. The withdrawal threshold is defined as the force that produces a withdrawal response in three of five consecutive applications within one trial. The threshold was determined in three trials per hindpaw with a 15 min interval between trials. An accelerating rotarod was used to evaluate motor coordination and balance. Five consecutive acceleration trials were performed with a 5 min interval between trials as described previously (Montana et al., 2009). Muscle strength was measured using an inverted screen test as previously described (Golden et al., 2010).
Osmicated teased sciatic nerve fibers.
Sciatic nerves were dissected and immersion fixed in 3% glutaraldehyde overnight. After washing with 0.1 m phosphate buffer, nerves were incubated in 1% osmium tetroxide plus 1.5% potassium ferricyanide in 0.5 m phosphate buffer for 1 h. Nerves were washed in PBS followed by incubation in 33, 66, and 100% glycerol/PBS for 6 h each. Nerves were then treated with 0.6% Sudan Black dissolved in 70% ethanol at 25°C for 30 min, rinsed with 70% ethanol and with water, and then placed back in 100% glycerol. Nerves were teased in 100% glycerol and coverslipped for imaging.
Statistical analysis.
All values are expressed as mean ± SEM. If not stated otherwise, p values were determined by unpaired, two-tailed Student's t test. In all applicable figures, an asterisk (*) indicates statistical significance and p values are indicated in the legends. All statistical analyses were performed using Microsoft Excel 2007.
Results
Schwann cell-specific disruption of the Tfam gene
To study how mitochondrial dysfunction affects SCs and their ability to support long-term axonal survival, we generated mice with disrupted mitochondria only in SCs (Tfam-SCKOs). For this purpose, we used a previously developed mouse with loxP-flanked Tfam alleles (TfamloxP) (Larsson et al., 1998) (Fig. 1a). TFAM is a nuclearly encoded mitochondrial protein that is essential for mtDNA maintenance, copy number regulation, and transcription (Larsson et al., 1998; Ekstrand et al., 2004). Previous reports have shown that the deletion of Tfam in TfamloxP homozygous mice that express cre-recombinase in a tissue of interest results in severe tissue-specific mtDNA depletion and mitochondrial respiratory chain deficiency (Larsson et al., 1998; Wang et al., 1999; Silva et al., 2000; Sorensen et al., 2001; Wredenberg et al., 2002). This makes the tissue-specific deletion of Tfam an effective way to induce mitochondrial dysfunction in a selected population of cells.
Figure 1.
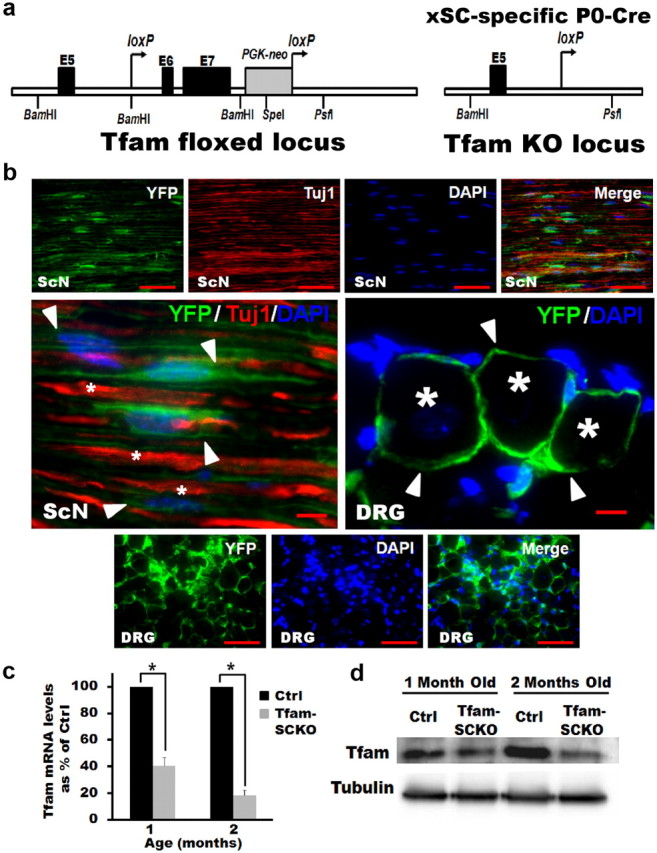
P0-Cre deletes Tfam from SCs specifically and efficiently in Tfam-SCKO mice. a, Diagram of the targeted Tfam locus in TfamloxP mice. When TfamloxP mice are crossed with mice that express Cre recombinase under the control of the P0 promoter, exons 6 and 7 of Tfam are excised to produce Tfam− mice. b, YFP fluorescence in SCs (arrowheads) of longitudinal sciatic nerve sections (ScN), as well as satellite SCs (arrowheads) in DRGs of Rosa26-YFP/P0-Cre mice. P0-Cre induces recombination specifically in SCs and excision-dependent YFP fluorescence is localized to regions of SC cytoplasm but not to axons (Tuj1 and stars; note non-overlapping Tuj1 and YFP staining) or DRG neuron cell bodies (stars). Scale bars: Top and bottom panels, 100 μm; middle magnified panel, 50 μm. c, qRT-PCR results showing efficient depletion of Tfam mRNA in Tfam-SCKO sciatic nerves. Tfam mRNA levels in the sciatic nerve of 1-month-old Tfam-SCKOs were decreased by 60% (p = 0.001, two-tailed Student's t test) compared with ctrl littermates. By 2 months of age Tfam mRNA levels were reduced by 85% (p = 0.002, two-tailed Student's t test) compared with ctrl littermates. Reported values are normalized to GAPDH. Error bars indicate SEM. n = 2 pools of 3 mice run in 3 independent experiments. d, Western blot showing efficient depletion of Tfam protein in the sciatic nerves of Tfam-SCKO mice compared with ctrl littermates by 2 months of age.
We mated TfamloxP mice to mice that express cre-recombinase under the control of the P0 promoter (P0-Cre) (Feltri et al., 1999). P0-Cre mice are well established to induce specific recombination of floxed genes in peripheral myelinating and nonmyelinating SCs starting at E13.5–E14.5, but not in any other central or peripheral neuron or glia (Feltri et al., 1999, 2002). We confirmed this selectivity by crossing P0-Cre mice with Cre-inducible Rosa26-YFP reporter animals. Indeed, yellow fluorescent protein (YFP) fluorescence was visible in most sciatic nerve SCs but not in axons or perineurial cells. Similarly, YFP could also be seen in most dorsal root ganglia SCs (satellite cells) but not in DRG neurons (Fig. 1b). In agreement with previous reports, therefore, P0-Cre induced selective recombination in most peripheral SCs.
The observed P0-Cre-mediated excision of Tfam in SCs was highly efficient. Analysis of Tfam mRNA and protein levels from Tfam-SCKO sciatic nerve extracts showed an 80% reduction compared with littermate controls (Fig. 1c,d). Considering that an estimated 15% of endoneurial cells are P0-Cre negative fibroblast and macrophages (King, 2005) and that nerve extracts also contain some perineurial and endothelial cells, the actual excision rate of Tfam in SCs was probably near complete. Given the high selectivity and efficiency of the P0-promoter driven expression of cre-recombinase, we conclude that the mating of TfamloxP to P0-Cre mice resulted in animals that lacked Tfam specifically in SCs.
SC mtDNA depletion and respiratory dysfunction in Tfam-SCKOs
To determine the functional effect of deleting Tfam in SCs, we next assessed mtDNA copy number and transcript level in Tfam-SCKO sciatic nerves. Consistent with the essential role of Tfam in the maintenance, replication, and transcription of mtDNA (Larsson et al., 1998; Ekstrand et al., 2004), as well as with previous reports describing tissue-specific Tfam knock-out mice (Larsson et al., 1998; Wang et al., 1999; Silva et al., 2000; Sorensen et al., 2001; Wredenberg et al., 2002), total mtDNA content was found to be significantly reduced in Tfam-SCKO nerves compared with control animals (Fig. 2a). Similarly, transcripts of two different mitochondrially encoded proteins (mt-ND2 and mt-Cox1) were also severely depleted in the sciatic nerves of Tfam-SCKO animals (Fig. 2b). As expected, no differences in the transcript levels of nuclearly encoded mitochondrial proteins were observed (Fig. 2b).
Figure 2.
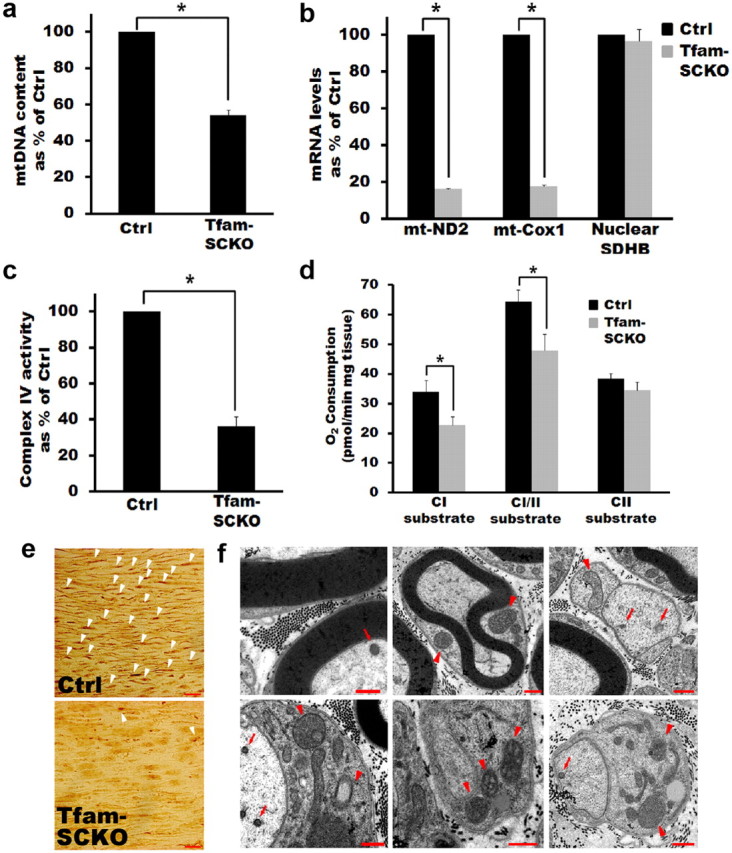
SC-specific excision of Tfam induces mtDNA and transcript depletion and respiratory dysfunction in Tfam-SCKOs. a, qRT-PCR results showing depletion of mtDNA content in the sciatic nerves of Tfam-SCKO mice compared with littermate ctrls. mtDNA content was significantly reduced by 50% (p = 0.01, two-tailed Student's t test) in 2-month-old Tfam-SCKO sciatic nerves compared with ctrl nerves. Reported values are normalized to nuclear DNA content. Error bars indicate SEM. n = 3 mice per genotype run in 2 independent experiments. b, qRT-PCR results showing depletion of mitochondrially encoded electron transport chain subunit transcripts (mt-ND2 and mt-Cox1) in Tfam-SCKO sciatic nerves. mRNA levels of these subunits were decreased 85% (p = 0.02, two-tailed Student's t test) in 2-month-old Tfam-SCKO sciatic nerves compared with ctrl littermates. In contrast, transcripts of nuclearly encoded electron transport chain subunits (nuclear SDHB) are not depleted in Tfam-SCKO sciatic nerves. Reported values are normalized to GAPDH. Error bars indicate SEM. n = 2 pools of 3 mice run in 3 independent experiments. c, Respiratory chain enzyme activity measured from mitochondria isolated from the sciatic nerves of 2-month-old Tfam-SCKO nerves and ctrl littermates. COX activity (complex IV), which contains critical mtDNA-encoded subunits, is reduced 65% (p < 0.01, two-tailed Student's t test) in Tfam-SCKO mice. Error bars indicate SEM. n = 3 pools of 2 mice per genotype. d, Mitochondrial respiration in 2-month-old Tfam-SCKO and littermate ctrl permeabilized sciatic nerves measured using high-resolution respirometry. Respiration induced by substrates delivering electrons to complex I (pyruvate plus malate) is reduced by 35% (p = 0.04, two-tailed Student's t test) in Tfam-SCKO sciatic nerves compared with littermate controls. Respiration induced by the convergent transport of electrons entering at complexes I and II (using pyruvate plus malate plus succinate as substrates) is decreased by 25% (p = 0.04, two-tailed Student's t test) in Tfam-SCKO sciatic nerves compared with littermate ctrls. Respiration induced by substrates delivering electrons to complex II alone (succinate), which is fully nuclearly encoded, was not significantly changed in Tfam-SCKO nerves compared with littermate ctrls. Error bars indicate SEM. n = 5 mice per genotype. e, COX enzymatic staining of 4-month-old ctrl and Tfam-SCKO sciatic nerves. The intense COX staining that localizes to SCs in ctrl nerves (arrowheads) is largely lost in Tfam-SCKO nerves, indicating mitochondrial dysfunction specifically in this cell type. f, Electron micrographs of Tfam-SCKO sciatic nerves show abundant abnormal enlarged mitochondria (arrowheads), often with distorted cristae. Aberrant mitochondria are mainly found in SCs, while axonal mitochondria show no morphological abnormalities (arrows). Pathological mitochondria are visible as early as 1 month in Tfam-SCKO nerves (top 3 panels) and become more abundant as these mice age (bottom panels from 4-month-old mice). Scale bars, 500 nm.
The mitochondrial genome encodes 13 subunits that are essential components of complexes I, III, IV, and V of the electron transport chain. The depletion of mtDNA and mitochondrially encoded transcripts following Tfam excision has been previously shown to induce severe respiratory chain deficiency (Larsson et al., 1998; Wang et al., 1999; Silva et al., 2000; Sorensen et al., 2001; Wredenberg et al., 2002). To examine the dysfunction of SC mitochondria in Tfam-SCKO mice, we first assessed respiratory chain enzyme activities in the sciatic nerves of 2-month-old animals. At this age, Tfam has already been efficiently excised in sciatic nerve SCs and mtDNA content and transcripts have been severely depleted (Figs. 1c,d, 2a,b). Mitochondria isolated from Tfam-SCKO sciatic nerves showed a 60% reduction in cytochrome oxidase (COX) activity (complex IV), which contains critical mtDNA-encoded subunits (Fig. 2c). As expected, the activity of succinate dehydrogenase (SDH) (complex II), which is fully nuclearly encoded, showed no significant change in mitochondria isolated from Tfam-SCKO sciatic nerves (data not shown).
To further confirm the disruption of SC mitochondria in Tfam-SCKO mice, we next used high-resolution respirometry to measure mitochondrial respiration under different substrates in permeabilized 2-month-old Tfam-SCKO sciatic nerves. Respiration induced by substrates delivering electrons to complex I (pyruvate plus malate) was significantly impaired in Tfam-SCKO sciatic nerves compared with littermate controls. Similarly, respiration induced by the convergent transport of electrons entering at complexes I and II (using pyruvate plus malate plus succinate as substrates), which best corresponds to the mitochondrial substrate supply in vivo (Gnaiger, 2009), was also significantly reduced in Tfam-SCKO sciatic nerves compared with littermate controls. These reductions in mitochondrial respiration in Tfam-SCKO nerves are likely explained by defects in complexes I, III, and IV as these complexes contain mtDNA-encoded subunits. Respiration induced by electrons entering at the level of complex II remained unchanged in Tfam-SCKO nerves, which is explained by the fact that complex II contains only nuclearly encoded subunits (Fig. 2d).
It should be emphasized that, while the functional deficits described above were measured from whole Tfam-SCKO sciatic nerves, they are a reflection of the mtDNA depletion and mitochondrial dysfunction taking place specifically in SCs. The high specificity of the P0-cre-mediated excision of Tfam ensures this (Fig. 1b). Moreover, COX enzymatic staining, which reflects respiratory chain activity in individual cells, showed reduced staining intensity in the cell bodies of SCs in Tfam-SCKO sciatic nerves (Fig. 2e). Finally, electron microscopy of Tfam-SCKO nerves revealed abundant abnormal, enlarged mitochondria, sometimes with distorted cristae, specifically in SC but not in axons (Fig. 2f). Together, our results confirm that, by deleting Tfam specifically in SCs, we were able to generate a mouse with disrupted mitochondrial metabolism only in peripheral glia.
Tfam-SCKOs develop a progressive degenerative peripheral neuropathy
Despite severe mtDNA and transcript depletion as well as respiratory chain dysfunction in their SCs, Tfam-SCKO animals were born at the expected Mendelian ratios and survived to at least 12 months of age (the longest a Tfam-SCKO was allowed to survive before it was killed). Tfam-SCKO animals developed normally and were indistinguishable from their control littermates up to 3 months of age. Starting at 3–4 months of age, however, Tfam-SCKOs showed signs of muscle weakness and gait abnormality. Tfam-SCKOs exhibited abnormal limb clasping, mild tremors, and impaired coordination, and developed a characteristic “swimming gait.” These deficits progressively worsened so that, by 8 months of age, most Tfam-SCKOs were often unable to support themselves on their hind legs (Fig. 3a). Such motor abnormalities are indicative of peripheral nerve disease and have been previously described in other mouse models of peripheral neuropathy (Le et al., 2005; Ryu et al., 2007; Baloh et al., 2009).
Figure 3.
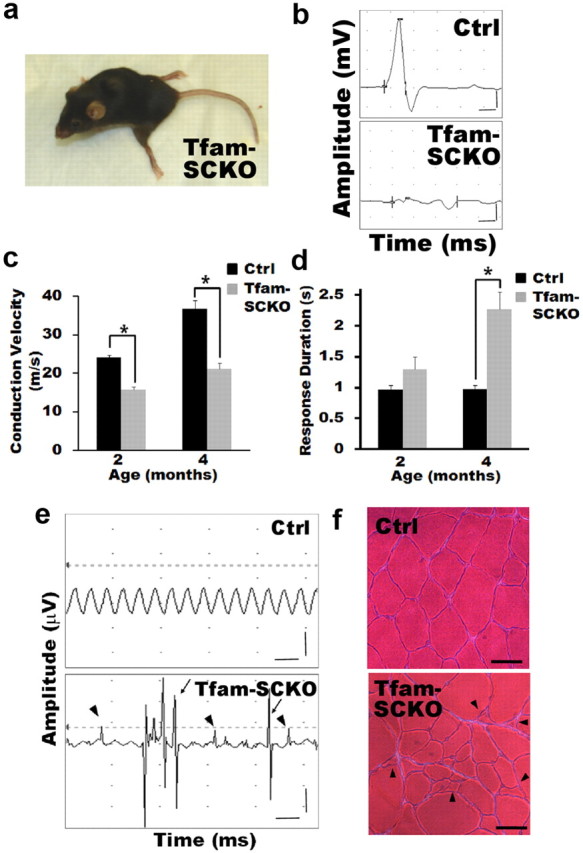
Tfam-SCKOs develop a progressive degenerative peripheral neuropathy. a, Photograph of end-stage Tfam-SCKO mouse (≈8 months of age). At this stage, Tfam-SCKOs display a characteristic swimming gait and are unable to support themselves on their hind legs. b, Typical nerve conduction trace from a 4-month-old Tfam-SCKO mouse showing marked temporal dispersion. Calibration: 1 ms, 1 mV. c, d, Electrophysiological studies of Tfam-SCKO mice show that, as early as 2 months of age, these mice have significantly reduced nerve conduction velocity (c) (p < 0.001, two-tailed Student's t test) compared with littermate ctrls. This deficit persists in 4-month-old animals (c) (p < 0.001, two-tailed Student's t test), which also have severe temporal dispersion as shown by an increased response duration (d) (p < 0.001, two-tailed Student's t test). These findings indicate segmental demyelination. Error bars indicate SEM. N = 4 mice per genotype at each age. e, Typical electromyography traces from a 4-month-old Tfam-SCKO mouse showing fibrillation (arrowheads) and fasciculations (arrows), common findings in diseases involving motor axon loss, indicative of muscle fiber denervation and motor unit degeneration/regeneration. These findings were never present in ctrl littermates. Calibration: 50 ms, 500 μV. f, Hematoxylin and eosin stain of the gastrocnemius muscle from a ctrl and a Tfam-SCKO mouse showing scattered and grouped muscle fiber atrophy (arrowheads). This is characteristic of motor axon degeneration. Scale bar, 50 μm.
Reduced peripheral nerve conduction velocity is a hallmark and diagnostic marker of diabetic as well as other mitochondria-related neuropathies (Arezzo and Zotova, 2002). To further characterize the peripheral nerve deficits in Tfam-SCKOs, we performed nerve conduction studies both on presymptomatic and symptomatic mice. Nerve conduction velocity was significantly reduced in Tfam-SCKO animals as early as 2 months of age and this deficit persisted over time (Fig. 3c). Moreover, Tfam-SCKO mice displayed marked temporal dispersion at 4 months of age (Fig. 3b,d). Both these findings are suggestive of a progressive demyelinating phenotype in Tfam-SCKO mice.
Interestingly, EMG of Tfam-SCKOs showed occasional fibrillation potentials and fasciculations at 2–3 months of age, which became prominent at 4 months and continued to worsen with increasing age (Fig. 3e) (data not shown). These EMG findings reflect loss of muscle fiber innervation and spontaneous firing of motor units, and are often seen in human nerve diseases involving motor axon loss. Thus, our EMG results indicated a progressive axonal degeneration phenotype in Tfam-SCKO mice. This was confirmed by muscle histology, which revealed abundant denervated atrophic muscle fibers (Fig. 3f). SC-specific mitochondrial dysfunction caused, therefore, a progressive peripheral neuropathy characterized by both demyelination as well as axonal degeneration.
Tfam-SCKOs display early preferential loss of small unmyelinated fibers
To examine in detail the peripheral nerve abnormalities in Tfam-SCKOs, we performed histological and electron microscopy analysis of Tfam-SCKO sciatic nerves at different ages. Tfam-SCKO nerves initially developed normally, and for the first 2–3 weeks of life there were no obvious differences between ctrl and Tfam-deficient nerves (see Notes). As early as 1 month of age, however, Tfam-SCKO nerves started to display signs of pathology. Interestingly, we found that these early abnormalities preferentially affected, and were initially limited to, small-caliber unmyelinated fibers (C-fibers) (Fig. 4a–e).
Figure 4.
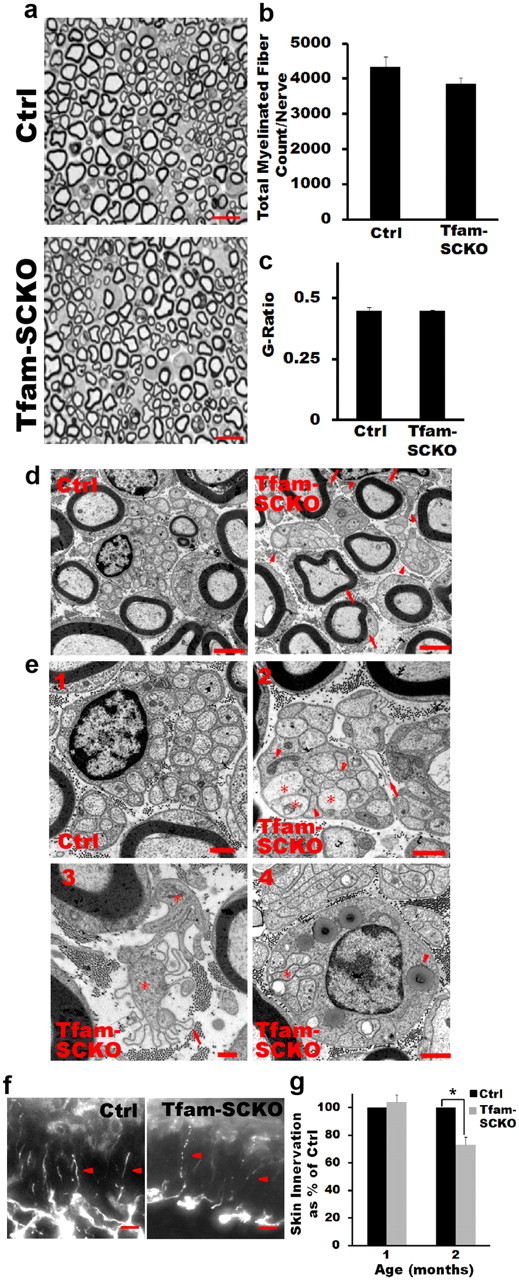
Tfam-SCKOs display early preferential loss of small unmyelinated fibers: a–c, Toluidine blue-stained plastic sections of 1-month-old Tfam-SCKO sciatic nerve and littermate ctrls (a) show that at this age there are no differences in the number of myelinated fibers (b) or the extent of myelination (g-ratio, axon area/fiber area) (c). Scale bar, 25 μm. Error bars indicate SEM. n = 4 mice per genotype. d, Electron micrographs of 1-month-old ctrl and Tfam-SCKO sciatic nerves show that, while large myelinated fibers are mostly normal in Tfam-SCKO mice at this age (arrows), the structure of Remak bundles (arrowheads) is largely abnormal. Scale bar, 2 μm. e1–e4, Compared with ctrl littermates (e1), unmyelinated axons in Tfam-SCKO Remak bundles are found touching one another (e2, e4, stars) and interspersed by pathological SC processes known as bands of Bungner (e2, arrowheads). Free-floating excess basement membrane (e2, e3, arrows) enclosing SC cytoplasm (e3, star) with few or no axons left, indicative of extensive degeneration of unmyelinated axons, is also visible. By 2 months of age, most nonmyelinating SCs are associated with unstructured, degenerating Remak bundles, containing abnormal axons (e4, star) and filled with phagocytic vesicles and membranous debris (e4, arrowhead). Scale bar, 500 nm. f, Representative photographs of epidermal fiber innervation (arrowheads) in the footpads of 2-month-old Tfam-SCKO and littermate ctrl mice. Scale bar, 10 μm. g, Quantification of fiber density in the epidermis shows a 30% decrease in Tfam-SCKO by 2 months of age (p = 0.02, two-tailed Student's t test), indicating early degeneration of small unmyelinated C-fibers. Results reported are normalized to per unit area. Error bars indicate SEM. n = 3 mice per genotype at each age.
Small-caliber fibers are normally associated with nonmyelinating SCs, such that a single SC wraps around multiple axons to form a Remak bundle. The Remak bundle is delimited by a basement membrane and individual axons are kept separate from one another inside it by a thin extension of SC cytoplasm (Fig. 4e1). At 1 month, the structure of Remak bundles in Tfam-SCKO nerves was clearly disrupted (Fig. 4d,e). Unmyelinated axons within a Remak bundle were often found touching one another and interspersed by pathological SC processes known as bands of Bungner (Fig. 4e2). Moreover, degeneration of unmyelinated axons was extensive and evidenced by free-floating, excess basement membrane surrounding empty spaces previously occupied by axon bundles (Fig. 4e2,e3). Precise quantification of small fiber loss in the nerves of Tfam-SCKO mice, however, was not possible because of the difficulty of distinguishing abnormal unmyelinated axons from SC processes forming bands of Bungner. Note that, at this age (1 month old), large-caliber myelinated fibers (A fibers) in Tfam-SCKO nerves appeared normal (Fig. 4a–c).
The unmyelinated fibers in Tfam-SCKO nerves quickly deteriorated, and by 2 months of age, most nonmyelinating SCs were associated with unstructured, degenerating Remak bundles. These bundles contained few intact axons and were often filled with phagocytic vesicles and membranous debris, indicative of axonal degeneration (Fig. 4e4). We confirmed the early degeneration of unmyelinated fibers observed in Tfam-SCKO sciatic nerves by quantifying C-fiber density in the epidermis. Somatic C-fibers are primarily high-threshold nociceptors that terminate in the epidermis (Kennedy and Wendelschafer-Crabb, 1993). In neuropathies characterized by loss of small unmyelinated fibers, skin denervation is one of the initial signs of pathology and correlates with disease severity (Ebenezer et al., 2007). While we did not observe differences in epidermal fiber density at 1 month of age, we found a significant decrease in skin innervation in 2-month-old Tfam-SCKO mice (Fig. 4f,g). This was again in contrast to large myelinated fibers, which at this age were just beginning to show initial signs of pathology (Fig. 5a). Overall numbers of dorsal root ganglion neurons in L3, whose axons project to the sciatic nerve, were comparable in 2-month-old Tfam-SCKO and ctrl mice (average total number of neurons: ctrl, 7048 ± 186; Tfam-SCKO, 7381 ± 211.3; n = 3 mice per genotype). Similarly, TUNEL and cleaved caspase 3 immunostaining of the dorsal root ganglion neurons whose axons project to the sciatic nerve (levels L3–L5) showed no signs of cell death in either 2- or 4-month-old Tfam-SCKOs (data not shown). The degeneration of small unmyelinated fibers in the nerves of Tfam-SCKO mice was, therefore, a primary pathological event and was not secondary to neuronal death.
Figure 5.
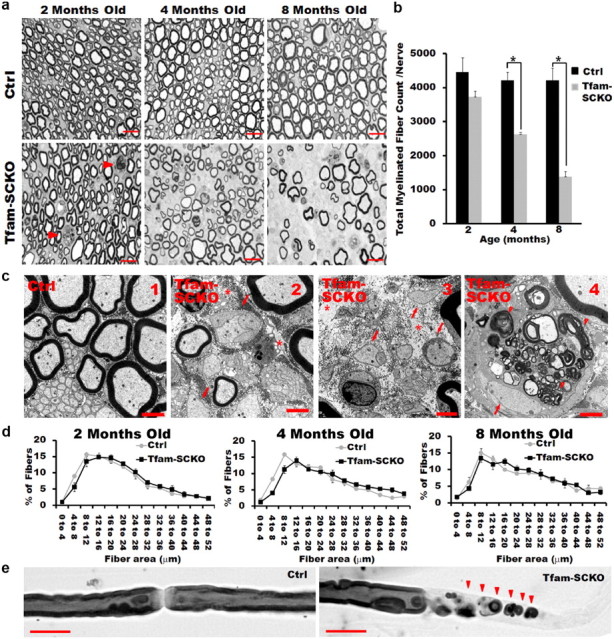
Degeneration of large-caliber myelinated fibers in older Tfam-SCKOs. a, b, Toluidine blue-stained plastic sections of Tfam-SCKO sciatic nerves and littermate ctrls at different ages (a) and quantification of total number of myelinated profiles per nerve (b). At 2 months, some initial signs of axonal degeneration are apparent (arrowhead) in Tfam-SCKO nerves, but at this age there are no differences in the number of myelinated fibers (b). By 4 months, there is prominent axonal degeneration, as shown by a significant decrease (p > 0.001, two-tailed Student's t test) in the total number of myelinated profiles in Tfam-SCKO nerves. Axonal degeneration worsens by 8 months of age (p > 0.001, two-tailed Student's t test). Scale bar, 25 μm. Error bars indicate SEM. n = 4 mice per genotype at each age. c1–c4, Electron micrographs of Tfam-SCKO nerves at different ages confirm that large myelinated fiber degeneration becomes prominent by 4 months of age (c2) and worsens by 8 months (c3), as shown by large portions of endoneurium completely devoid of axons (stars). By 4 months of age, actively demyelinating and degenerating axons filled with membranous debris (arrowheads) are also common in Tfam-SCKO nerves (c4). By this age, segmental demyelination as shown by large-caliber axons without myelin is also prominent (c2–c4, arrows). This type of pathology is not visible in ctrl nerves (c1). Scale bar, 2 μm. d, Fiber size distribution analysis in Tfam-SCKO and ctrl littermate nerves at different ages. No differences in fiber size distribution are evident despite severe axonal degeneration, indicating that myelinated fibers of all calibers are susceptible to degeneration. Error bars indicate SEM. n = 4 mice per group at each age. e, Osmicated teased fibers from 4-month-old Tfam-SCKO nerves confirm the presence of segmental demyelination. Scale bar, 25 μm.
Together, our results show that Tfam-SCKO mice initially suffered early preferential loss of small unmyelinated fibers, suggesting that these fibers are most sensitive to SC mitochondrial dysfunction. The temporal progression of fiber loss in Tfam-SCKO nerves is particularly interesting because early preferential loss of small unmyelinated fibers has been reported in polyneuropathies associated with diabetes and HIV/AIDS (Kennedy et al., 1996; McArthur et al., 2005), diseases in which mitochondrial dysfunction is known to occur (Dalakas et al., 2001; Fernyhough et al., 2003).
Loss of large-caliber myelinated fibers in older Tfam-SCKOs
While Tfam-SCKOs suffered an early preferential loss of small unmyelinated fibers, these mice also developed deficits in large-caliber myelinated axons as they aged. Toluidine blue-stained plastic sections of 2-month-old Tfam-SCKO sciatic nerves showed early signs of axonal degeneration (Fig. 5a). This observation was confirmed by electron microscopic examination, which revealed a few axons undergoing active demyelination and Wallerian degeneration (data not shown). Note, however, that at this age there was not a significant decrease in the number of myelinating profiles in Tfam-SCKO nerves (Fig. 5b).
Severe axonal loss became prominent in Tfam-SCKOs by 4 months (Fig. 5a–c). At this age, the number of myelinating profiles in Tfam-SCKO nerves was greatly reduced compared with control littermates (Fig. 5b). Degeneration of myelinated axons continued to progress as Tfam-SCKO mice aged, and by 8 months of age the number of myelinating profiles in these animals was <40% that of control littermates (Fig. 5a–c3). Electron micrographs confirmed the severity of the axonal degeneration in Tfam-SCKO nerves, revealing large portions of endoneurium completely devoid of axons (Fig. 5c). Similarly, numerous axons undergoing degeneration were also visible. Detailed morphometric analysis (Hunter et al., 2007) showed no differences in the size distribution of myelinated axons in Tfam-SCKO nerves compared with control littermates at different ages, indicating that myelinated fibers of all calibers were susceptible to degeneration (Fig. 5d). As in the case of small unmyelinated fibers, the degeneration of myelinated fibers was not secondary to neuronal death since no TUNEL- or cleaved caspase 3-positive motor neurons were observed in the spinal cords of either 2- or 4-month-old Tfam-SCKOs (data not shown).
At 4 months, demyelination was also widespread in Tfam-SCKO nerves. At this age, large-caliber axons without any myelin were a prominent pathological feature (Fig. 5c). Examination of osmicated teased fibers from 4-month-old Tfam-SCKO nerves confirmed the presence of segmental demyelination (Fig. 5e). Consistent with demyelination, SCs actively digesting myelin debris were present in Tfam-SCKO nerves at this age (Fig. 5c4). Occasionally, axons surrounded by abnormally thin myelin were also observed. Thin myelin was, however, not characteristic, and in fact there was no difference in the overall g-ratio between Tfam-SCKO and control nerves at different ages (data not shown). Overall, these results show that extensive axonal degeneration and demyelination ensue when SC mitochondria are disrupted. Normal SC mitochondrial function is therefore essential for maintaining the interaction between myelinating SCs and axons.
Tfam-SCKOs have behavioral deficits consistent with early loss of unmyelinated fibers followed by extensive degeneration of myelinated axons
To determine the physiological effects and confirm the temporal progression of the pathology observed in Tfam-SCKO nerves, we performed a battery of sensorimotor tests in 2- and 4-month-old animals. Behavioral tests that primarily assess the function of small unmyelinated C-fibers were expected to show deficits in both 2- and 4-month-old Tfam-SCKO mice. However, behavioral paradigms looking mainly at the function of large myelinated fibers were predicted to show differences only in 4-month-old Tfam-SCKO mice.
Small unmyelinated C-fibers have a primary role in nociception. To examine the physiological effect of the early C-fiber loss observed in Tfam-SCKOs, we assessed their sensitivity to a noxious heat stimulus. When a noxious radiant heat stimulus was applied to the hindpaw (Hargreaves test), Tfam-SCKOs displayed a longer withdrawal latency than their control littermates. As expected, this reduced sensitivity to noxious heat stimuli was detectable in 2-month-old animals and persisted as these mice aged (Fig. 6a).
Figure 6.

Tfam-SCKOs have behavioral deficits consistent with early loss of unmyelinated fibers followed by extensive degeneration of myelinated axons. a, Two-month-old Tfam-SCKOs are less sensitive to an applied noxious heat stimulus than ctrl littermates (p < 0.01, two-tailed Student's t test), as shown by an increased withdrawal latency, which indicates loss of unmyelinated C-fiber nociceptors. This deficiency persists in 4-month-old Tfam-SCKO mice (p < 0.01, two-tailed Student's t test). b–d, At 2 months of age, consistent with the early preservation of large myelinated motor axons, Tfam-SCKOs do not display significant motor deficits as shown by the inverted screen test (b) or accelerating rotarod (c). Four-month-old Tfam-SCKOs, however, spend significantly less time on an inverted screen (p < 0.01, two-tailed Student's t test) (b) and accelerating rotarod (p < 0.01, two-tailed Student's t test) (d), motor deficits that indicate the loss of large myelinated fibers by this age. Error bars indicate SEM. N = 4 mice per genotype at each age.
We also tested innocuous mechanical sensitivity in 2-month-old Tfam-SCKOs using the von Frey test. Innocuous mechanical sensitivity is another form of somatosensation, but, unlike nociception, it is primarily mediated by myelinated fibers. Interestingly, 2-month-old Tfam-SCKO animals showed no deficits in the von Frey test compared with wild-type littermate controls (average withdrawal threshold: ctrl, 0.86 ± 0.08 g; Tfam-SCKO, 1.0 ± 0.05 g; n = 4 mice per genotype). The results from these somatosensory studies supported the pathological observations indicating early preferential loss of small unmyelinated fibers in Tfam-SCKO mice.
Given that the axons of motor neurons are large myelinated fibers, we next determined whether the pathology observed in Tfam-SCKO nerves caused any abnormalities in motor function. As predicted, no motor deficits were detected in 2-month-old Tfam-SCKOs (Fig. 6b,c). Four-month-old Tfam-SCKO animals, however, spent significantly less time on an accelerating rotarod (Fig. 6d) as well as on an inverted screen (Fig. 6b), indicating abnormal coordination and muscle weakness. The results from these motor studies are consistent with the observation that large-caliber axons are not affected in Tfam-SCKO mice until later in the disease. Together, our results confirm that Tfam-SCKO mice initially suffered early preferential loss of small unmyelinated fibers followed by extensive loss of large-caliber myelinated axons.
SC mitochondrial dysfunction does not compromise SC survival
To determine whether the axonal loss and demyelination observed in Tfam-SCKO animals was simply the result of SC death, we assessed changes in SC numbers in the nerves of these animals. Remarkably, we found no differences in the number of SC nuclei between Tfam-SCKO sciatic nerve and littermate controls up to 8 months of age (Fig. 7a,b). Moreover, by crossing Tfam-SCKO mice to Cre-inducible Rosa26-YFP reporter animals, we were able to confirm that, at all ages examined, Tfam-deficient SCs present in Tfam-SCKO nerves (identified by their excision-dependent YFP fluorescence) remained viable (Fig. 7a,c). These results indicate that the survival of Tfam-deficient SCs was not compromised despite severe mtDNA depletion and respiratory chain abnormalities.
Figure 7.
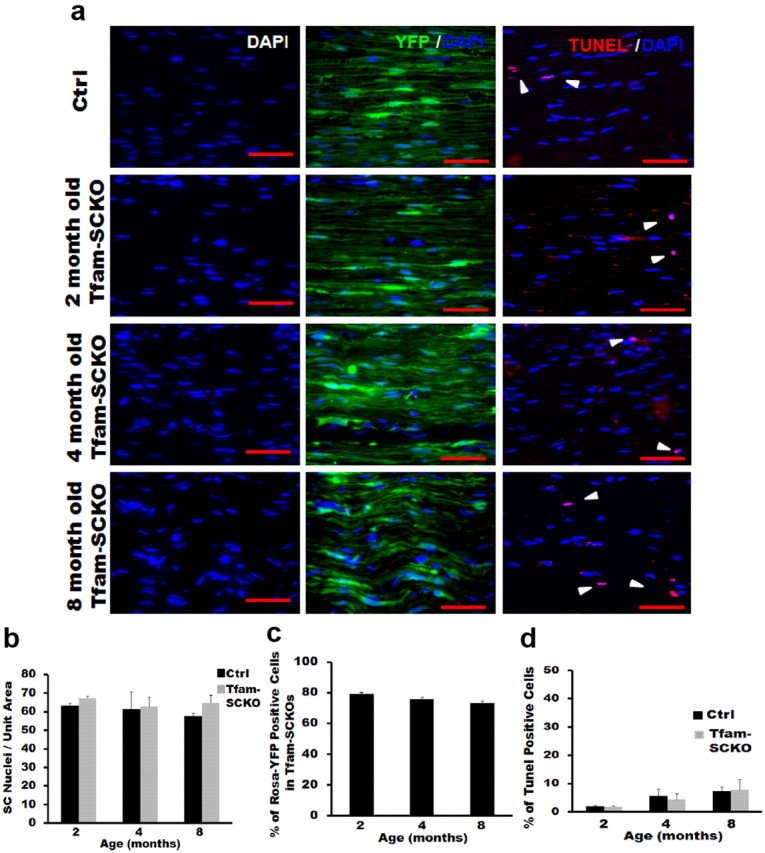
SC mitochondrial dysfunction does not compromise SC survival. a, b, Nuclear staining (a, DAPI) and quantification (b) shows that there is no difference in the number of SC nuclei between Tfam-SCKO and ctrl sciatic nerves at different ages. a, c, Quantification of Rosa-YFP immunofluorescence (a, YFP/DAPI; c) in Tfam-SCKO/Rosa-YFP nerves at different ages shows that the majority of SCs are Tfam-deficient at all ages. a, d, Quantification of TUNEL immunohistochemistry (a, TUNEL/DAPI; d) of Tfam-SCKO and ctrl littermate shows no differences in the number of TUNEL-positive cells at different ages, indicating that SCs are able to survive despite the disruption in their mitochondria. Scale bar, 100 μm. Error bars indicate SEM. n = 3 mice per genotype at each age.
Consistent with the viability of Tfam-SCKO SCs, we did not detect any differences in TUNEL staining, a marker of cell death, between Tfam-SCKO sciatic nerve and controls at different ages (Fig. 7a,d). Similarly, staining with an antibody that recognizes the proliferation marker phosphohistone 3 showed no changes in proliferation that could compensate for the potential loss of Tfam-deficient SCs in Tfam-SCKO nerves (data not shown). The pathology observed in Tfam-SCKOs, therefore, was not due to the loss of SCs. Together, our findings indicate that normal mitochondrial function is not required for SC survival, but is essential for maintenance of axonal survival and normal peripheral nerve function.
Tfam-deficient SCs support axonal regeneration but fail to remyelinate following injury
Schwann cells play a critical role in promoting axonal regeneration following peripheral nerve injury. Upon degeneration of peripheral axons, SCs in the distal stump undergo an orchestrated process that helps support axonal regeneration (Geuna et al., 2009). To determine whether disrupting mitochondrial function affects the ability of SCs to support axonal regeneration, we studied the response of these cells after peripheral nerve crush injury in 2-month-old ctrl and Tfam-SCKO mice. At this age, the peripheral nerves of Tfam-SCKOs show modest pathology that is mainly limited to unmyelinated fibers yet mitochondrial function has already been severely disrupted (see above).
We first looked at the initial response of SCs to axonal degeneration by assessing changes in SC proliferation and in their ability to break down existing myelin. Within the first few days after losing contact with axons, SCs dedifferentiate and start to proliferate (Geuna et al., 2009). Immunostaining of nerves harvested 4 d after injury distal to the site of crush using a phosphohistone 3 antibody showed no impairment in the ability of Tfam-deficient SCs to proliferate following axonal loss (Fig. 8a,b). Nerve regeneration also depends on the ability of SCs to break down existing myelin and remove the resulting debris. This process starts immediately after injury and takes place over a period of 3–6 weeks (Geuna et al., 2009). We did not observe differences in the amount of myelin debris present in ctrl and Tfam-SCKO nerves 3 weeks after injury, suggesting that disruption of mitochondrial function in SCs does not interfere with their ability to break down and remove myelin following axonal degeneration (Fig. 8c).
Figure 8.
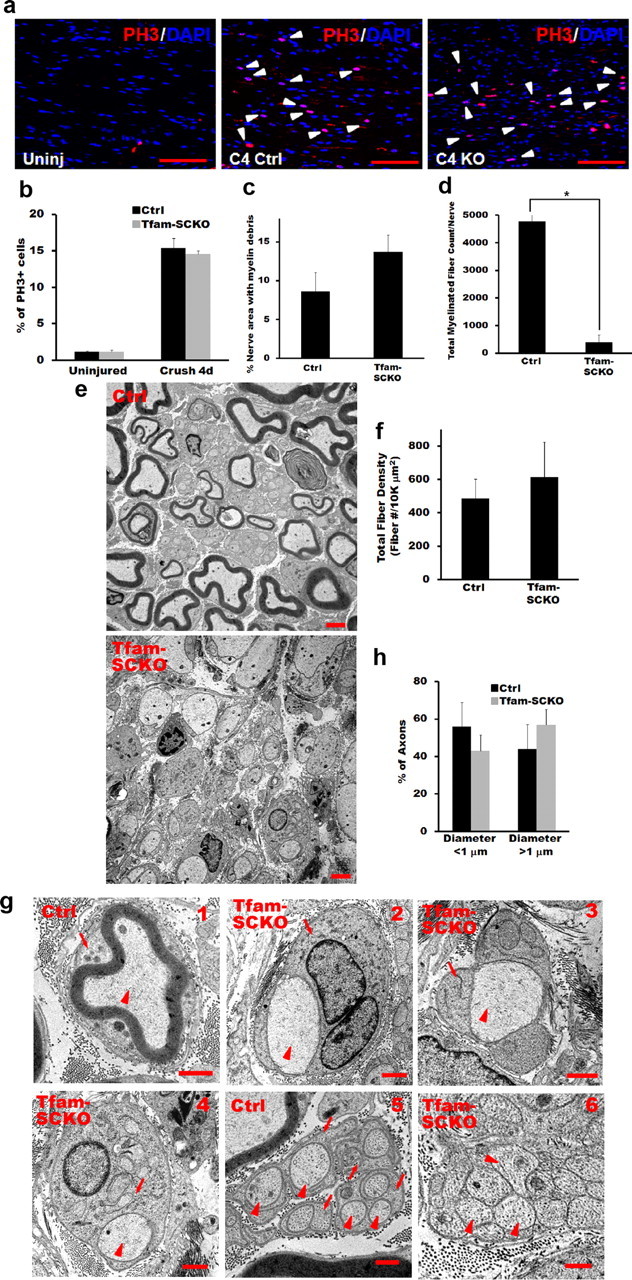
Tfam-deficient SCs support axonal regeneration but fail to remyelinate following injury. a, b, Phosphohistone 3 (PH3) immunostaining (a) and quantification (b) show that there is no difference in the number of proliferating SCs (arrowheads) between Tfam-SCKO and ctrl sciatic nerves 4 d after crush injury. Scale bar, 100 μm. Error bars indicate SEM. n = at least 3 mice per genotype. c, Quantification of the amount of myelin debris present in ctrl and Tfam-SCKO nerves 3 weeks after crush injury shows no impairment in the ability of Tfam-deficient SCs to break down existing myelin. Error bars indicate SEM. n = 3 mice per genotype. d, Quantification of the number of myelinated profiles in ctrl and Tfam-SCKO nerves 3 weeks after crush distal to the site of injury shows a significant decrease (p < 0.001, two-tailed Student's t test) in the number of myelinated profiles formed by Tfam-deficient SCs following injury. Error bars indicate SEM. n = 3 mice per genotype. e, Electron micrographs of ctrl and Tfam-SCKO nerves 3 weeks after crush depicting the inability of Tfam-deficient SCs to form mature myelin even though they are capable of supporting axonal regeneration. Scale bar, 2 μm. f, Quantification of the total number of fibers (myelinated and unmyelinated) distal to the site of injury 3 weeks after crush shows no differences between Tfam-SCKO and ctrl nerves, confirming that Tfam-deficient SCs are able to support axonal regeneration. Error bars indicate SEM. n = 3 mice per genotype. g, Electron micrographs of ctrl (g1) and Tfam-SCKO (g2–g4) large-caliber fibers distal to the site of injury 3 weeks after crush showing that, while Tfam-deficient SCs are capable of ensheathing axons and forming one-to-one associations with them (arrowheads, axons; arrows, SCs), they are not able to form mature myelin. Regenerating Remak bundles are present in ctrl nerves (g5) but are absent in Tfam-SCKO nerves (g6; arrowheads, axons; arrows, SCs). h, Proportion of axons >1 μm is similar between Tfam-SCKO and ctrl nerves following injury and cannot explain the absence of myelin in Tfam-SCKO nerves.
We next studied the ability of Tfam-deficient SCs to support axonal regeneration by analyzing toluidine blue-stained plastic sections of Tfam-SCKO and ctrl nerves collected distal to the site of crush 3 weeks after injury. At this time, we found that substantial axonal regeneration and SC remyelination had already taken place in ctrl nerves (Fig. 8d). In Tfam-SCKO nerves, however, we observed a striking decrease in the number of myelinated fibers (Fig. 8d). Interestingly, examination of Tfam-SKO nerves by electron microscopy revealed that this decrease in myelinated profiles was not due to the inability of Tfam-deficient SCs to support axonal regrowth. In fact, the total number of fibers (myelinated and unmyelinated) distal to the crush site was equivalent between ctrl and Tfam-SCKO nerves 3 weeks after injury (Fig. 8e,f). Moreover, the majority of regenerated axons in Tfam-SCKO nerves 3 weeks after injury were associated with SCs in the typical 1:1 ratio (Fig. 8g1–g4). Tfam-deficient SCs, however, failed to form mature myelin. Given that the proportion of axons of large enough caliber to induce myelination (>1 μm in diameter) was not different between ctrl and Tfam-SCKOs (Fig. 8h), the inability of Tfam-deficient SCs to myelinate is likely a direct consequence of their disrupted mitochondrial metabolism.
Regenerated Remak bundles were also absent in Tfam-SCKO nerves compared with ctrl littermates 3 weeks after crush injury. Instead, we observed pathological bundles of unorganized unmyelinated axons interspersed by bands of Bungner (Fig. 8g5,g6), similar to those present in 2-month-old Tfam-SCKO mice before injury. We conclude that disrupting SC mitochondrial function does not affect the ability of these glia to dedifferentiate, proliferate, or support axonal regeneration following injury. Normal SC mitochondrial metabolism, however, is essential for SCs to form mature myelin and Remak bundles, suggesting that the formation and maintenance of these mature structures poses a high metabolic challenge.
Discussion
In the present study, we describe the generation and characterization of mice with disrupted mitochondrial function only in SCs. We found that the induction of SC-specific mitochondrial dysfunction did not affect the survival of these cells but resulted in a severe, progressive peripheral neuropathy characterized by extensive axonal degeneration. SC mitochondrial dysfunction, therefore, disrupts the axoglial interactions required for the long-term support of axons and is a likely contributor to the clinical impairment of patients suffering from peripheral neuropathies.
Mitochondrial dysfunction is a common cause of peripheral neuropathies. An extensive body of literature has now addressed how neuronal/axonal mitochondria may contribute to the pathology observed in these diseases (Niemann et al., 2006; Baloh, 2008). The results described above show for the first time that disrupted mitochondrial function specifically in SCs can itself also cause axonal degeneration and peripheral nerve disease. These findings are consistent with observations in patients suffering from polyneuropathies associated with diabetes, HIV/AIDS, and mitochondrial disorders, in which most nerve mitochondrial abnormalities are localized to SCs (Schroder, 1993; Kalichman et al., 1998).
Moreover, the Tfam-SCKO mice generated in our study recapitulated a number of critical pathological features of human peripheral neuropathies. Disrupting mitochondrial metabolism specifically in SCs was sufficient to induce both axonal degeneration as well as demyelination. Tfam-SCKOs also displayed distal weakness and sensory deficits, two common problems in human patients. Finally, Tfam-SCKOs suffered preferential early loss of unmyelinated C-fibers followed by the degeneration of large-caliber myelinated axons. This was of particular interest because preferential loss of small-caliber unmyelinated fibers is characteristic of peripheral and optic neuropathies associated with systemic diseases such as diabetes or with exposure to environmental toxins (Kennedy et al., 1996; Sadun, 2002; McArthur et al., 2005). Together, our results indicate that SC mitochondria are underappreciated contributors to the abnormalities observed in peripheral neuropathies. Furthermore, key pathological features commonly encountered in human peripheral nerve disease can potentially be explained by SC-specific mitochondrial dysfunction.
The findings described in the present paper are consistent with a growing body of literature that implicates glial dysfunction in a number of neurodegenerative diseases traditionally thought to be cell autonomous. This is best exemplified by amyotrophic lateral sclerosis (ALS), a disease characterized by premature death of motor neurons that is sometimes linked to dominant mutations in the antioxidant protein superoxide dismutase 1 (SOD1) (Rosen et al., 1993). While neuronal expression of mutant SOD1 in transgenic mouse models of ALS was found to be important for the initial timing of disease onset (Boillee et al., 2006; Yamanaka et al., 2008; Wang et al., 2009), expression in microglia cells (Boillee et al., 2006; Wang et al., 2009) and astrocytes (Yamanaka et al., 2008) was found to significantly contribute to disease progression. Similarly, in a mouse model of Parkinson's disease, oligodendrocyte-specific overexpression of the disease-related protein α-synuclein resulted in neurodegeneration (Yazawa et al., 2005). A central role for glia in diseases such as Huntington's, spinocerebellar ataxia, or Alzheimer's has also been reported (for review, see Ilieva et al., 2009). The development of effective therapies for peripheral neuropathies and neurodegenerative disorders, therefore, will need to address the contribution of SCs and other glia to disease progression.
An unexpected finding of our work was that severe mtDNA depletion and respiratory chain abnormalities in the SCs of Tfam-SCKO mice did not affect their survival. This observation suggests that SCs have an adaptable energy metabolism and that their survival and ATP production are not fully dependent on mitochondrial function. Previous studies looking at the effect of Tfam deletion in a number of tissues have indeed reported cell-specific metabolic changes that take place following the Tfam deletion-induced disruption of the electron transport chain. For example, following the muscle-specific deletion of Tfam, myocytes increased their mitochondrial mass to compensate for any deficiencies in their respiratory chain (Wredenberg et al., 2002). Cardiomyocytes, however, were reported to switch to a primarily glycolytic metabolism in response to the heart-specific excision of Tfam (Hansson et al., 2004). Some of these adaptations likely account for the limited or largely delayed cell death observed in several tissue-specific Tfam KOs (Larsson et al., 1998; Wang et al., 1999; Silva et al., 2000; Sorensen et al., 2001; Wredenberg et al., 2002). Specific metabolic adaptations following Tfam depletion-induced mitochondrial dysfunction may also explain why SC viability was not affected in Tfam-SCKO mice.
SCs have, in fact, been hypothesized to be largely glycolytic and rely on the nonoxidative catabolism of glucose to meet their energy needs (Pellerin and Magistretti, 2003). This idea is supported by the observation that, in explant systems, SCs take up a disproportionately large amount of glucose (Vega et al., 2003). The fact that disruption of mitochondrial energy metabolism in Tfam-SCKO mice did not affect SC survival provides indirect support for this hypothesis. Our current knowledge of the basic energy metabolism and requirements of SCs, however, is still very rudimentary. Future work aimed at understanding the metabolic changes that take place in SCs following Tfam deletion-induced respiratory chain deficiency should provide valuable insight into both the biology and pathology of SCs.
Following peripheral nerve injury, SCs undergo a process of dedifferentiation, proliferation, and formation of tubular structures known as bands of Bungner, which, together with their production of neurotrophic factors, help support axonal regeneration (Geuna et al., 2009). Interestingly, we found that Tfam depletion-induced mitochondrial dysfunction did not interfere with the ability of SCs to perform these duties and support axonal regrowth. However, Tfam-deficient SCs failed to redifferentiate and form mature Remak bundles and myelin sheaths after having promoted axonal regeneration. The high metabolic challenge posed by the reconstruction of these mature structures, therefore, appears to require an undisturbed SC mitochondrial anaplerotic metabolism. Elucidation of deficiencies in the intermediate metabolism of Tfam-SCKOs could thus provide new targets whose manipulation may promote the remyelination and the restoration of normal nerve function in patients suffering from mitochondrial-related peripheral neuropathies.
SCs have a primary role in supporting axonal function and survival (Chen et al., 2003; Reddy et al., 2003; Meyer zu Horste et al., 2007), yet the mechanisms and cellular machinery underlying SC preservation of axons still remain largely unknown. Others have hypothesized that glial support requires axonal ensheathment as well as some myelin-related proteins. Trophic support of axons through neurotrophic factors has also been postulated as a potential mechanism behind glia-mediated axonal maintenance. Metabolic support of axons by ensheathing cells is also a likely but poorly understood mechanism of axonal preservation (Nave and Trapp, 2008). For example, SCs may support axons through the transfer of metabolites or by helping axons eliminate certain toxic species, as is the case between CNS neurons and astrocytes (Benarroch, 2005). Our results show that SC mitochondria are essential for the SC-mediated support of axonal function and survival. Given the central role of mitochondria in cellular energy and anaplerotic metabolism, we speculate that the axonal degeneration in Tfam-SCKOs reflects a disruption of SC-mediated metabolic support of axons. The preferential loss of small unmyelinated fibers early in the Tfam-SCKO disease process would then reflect a greater dependence of these fibers on SC metabolic support. Consistent with this hypothesis, unmyelinated axons have been found to be more energetically demanding than myelinated axons and are estimated to consume 2.5- to 10-fold more energy per action potential generated (Wang et al., 2008).
In summary, Tfam-SCKOs are, to our knowledge, the first SC-specific metabolic mutant that recapitulates some of the pathology of human peripheral neuropathies. These mice demonstrate that SC-specific mitochondrial dysfunction can be sufficient to cause both demyelination and axonal degeneration. Our study also provides evidence that SC mitochondria help maintain the axoglial interactions required for the long-term support of axons. A greater understanding of SC mitochondria and how to prevent their dysfunction is, therefore, relevant to the treatment of patients suffering from peripheral neuropathies.
Notes
Supplemental material for this article is available at http://www.genetics.wustl.edu/jmlab/wp-content/uploads/2011/05/Viader-et-al_Schwann-cell-mitochondrial-metabolism-supports-long-term-axonal-survival-and-peripheral-nerve-function_Supplementary-Figure-1.pdf. Supplemental Figure 1 shows that Tfam-SCKOs initially develop and myelinate normally. This material has not been peer reviewed.
Footnotes
This work was supported by NIH Neuroscience Blueprint Center Core Grant P30 NS057105 (Washington University), the Hope Center for Neurological Disorders, NIH Grants NS040745 (J.M.), AG13730 (J.M.), NS055980 (R.H.B.), DK19645 (R.E.S.), and R21NS059566 (J.P.G.), and Muscular Dystrophy Association Grants 10040 (J.M.) and 135428 (R.H.B.). We thank Michael A. Kiebish for providing valuable technical advice and helpful discussions, Amy Strickland and Nina Panchenko for experimental assistance, Nils-Goran Larsson for the TfamloxP mice and comments on this manuscript, Lawrence Wrabetz and Albee Messing for the P0-Cre mice, and members of the Milbrandt Laboratory for their comments on this manuscript and helpful discussions.
A.V., J.P.G., R.H.B., R.E.S., D.A.H., J.M., and Washington University may derive benefit from a licensing agreement with Sirtris Pharmaceuticals, which did not provide any support for this work.
References
- Arezzo JC, Zotova E. Electrophysiologic measures of diabetic neuropathy: mechanism and meaning. Int Rev Neurobiol. 2002;50:229–255. doi: 10.1016/s0074-7742(02)50079-9. [DOI] [PubMed] [Google Scholar]
- Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Strickland A, Ryu E, Le N, Fahrner T, Yang M, Nagarajan R, Milbrandt J. Congenital hypomyelinating neuropathy with lethal conduction failure in mice carrying the Egr2 I268N mutation. J Neurosci. 2009;29:2312–2321. doi: 10.1523/JNEUROSCI.2168-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc. 2005;80:1326–1338. doi: 10.4065/80.10.1326. [DOI] [PubMed] [Google Scholar]
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G. Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci. 2003;6:1186–1193. doi: 10.1038/nn1139. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Semino-Mora C, Leon-Monzon M. Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2′3′-dideoxycytidine (ddC) Lab Invest. 2001;81:1537–1544. doi: 10.1038/labinvest.3780367. [DOI] [PubMed] [Google Scholar]
- Ebenezer GJ, Hauer P, Gibbons C, McArthur JC, Polydefkis M. Assessment of epidermal nerve fibers: a new diagnostic and predictive tool for peripheral neuropathies. J Neuropathol Exp Neurol. 2007;66:1059–1073. doi: 10.1097/nen.0b013e31815c8989. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–123. [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernyhough P, Huang TJ, Verkhratsky A. Mechanism of mitochondrial dysfunction in diabetic sensory neuropathy. J Peripher Nerv Syst. 2003;8:227–235. doi: 10.1111/j.1085-9489.2003.03028.x. [DOI] [PubMed] [Google Scholar]
- Geuna S, Raimondo S, Ronchi G, Di Scipio F, Tos P, Czaja K, Fornaro M. Chapter 3: Histology of the peripheral nerve and changes occurring during nerve regeneration. Int Rev Neurobiol. 2009;87:27–46. doi: 10.1016/S0074-7742(09)87003-7. [DOI] [PubMed] [Google Scholar]
- Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Golden JP, Hoshi M, Nassar MA, Enomoto H, Wood JN, Milbrandt J, Gereau RW, 4th, Johnson EM, Jr, Jain S. RET signaling is required for survival and normal function of nonpeptidergic nociceptors. J Neurosci. 2010;30:3983–3994. doi: 10.1523/JNEUROSCI.5930-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspan JB, Marchionni MA, Reeves M, Coulaloglou M, Scherer SS. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci. 1996;16:6107–6118. doi: 10.1523/JNEUROSCI.16-19-06107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci U S A. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RA. Peripheral neuropathy. BMJ. 2002;324:466–469. doi: 10.1136/bmj.324.7335.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DA, Moradzadeh A, Whitlock EL, Brenner MJ, Myckatyn TM, Wei CH, Tung TH, Mackinnon SE. Binary imaging analysis for comprehensive quantitative histomorphometry of peripheral nerve. J Neurosci Methods. 2007;166:116–124. doi: 10.1016/j.jneumeth.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalichman MW, Powell HC, Mizisin AP. Reactive, degenerative, and proliferative Schwann cell responses in experimental galactose and human diabetic neuropathy. Acta Neuropathol. 1998;95:47–56. doi: 10.1007/s004010050764. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Wendelschafer-Crabb G. The innervation of human epidermis. J Neurol Sci. 1993;115:184–190. doi: 10.1016/0022-510x(93)90223-l. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Wendelschafer-Crabb G, Johnson T. Quantitation of epidermal nerves in diabetic neuropathy. Neurology. 1996;47:1042–1048. doi: 10.1212/wnl.47.4.1042. [DOI] [PubMed] [Google Scholar]
- King RH. Nerve trunks and spinal roots. In: Dyck PJ, Thomas PK, editors. Peripheral neuropathy. Ed 4. Philadelphia: Elsevier Saunders; 2005. pp. 35–42. [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Svaren J, LaPash C, Araki T, Schmidt RE, Milbrandt J. Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci. 2005;8:932–940. doi: 10.1038/nn1490. [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, Jenkins CM, Guan S, Moon SH, Pietka T, Nassir F, Schappe T, Moore K, Han X, Abumrad NA, Gross RW. Genetic ablation of calcium-independent phospholipase A2γ prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J Biol Chem. 2010;285:36495–36510. doi: 10.1074/jbc.M110.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol. 2007;61:61–72. doi: 10.1002/ana.21026. [DOI] [PubMed] [Google Scholar]
- Montana MC, Cavallone LF, Stubbert KK, Stefanescu AD, Kharasch ED, Gereau RW., 4th The metabotropic glutamate receptor subtype 5 antagonist fenobam is analgesic and has improved in vivo selectivity compared with the prototypical antagonist 2-methyl-6-(phenylethynyl)-pyridine. J Pharmacol Exp Ther. 2009;330:834–843. doi: 10.1124/jpet.109.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron. 2001;30:355–368. doi: 10.1016/s0896-6273(01)00282-3. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:217–242. doi: 10.1385/nmm:8:1-2:217. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. How to balance the brain energy budget while spending glucose differently. J Physiol. 2003;546:325. doi: 10.1113/jphysiol.2002.035105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP. Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron. 2003;40:563–580. doi: 10.1016/s0896-6273(03)00682-2. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Wang JY, Le N, Baloh RH, Gustin JA, Schmidt RE, Milbrandt J. Misexpression of Pou3f1 results in peripheral nerve hypomyelination and axonal loss. J Neurosci. 2007;27:11552–11559. doi: 10.1523/JNEUROSCI.5497-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadun AA. Metabolic optic neuropathies. Semin Ophthalmol. 2002;17:29–32. doi: 10.1076/soph.17.1.29.10290. [DOI] [PubMed] [Google Scholar]
- Schroder JM. Neuropathy associated with mitochondrial disorders. Brain Pathol. 1993;3:177–190. doi: 10.1111/j.1750-3639.1993.tb00742.x. [DOI] [PubMed] [Google Scholar]
- Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci. 2001;21:8082–8090. doi: 10.1523/JNEUROSCI.21-20-08082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega C, Martiel JL, Drouhault D, Burckhart MF, Coles JA. Uptake of locally applied deoxyglucose, glucose and lactate by axons and Schwann cells of rat vagus nerve. J Physiol. 2003;546:551–564. doi: 10.1113/jphysiol.2002.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- Wang L, Sharma K, Grisotti G, Roos RP. The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol Dis. 2009;35:234–240. doi: 10.1016/j.nbd.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Shultz JR, Burish MJ, Harrison KH, Hof PR, Towns LC, Wagers MW, Wyatt KD. Functional trade-offs in white matter axonal scaling. J Neurosci. 2008;28:4047–4056. doi: 10.1523/JNEUROSCI.5559-05.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–859. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]