

Abstract
A subunit vaccine using a defined antigen(s) may be one effective solution for controlling leishmaniasis. Because of genetic diversity in target populations, including both dogs and humans, a multiple-antigen vaccine will likely be essential. However, the cost of a vaccine to be used in developing countries must be considered. We describe herein a multiantigen vaccine candidate comprised of antigens known to be protective in animal models, including dogs, and to be recognized by humans immune to visceral leishmaniasis. The polyprotein (KSAC) formulated with monophosphoryl lipid A, a widely used adjuvant in human vaccines, was found to be immunogenic and capable of inducing protection against Leishmania infantum, responsible for human and canine visceral leishmaniasis, and against L. major, responsible for cutaneous leishmaniasis. The results demonstrate the feasibility of producing a practical, cost-effective leishmaniasis vaccine capable of protecting both humans and dogs against multiple Leishmania species.
INTRODUCTION
Leishmaniasis represents a spectrum of diseases caused by protozoan parasites of the genus Leishmania. According to the WHO Health Report 2004, the disease is endemic in 88 countries, putting 350 million people at risk and resulting in 12 million cases. Current morbidity and mortality surveys estimate there are 2 million new cases of leishmaniasis and 59,000 deaths annually. Leishmaniasis can be classified into three general types of disease: cutaneous leishmaniasis (CL), mucosal leishmaniasis (ML), and visceral leishmaniasis (VL), based on the clinical manifestations of the disease. These three forms of leishmaniasis are caused by different parasite species, including Leishmania major (CL), Leishmania braziliensis (ML), and Leishmania donovani and Leishmania infantum (VL).
Although there is no commercially available vaccine for human leishmaniasis, extensive preclinical and promising clinical results indicate that control of the disease by preventative vaccination will likely become a viable approach (10, 32, 33). Evidence that people cured from VL develop protection mediated by Th1-type cellular responses against new infections (8, 19) is also supportive of the possibility of a vaccine for disease prevention. First-generation vaccines using either parasite lysates or killed parasites have occasionally been shown to be safe and immunogenic and in some cases have exhibited protective efficacy against leishmaniasis (1, 21, 40). However, whole inactivated Leishmania parasites will not consistently provide the safe, effective, stable, and reliable source of antigens that is needed for a large-scale vaccination program against leishmaniasis because of the expense of running both a good-manufacturing-practice (GMP) parasite production facility and the subsequent antigen purification process and because the antigens represented in each batch of parasite culture will be affected by cell culture conditions and antigen preparation processes.
Fortunately, there has been progress in characterizing defined Leishmania antigens that provide beneficial immune responses (10, 33). Those antigens, which include a Leishmania homolog of receptors for activated C kinase (LACK), GP63, thiol-specific antigen (TSA), hydrophilic acylated surface protein B (HASPB), sterol 24-c-methyltransferase (SMT), kinetoplastid membrane protein 11 (KMP11), A2, and cysteine proteinase B (CPB), confer protection against disease in different animal models of leishmaniasis (3, 14, 16, 28, 36, 37, 44, 46). Because of the genetic polymorphism in the mammalian immune system, a vaccine composed of multiple antigens rather than a single gene product is more likely to elicit a protective immune response against leishmaniasis in a broad spectrum of individuals. We have previously produced the polyprotein antigen Leish-111f, which is composed of TSA (46), stress inducible protein 1 (LmSTI1) (7, 47), and a homolog of the eukaryotic translation initiation factor eIF4A (LeIF) (41, 42) and have shown that immunization with this polyprotein formulated in an adjuvant protects mice against L. major (11). Although the vaccine candidate provides some protection against L. infantum infection (9), Leish-111f was originally designed and optimized to target CL. In this study we focused on several proteins previously demonstrated to be protective against VL in animal models, including dogs, and recognized in humans cured from VL (22). The proteins KMP-11 (3), SMT (16), A2 (13, 14), and CPB (34, 36) were genetically fused to produce a single multiepitope product developed with the goal of achieving a cost-effective product with maximum efficacy. We evaluated protective efficacy of the resulting polyprotein in two different experimental murine leishmaniases, CL and VL.
MATERIALS AND METHODS
Animals and parasites.
All mice were maintained in the Infectious Disease Research Institute (IDRI) animal care facility under specific-pathogen-free conditions and were treated in accordance with the regulations and guidelines of the IDRI Animal Care and Use Committee. Female BALB/c and C57BL/6 mice (6 to 8 weeks old) were purchased from Charles River Laboratories (Wilmington, MA). Promastigotes of L. infantum (MHOM/BR/82/BA-2) were cultured as previously described (16). The L. major Friedlin strain clone V1 was kindly provided by David Sacks (NIH) and maintained as previously described (38).
Production of KSAC.
Nucleotide sequences encoding individual components were PCR amplified using Platinum Pfx DNA polymerase (Invitrogen) with genomic DNA from either L. infantum or L. donovani promastigotes. Primers, as listed in Table 1, were designed to amplify nucleotides (nt) 1 to 276 of KMP11 (GenBank accession no. XM_001468995.1), nt 4 to 1059 of SMT (GenBank accession no. XM_001469795.1), nt 150 to 779 of A2 (GenBank accession no. S69693.1), and nt 738 to 1687 of CPB (GenBank accession no. AJ420286.1). Each of the sets of primers included unique restriction enzyme sites. After digestion of the amplified DNAs with the corresponding restriction enzymes, products were ligated to create a polyprotein gene construct with the sequence (5′ to 3′) of KMP11, SMT, A2, and CPB and inserted into the pET-29 plasmid. The KSAC pET-29 construct was transformed into Escherichia coli HMS174 (DE3).
Table 1.
PCR primers used for this study
| Name | Sequence |
|---|---|
| KMP11-5NdeI | CAATTACATATGGCCACCACGTACGAGGAG |
| KMP11-3SpeI-HindIII | CAATTAAAGCTTTTAACTAGTCTTGGACGGGTACTGCGCAGC |
| SMT-5SpeI | CAATTAACTAGTTCCGCCGGTGGCCGTGAGACC |
| SMT-3BamHI- HindIII | CAATTAAAGCTTTTAGGATCCAGCCTGCTTGGACGGCTTGCG |
| A2-5BamHI | CAATTAGGATCCGAGCCGCACAAGGCGGCCGTT |
| A2-3BamHI-EcoRI- HindIII | CAATTAAAGCTTTTAGAATTCGGATCCAGACACCGGAGAAACGTCAAC |
| CPB-5EcoRI | CAATTAGAATTCGATGCGGTGGACTGGCGC |
| CPB-3HindIII | CAATTAAAGCTTTTACGTGTACTGGCAGGTGTTCAT |
For KSAC protein expression, a single colony was inoculated into one liter of 2× yeast extract-tryptone medium containing 30 μg/ml kanamycin and grown at 37°C with shaking (225 rpm) until optical densities reached 0.4 to 0.6. Gene expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside at 37°C for 3 h. Cells were harvested by centrifugation (2,000 × g, 15 min), and pellets were stored at −80°C.
The cell pellet was thawed and resuspended in 40 ml of cold lysis buffer (LyB) (50 mM NaCl, 10 mM Tris-HCl, pH 8.0) and passed four times through an M-110S microfluidizer (Microfluidics, Newton, MA) set at 40 lb/in2. The lysate was centrifuged at 10,000 × g for 30 min at 4°C to pellet the inclusion body (IB). The IB was washed one time with 40 ml LyB containing 1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) and once with 40 ml 30% isopropanol with centrifugation (10,000 × g for 30 min, 4°C) after each step. The washed IB pellet was stored at −80°C.
The IB material was solubilized in 30 ml buffer A (8 M urea, 20 mM Bis-Tris-propane, pH 7.0) at ambient temperature for 3 h, and insoluble material was removed by centrifugation at 10,000 × g for 30 min at 4°C. Protein purification was carried out using an Äkta purifier system (GE Lifesciences). The solubilized IB fraction was loaded onto an anion exchange Q Sepharose Fast Flow resin (QFF) (Amersham Biosciences) column equilibrated with buffer A, the column was washed with 6 column volumes of buffer A, and KSAC was eluted with 50 mM NaCl-buffer A at a flow rate of 14 ml/min. Those fractions containing KSAC but having minimal contamination by E. coli proteins (confirmed by Western blotting) were pooled and dialyzed at 4°C against buffer B (20 mM Tris-HCl, pH 7.5). The protein solution was adjusted to 1 M NaCl and loaded onto a Macro-Prep methyl hydrophobic interaction chromatography (HIC) column (Bio-Rad, Hercules, CA) equilibrated with 1 M NaCl-buffer B. After the column was washed with five volumes of 1 M NaCl-buffer B and three volumes of 0.75 M NaCl-buffer B, KSAC was eluted with 0.3 M NaCl-buffer B, dialyzed against buffer B, and used for further studies.
Purity of the protein was assessed by SDS-PAGE and Coomassie blue staining. The concentration of the purified protein was calculated using the A280 value and the extinction coefficient (1 A280=0.95 mg/ml). Endotoxin levels of each purified protein lot were measured by using a Limulus amoebocyte lysate test (Cambrex Corporation, East Rutherford, NJ).
Other protein preparations.
Recombinant of KMP11, SMT, A2, and CPB proteins were produced as previously described (16, 22). L110f, which is an optimized derivative of Leish-111f, was produced as previously described (5). Lysates of L. infantum, L. donovani, and L. major were prepared as previously described (15, 17). All the proteins had endotoxin levels of less than 100 endotoxin units (EU)/mg, determined by the Limulus amoebocyte lysate test.
Immunization of mice.
Mice were immunized with either 10 μg of recombinant antigen (either SMT, L110f, or KSAC) or a mixture of four proteins (2.5 μg each of KMP11, SMT, A2, and CPB). For these immunizations, 20 μg of MPL-SE (GlaxoSmithKline Biologicals, Rixensart, Belgium) was used as an adjuvant; the formulated vaccine was given in a volume of 0.1 ml. Control groups received either saline or MPL-SE alone (20 μg). The mice were immunized three times subcutaneously at the base of the tail at 3-week intervals.
Antibody ELISA.
Mice were bled 1 week after the last vaccination. Mouse antibody enzyme-linked immunosorbent assay (ELISA) was carried out as previously described (16). Either lysate antigen (1 μg/well) or recombinant protein (200 ng/well) was used for coating the ELISA plate. Reciprocal endpoint titers to individual antigens were calculated with the GraphPad Prism 5 software program (GraphPad Software, Inc., La Jolla, CA) using the OD of 0.1 as a cutoff value. Endpoint titers of samples were recorded as <100 if OD values of the samples were lower than the cutoff value at 1:100.
Cytokine assay.
Spleens were collected 2 weeks after the last immunization (three mice per group) and analyzed for antigen-specific gamma interferon (IFN-γ) production 3 days after stimulation, as previously described (15).
Intracellular staining and flow cytometry.
Single-cell suspensions were prepared from individual spleens isolated from C57BL/6 mice 2 weeks after the last immunization. Cytokine production by the splenocytes were analyzed upon antigen recall, as previously described (16).
Challenge of mice with Leishmania spp.
C57BL/6 mice were challenged with 5 × 106 promastigotes of L. infantum by intravenous injection into the tail vein 3 weeks after the last vaccination, and the liver and spleen parasite burdens were quantified 4 weeks postinfection by limiting dilution, as previously described (16). The challenge of BALB/c mice with L. major was performed by intradermal injection into both the right and left ears 3 weeks after the last vaccination, followed by measurement of lesion sizes and determination of ear parasite burdens at 8 weeks postinfection, as previously described (15).
RESULTS
Construction and immunological characterization of KSAC.
The KSAC expression construct was prepared in order to express a fusion of KMP11, SMT, A2, and CPB (Fig. 1 A). Within the KSAC fusion, KMP11 was full length and the SMT region was missing its N-terminal methionine. A2, on the other hand, lacked its first 26 amino acids; these amino acids were predicted by the SignalP 3.0 software program (4) to comprise the signal peptide. The partial CPB sequence (amino acids [aa] 128 to 443) in our KSAC construct is the same one shown to be protective in previous studies (34, 36). KSAC was expressed in E. coli and purified to homogeneity using two orthogonal chromatographic steps. The apparent molecular mass of the protein (105 kDa as determined by reducing SDS-PAGE) agreed well with the predicted value (Fig. 1B). Due to the several Cys residues present in the fusion protein, KSAC has the tendency to form disulfide-bonded multimers under nonreducing conditions with apparent molecular masses at least twice that of the monomer (Fig. 1B). The purified KSAC was free of detectable E. coli protein contamination based on Western blotting (data not shown) and had a low endotoxin content (38 EU/mg protein).
Fig. 1.

Purified recombinant KSAC. (A) A schematic illustration of the KSAC construct. (B) A Coomassie blue-stained SDS-4 to 20% polyacrylamide gradient gel loaded with purified proteins (10 μg per lane) under reducing (R) or nonreducing (NR) conditions. Sizes of a protein marker (M) are shown in kDa.
To examine the ability of this antigen to elicit antigen-specific immune responses, C57BL/6 mice immunized with KSAC plus MPL-SE were examined for serum antibody titers to KSAC as well as the four component antigens. For comparison, mice immunized with a mixture of the four component antigens (K/S/A/C) plus MPL-SE were also examined. Injection with either saline alone or MPL-SE alone did not induce antibody responses to KSAC (reciprocal endpoint titers of <100). In contrast, mice vaccinated with either KSAC plus MPL-SE or K/S/A/C plus MPL-SE showed high levels of antibodies to KSAC (Fig. 2). When responses to individual component antigens were examined, different patterns were observed in these two vaccination groups. When K/S/A/C was used for vaccination, antibody responses to SMT and CPB were dominant compared to responses to KMP11 and A2. In contrast, comparable levels of antibodies were detected to all the four antigens in mice vaccinated with KSAC.
Fig. 2.
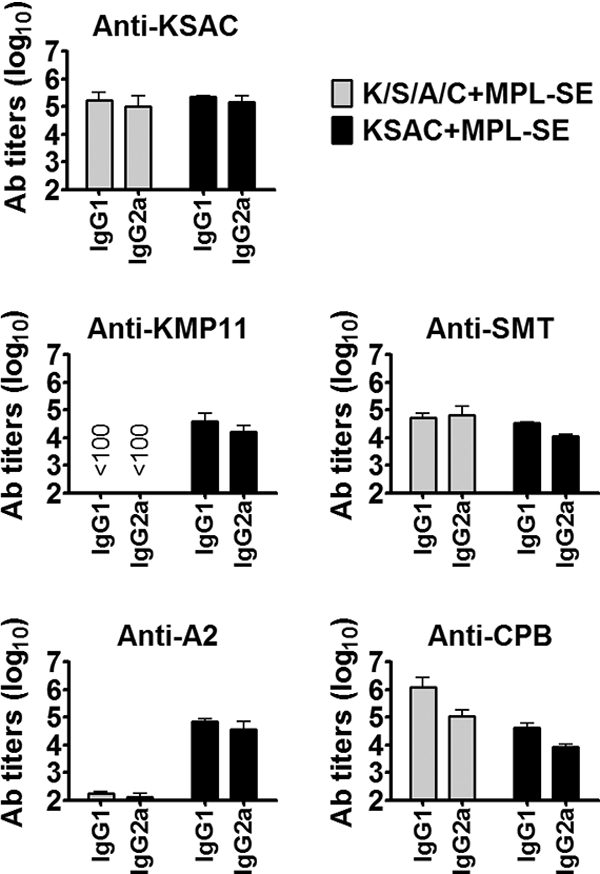
Induction of antibody responses by KSAC immunization. C57BL/6 mice immunized three times with either saline, MPL-SE alone, K/S/A/C plus MPL-SE, or KSAC plus MPL-SE were examined for antibody responses to KSAC as well as the four component antigens. Mice in the saline- and MPL-SE-alone groups possessed no detectable antibodies to any of the antigens tested (data not shown). Means and SEM of reciprocal endpoint titers for the K/S/A/C-plus-MPL-SE and KSAC-plus-MPL-SE groups are shown.
To examine the ability of this antigen to induce the desirable Th1 type of immune response, splenocytes of C57BL/6 mice immunized with KSAC plus MPL-SE were examined for production of the cytokine IFN-γ after ex vivo antigen stimulation (Fig. 3 A). Injection of saline or MPL-SE alone did not induce any significant antigen-specific responses. Spleen cells from mice receiving either KSAC plus MPL-SE or K/S/A/C plus MPL-SE responded to vaccine antigens with elevated IFN-γ production when stimulated ex vivo with KSAC. These spleen cells were also capable of producing IFN-γ upon recall with lysate antigens of L. infantum and L. donovani. When individual components of KSAC were used for in vitro recall, SMT and CPB induced high levels of IFN-γ, whereas little if any IFN-γ was detected with KMP11 or A2 stimulation. Although the IFN-γ response to A2 was higher in the K/S/A/C group than in the KSAC group, it was not statistically significant. The general response pattern noted above was not unique to C57BL/6 mice; a similar pattern was observed in BALB/c mice (Fig. 3B). Once again, SMT and CPB were observed to be the dominant antigen components of KSAC.
Fig. 3.
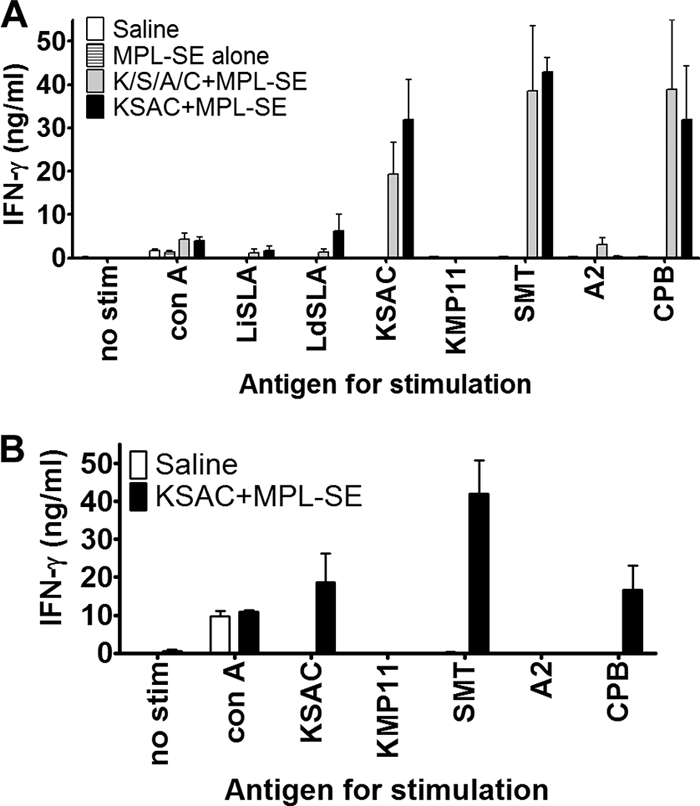
Immunogenicity of KSAC in C57BL/6 and BALB/c mice. C57BL/6 (A) or BALB/c (B) mice were immunized three times with either saline, MPL-SE alone, K/S/A/C plus MPL-SE, or KSAC plus MPL-SE, as described in Materials and Methods. IFN-γ production after ex vivo antigen stimulation of splenocytes from those immunized mice was determined by sandwich ELISA. Means and SEM of data for three mice per group are shown.
To further investigate the quality of cellular responses induced by the KSAC/MPL-SE vaccine, splenocytes were analyzed for the frequency of CD4+ and CD8+ T cells producing the Th1-type cytokine tumor necrosis factor alpha (TNF-α), interleukin 2 (IL-2), or IFN-γ in response to incubation with KSAC (Fig. 4 A). The flow cytometric data show that antigen-specific CD4+ T cells but not CD8+ T cells were induced by KSAC/MPL-SE vaccination (Fig. 4B). When the CD4+ T-cell population was subdivided into categories based on TNF-α, IL-2, and IFN-γ expression (a total of seven distinct populations), many of the Ag-specific T cells were found to be multifunctional Th1 cells. In fact, that category of Th1 cells simultaneously producing TNF-α, IL-2, and IFN-γ was found to be the largest cell population of the seven (Fig. 4C).
Fig. 4.
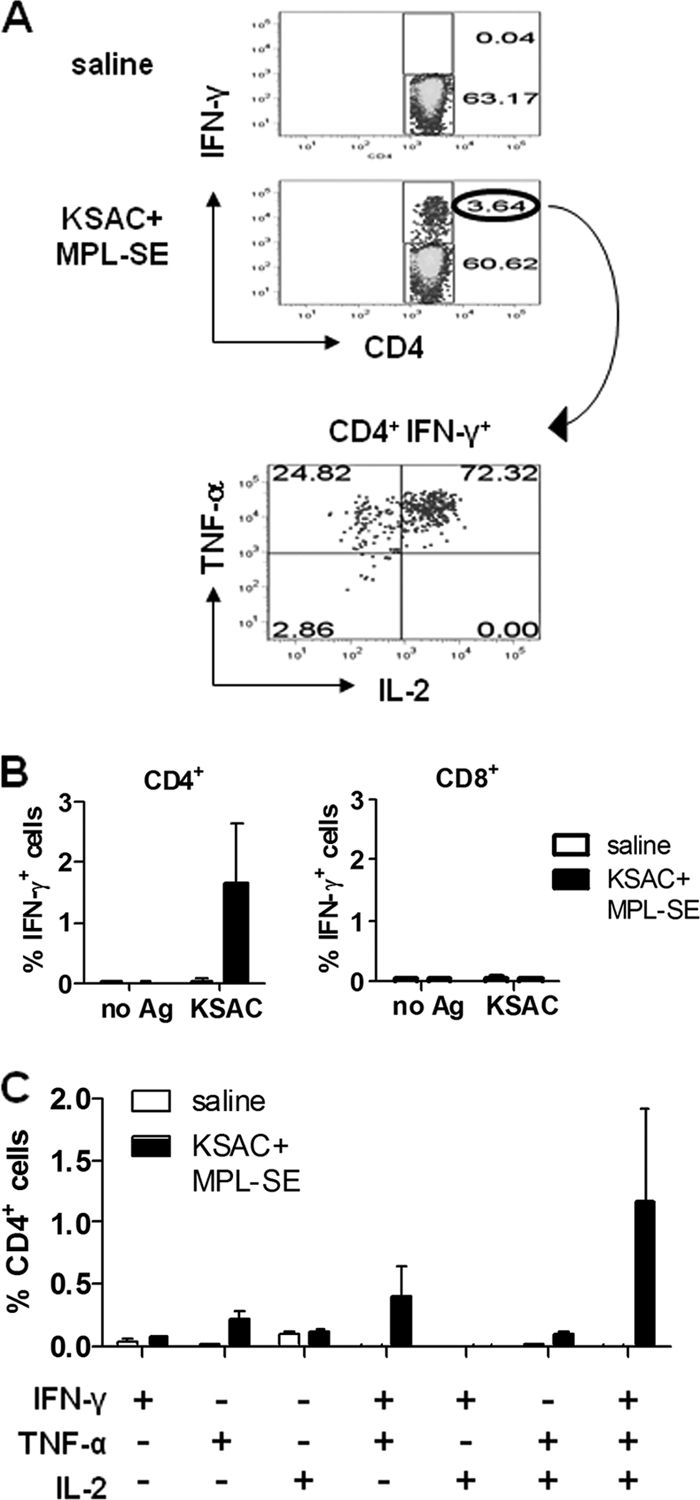
Induction of multifunctional Th1 cells by the KSAC/MPL-SE vaccine. (A) The flow cytometric gating strategy is shown for CD4+ T cells from the spleens of vaccinated C57BL/6 mice that produce IFN-γ, TNF-α, and IL-2 upon antigen recall. (B) Percentage of IFN-γ-producing CD4+ or CD8+ cells in the spleens of mice immunized with either saline or KSAC/MPL-SE after in vitro culture with or without KSAC. Means and SEM of data for three mice per group are shown. (C) Single-cell analysis of CD4+ T cells producing multiple Th1-type cytokines upon recall with KSAC. Means and SEM of data for three mice per group are shown.
The KSAC/MPL-SE vaccine protects against L. infantum and L. major.
The protective efficacy of the KSAC/MPL-SE vaccine against experimental challenge with Leishmania spp. was evaluated. To examine the efficacy of the vaccine against VL, immunized C57BL/6 mice were challenged by intravenous injection of 5 × 106 promastigotes of L. infantum. At this challenge dose, separate experiments showed the peak liver parasite burden in C57BL/6 mice occurred at 4 weeks of infection (mean = ∼107 amastigotes/liver) and between 4 and 8 weeks in the spleen (∼106 amastigotes/organ; unpublished data). These kinetics of infection regarding parasite burdens were consistent with those observed in other inbred mice (23). Thus, we chose 4 weeks as a time point for determining parasite numbers in spleens and livers of challenged mice. A statistically significant reduction in parasite numbers was seen in the livers of mice immunized with KSAC plus MPL-SE compared with those receiving saline or MPL-SE (Fig. 5 A). The protection level for vaccination with KSAC plus MPL-SE (93% reduction in parasite burden) was comparable to that with K/S/A/C plus MPL-SE (90% reduction). While the finding was not statistically significant, KSAC gave better protection against L. infantum infection than the single-component SMT (81% reduction) and our first-generation polyprotein vaccine antigen L110f (77% reduction). In a separate study, the protection in the spleen was examined for the KSAC/MPL-SE vaccine. Parasite numbers in the spleens of the vaccinated mice were significantly lower than those for unvaccinated mice (66% reduction). In multiple independent experiments, KSAC plus MPL-SE consistently protected mice against L. infantum infection as well as or better than MPL-SE combined with SMT alone or L110f (Fig. 5 and data not shown).
Fig. 5.

Immunization with KSAC/MPL-SE protects mice from L. infantum and L. major challenge. (A) In experiment 1, C57BL/6 mice were immunized with saline, MPL-SE alone, or the indicated vaccine and challenged with L. infantum. The number of parasites in each liver was measured by limiting dilution 4 weeks after the infection. In experiment 2, C57BL/6 mice were immunized with saline or KSAC plus MPL-SE and challenged with L. infantum. The numbers of parasites in both the liver and spleen were measured by limiting dilution 4 weeks after the infection. (B) BALB/c mice immunized with saline or KSAC/MPL-SE were challenged with L. major intradermally in both the right and left ears. Lesion sizes of mice immunized with saline (○) or KSAC/MPL-SE (•) were measured every week up to 8 weeks, at which time the parasite numbers in the ear lesions were determined by limiting dilution. Means and SEM of data for five or six mice in each group are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by unpaired t test compared with the saline group. These data are representative of at least three independent experiments with similar results.
KSAC/MPL-SE was assessed for the ability to also protect against L. major infection. Unimmunized control mice or vaccinated BALB/c mice were challenged with L. major promastigotes by intradermal injection into the ear. Control mice developed ear lesions, and the size of the lesions increased over time (Fig. 5B, left). The lesions in the KSAC/MPL-SE group were significantly smaller than those of control group mice over the 8-week period after infection. The smaller lesions found in the vaccinated mice were associated with a significantly lower number of parasites (99.7% reduction) at the lesion site (Fig. 5B, right). To explore the immune responses associated with vaccine-induced protection against L. major infection, unvaccinated mice or those vaccinated with KSAC/MPL-SE were examined for the production of anti-SLA antibodies and the cytokines IFN-γ and IL-4 8 weeks after infection. The IgG1-dominant antibody response to SLA observed in the unvaccinated mice after L. major infection was strongly suppressed in mice protected by KSAC/MPL-SE vaccination (Fig. 6 A), reflecting the observed shift to a Th1 response with vaccination. Analysis of splenic T-cell responses to SLA following infection demonstrated that vaccine-protected mice produced significantly less IL-4 than unvaccinated mice, and the IFN-γ/IL-4 ratio in response to SLA clearly shifted toward Th1 in the vaccinated and protected group (Fig. 6B).
Fig. 6.
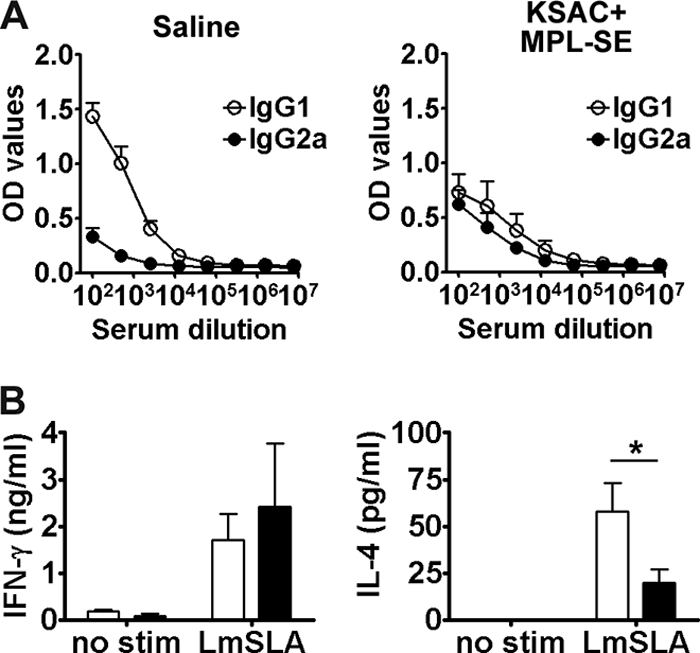
Th1/Th2 balance of postchallenge immunity in the vaccinated/protected mice. Eight weeks after challenge with L. major, BALB/c mice pretreated with either saline or KSAC/MPL-SE were examined for their immune responses to crude parasite antigen (LmSLA). (A) IgG1 and IgG2a levels for LmSLA were measured by ELISA (OD values are shown). Mean and SEM of five mice are shown. (B) IFN-γ and IL-4 produced by spleen cells were analyzed upon antigen recall (open bars, saline placebo; closed bars, KSAC/MPL-SE vaccine). *, P < 0.05 by unpaired t test compared with results for the saline group. This is representative of three independent experiments with similar results.
DISCUSSION
We have demonstrated herein that KSAC/MPL-SE is protective against L. infantum, which causes zoonotic VL in humans and dogs. VL is the most severe form of leishmaniasis, and development of a vaccine against VL is a high priority. The primary objective of the present study was to develop a subunit vaccine against VL with high efficacy and broad coverage of human or animal populations. Although Leish-111f/L110, our first-generation polyprotein vaccine antigens, are promising, i.e., they are protective against both CL and VL in mouse models (9, 11), show therapeutic vaccine efficacy for canine VL (26, 45), and are showing some promise in human clinical trials (25, 31), the vaccine failed to protect dogs from VL in a field trial (18), and it may be possible to obtain a new construct with improved efficacy. KSAC could be one such antigen, because it consistently protected against L. infantum infection as well as or better than Leish-111f/L110, as shown in this study, and also protected against L. major infection better than L110f using a sandfly challenge model (R. Gomes, C. Teixeira, F. Oliveira, P. G. Lawyer, D.-E. Elnaiem, Y. Goto, A. Bhatia, S. Bertholet, R. N. Coler, R. F. Howard, S. G. Reed, S. Kamhawi, and J. G. Valenzuela, unpublished data).
Vaccination with KSAC plus MPL-SE induces antigen-specific multifunctional Th1 cells and protects mice against challenge with L. infantum, as well as L. major, which causes CL. Darrah et al. have reported that there is a correlation between the induction of multifunctional Th1 cells, which are individually capable of producing more than one Th1 cytokine, and protection against L. major infection (12). The three Th1-related cytokines TNF-α, IL-2, and IFN-γ are involved in protection against VL (29, 30, 43). As TNF-α synergizes with IFN-γ in killing Leishmania parasites (24), induction of Ag-specific T cells capable of producing multiple cytokines upon Ag recall might be more beneficial for control of Leishmania infection than those producing a single cytokine, and such induction may be a good indicator of whether a vaccine composed of Ag and adjuvant is protective against leishmaniasis. The presence of such multifunctional Th1 cells may also have roles in suppressing Th2 responses that are, unless otherwise modulated, generated upon challenge. During L. major infection, postchallenge responses in the KSAC/MPL-SE-vaccinated mice were associated with an improved Th1/Th2 balance for parasites, as represented by higher IFN-γ/IL-4 ratios and decreased IgG1 responses to SLA. These findings are consistent with previous reports that IL-4, as a Th2 cytokine, is associated with disease progression during L. major infection in BALB/c mice (20, 39).
It is not surprising that the polyprotein fusion KSAC is protective against L. infantum, because individual antigens comprising the fusion proteins were selected based on their protective efficacy against VL (3, 14, 16, 36). Although a number of defined antigens have been identified as protective against leishmaniasis in experimental animal models, any single antigen may not be sufficient to efficiently protect the human population due to differences in their immunological backgrounds: Although inbred mice with uniform genotypes are often used in experimental models, humans and dogs have divergent genotypes. In mouse models, SMT is the most immunogenic component of KSAC and, in our hands, is the most protective among the four components of KSAC when tested individually (22). In contrast, SMT is not always the strongest antigen in humans cured from VL (22). Thus, it is anticipated that one component may not be sufficient to serve as a protective vaccine antigen for every member of a genetically diverse population. In such cases, it is important to have additional protective components. In the mouse models used in this study, T cells were not as reactive to A2 and KMP11 as they were to SMT and CPB. Although one possibility is that A2 and KMP11 are not properly encoded in the KSAC construct, this can be ruled out based on the antibody ELISA data; mice immunized with KSAC plus MPL-SE showed antibody responses to A2 and KMP11. When the A2 sequence in the KSAC polyprotein was used as a single antigen for immunizing mice, it could induce detectable levels of T-cell responses (data not shown), suggesting the sequence itself is not a problem. A2 or KMP11 antigens were also not strongly recognized by T cells from humans cured from VL (22), possibly suggesting lower antigenicity/immunogenicity than SMT or CPB in some species. However, these antigens may still be useful anti-VL vaccine antigens in other animals. In fact, protective efficacy of KMP11 delivered as DNA has been shown in a hamster model (3). It is also a fact that KMP11 as a single antigen was not able to induce even an antibody response in mice when injected together with MPL-SE, while we could produce polyclonal antibody to this antigen in rabbits by injection with KMP-11 plus Freund's adjuvant (data not shown). In a related fashion, the immunogenicity and protective efficacy of A2 have been demonstrated recently in dogs, which are important reservoirs of Leishmania parasites in some areas of endemicity (13). Therefore, it will be intriguing to evaluate this vaccine for VL in a variety of affected mammalian hosts, including dogs and humans in future studies.
Due to the value of having more than one antigen in a subunit vaccine as a means to circumvent differing immune recognition within a heterogeneous population, a vaccination approach using multiple antigens has been taken by our group and others, using either polyprotein fusions or mixtures of multiple gene products (2, 9, 11, 27, 34). An advantage of a polyprotein over a mixture is the reduced manufacturing cost, provided that protective efficacies are equivalent between the two, as seen in this study. Using KSAC as an example, if the four individual components are produced separately, the cost of producing proteins individually will be much higher than for the KSAC fusion protein, which we have produced and purified on a large scale. Therefore, the use of this fusion strategy may provide benefits over the use of a protein cocktail in terms of simplicity and cost and would be a practical format in clinical use.
Because multiple Leishmania species causing different forms of leishmaniasis are endemic to some areas and because manufacturing and distribution of a single, broadly useful vaccine would be cost-effective, vaccines that are protective against multiple Leishmania species would be ideal. The KSAC/MPL-SE vaccine induced protection not only against L. infantum but also against L. major, which causes CL. This broad protection is likely attributable to observations that, except for the L. donovani complex-specific antigen A2, the other antigenic components of KSAC are conserved between Leishmania species and have been demonstrated to be protective against L. major infection (6, 15, 35).
In summary, we produced the polyprotein vaccine candidate designed to target VL and demonstrated its protective efficacy against both L. infantum and L. major. As vaccine development for leishmaniasis is advancing worldwide with some promise, it is important to have a clinical perspective, even during preclinical vaccine development. Factors relevant to clinical development, manufacturing, and widespread use are taken into consideration for the antigens tested in this study, and we believe that this polyprotein, or other Leishmania antigens/vaccines developed with these strategies, will be the lead vaccine antigens for leishmaniasis in dogs and humans beginning in the near future.
ACKNOWLEDGMENTS
We are grateful to Rhea Coler, Sylvie Bertholet, Darrick Carter, Malcolm Duthie, Franco Piazza, and Anna Marie Beckmann for valuable discussions, Jeff Guderian and Garrett Poshusta for technical assistance, and Winston Wicomb and Jeff Doepping for maintaining the IDRI animal facility.
This work was supported by National Institutes of Health grant AI25038 and a grant from the Bill and Melinda Gates Foundation (no. 39129).
Footnotes
Published ahead of print on 1 June 2011.
REFERENCES
- 1. Armijos R. X., Weigel M. M., Aviles H., Maldonado R., Racines J. 1998. Field trial of a vaccine against New World cutaneous leishmaniasis in an at-risk child population: safety, immunogenicity, and efficacy during the first 12 months of follow-up. J. Infect. Dis. 177: 1352– 1357 [DOI] [PubMed] [Google Scholar]
- 2. Badaro R., et al. 2006. Immunotherapy for drug-refractory mucosal leishmaniasis. J. Infect. Dis. 194: 1151– 1159 [DOI] [PubMed] [Google Scholar]
- 3. Basu R., et al. 2005. Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1- and Th2-like responses in visceral leishmaniasis. J. Immunol. 174: 7160– 7171 [DOI] [PubMed] [Google Scholar]
- 4. Bendtsen J. D., Nielsen H., von Heijne G., Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340: 783– 795 [DOI] [PubMed] [Google Scholar]
- 5. Bertholet S., et al. 2009. Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine 27: 7036– 7045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhaumik S., Basu R., Sen S., Naskar K., Roy S. 2009. KMP-11 DNA immunization significantly protects against L. donovani infection but requires exogenous IL-12 as an adjuvant for comparable protection against L. major. Vaccine 27: 1306– 1316 [DOI] [PubMed] [Google Scholar]
- 7. Campos-Neto A., et al. 2001. Protection against cutaneous leishmaniasis induced by recombinant antigens in murine and nonhuman primate models of the human disease. Infect. Immun. 69: 4103– 4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carvalho E. M., Badaro R., Reed S. G., Jones T. C., Johnson W. D., Jr 1985. Absence of gamma interferon and interleukin 2 production during active visceral leishmaniasis. J. Clin. Invest. 76: 2066– 2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coler R. N., Goto Y., Bogatzki L., Raman V., Reed S. G. 2007. Leish-111f, a recombinant polyprotein vaccine that protects against visceral Leishmaniasis by elicitation of CD(4+) T cells. Infect. Immun. 75: 4648– 4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coler R. N., Reed S. G. 2005. Second-generation vaccines against leishmaniasis. Trends Parasitol. 21: 244– 249 [DOI] [PubMed] [Google Scholar]
- 11. Coler R. N., et al. 2002. Immunization with a polyprotein vaccine consisting of the T-cell antigens thiol-specific antioxidant, Leishmania major stress-inducible protein 1, and Leishmania elongation initiation factor protects against leishmaniasis. Infect. Immun. 70: 4215– 4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darrah P. A., et al. 2007. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 13: 843– 850 [DOI] [PubMed] [Google Scholar]
- 13. Fernandes A. P., et al. 2008. Protective immunity against challenge with Leishmania (Leishmania) chagasi in beagle dogs vaccinated with recombinant A2 protein. Vaccine 26: 5888– 5895 [DOI] [PubMed] [Google Scholar]
- 14. Ghosh A., Zhang W. W., Matlashewski G. 2001. Immunization with A2 protein results in a mixed Th1/Th2 and a humoral response which protects mice against Leishmania donovani infections. Vaccine 20: 59– 66 [DOI] [PubMed] [Google Scholar]
- 15. Goto Y., et al. 2009. Leishmania infantum sterol 24-c-methyltransferase formulated with MPL-SE induces cross-protection against L. major infection. Vaccine 27: 2884– 2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goto Y., Bogatzki L. Y., Bertholet S., Coler R. N., Reed S. G. 2007. Protective immunization against visceral leishmaniasis using Leishmania sterol 24-c-methyltransferase formulated in adjuvant. Vaccine 25: 7450– 7458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goto Y., et al. 2009. Distinct antigen recognition pattern during zoonotic visceral leishmaniasis in humans and dogs. Vet. Parasitol. 160: 215– 220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gradoni L., et al. 2005. Failure of a multi-subunit recombinant leishmanial vaccine (MML) to protect dogs from Leishmania infantum infection and to prevent disease progression in infected animals. Vaccine 23: 5245– 5251 [DOI] [PubMed] [Google Scholar]
- 19. Haldar J. P., Ghose S., Saha K. C., Ghose A. C. 1983. Cell-mediated immune response in Indian kala-azar and post-kala-azar dermal leishmaniasis. Infect. Immun. 42: 702– 707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Himmelrich H., et al. 2000. In BALB/c mice, IL-4 production during the initial phase of infection with Leishmania major is necessary and sufficient to instruct Th2 cell development resulting in progressive disease. J. Immunol. 164: 4819– 4825 [DOI] [PubMed] [Google Scholar]
- 21. Khalil E. A., et al. 2000. Autoclaved Leishmania major vaccine for prevention of visceral leishmaniasis: a randomised, double-blind, BCG-controlled trial in Sudan. Lancet 356: 1565– 1569 [DOI] [PubMed] [Google Scholar]
- 22. Kumar R., et al. 2010. Evaluation of ex vivo human immune response against candidate antigens for a visceral leishmaniasis vaccine. Am. J. Trop. Med. Hyg. 82: 808– 813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leclercq V., Lebastard M., Belkaid Y., Louis J., Milon G. 1996. The outcome of the parasitic process initiated by Leishmania infantum in laboratory mice: a tissue-dependent pattern controlled by the Lsh and MHC loci. J. Immunol. 157: 4537– 4545 [PubMed] [Google Scholar]
- 24. Liew F. Y., Li Y., Millott S. 1990. Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J. Immunol. 145: 4306– 4310 [PubMed] [Google Scholar]
- 25. Llanos-Cuentas A., et al. 2010. A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1+MPL-SE vaccine when used in combination with sodium stibogluconate for the treatment of mucosal leishmaniasis. Vaccine 28: 7427– 7435 [DOI] [PubMed] [Google Scholar]
- 26. Miret J., et al. 2008. Evaluation of an immunochemotherapeutic protocol constituted of N-methyl meglumine antimoniate (Glucantime) and the recombinant Leish-110f + MPL-SE vaccine to treat canine visceral leishmaniasis. Vaccine 26: 1585– 1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moreno J., et al. 2007. Immunization with H1, HASPB1 and MML Leishmania proteins in a vaccine trial against experimental canine leishmaniasis. Vaccine 25: 5290– 5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mougneau E., et al. 1995. Expression cloning of a protective Leishmania antigen. Science 268: 563– 566 [DOI] [PubMed] [Google Scholar]
- 29. Murray H. W., Jungbluth A., Ritter E., Montelibano C., Marino M. W. 2000. Visceral leishmaniasis in mice devoid of tumor necrosis factor and response to treatment. Infect. Immun. 68: 6289– 6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murray H. W., Miralles G. D., Stoeckle M. Y., McDermott D. F. 1993. Role and effect of IL-2 in experimental visceral leishmaniasis. J. Immunol. 151: 929– 938 [PubMed] [Google Scholar]
- 31. Nascimento E., et al. 2010. A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1+MPL-SE vaccine when used in combination with meglumine antimoniate for the treatment of cutaneous leishmaniasis. Vaccine 28: 6581– 6587 [DOI] [PubMed] [Google Scholar]
- 32. Noazin S., et al. 2008. First generation leishmaniasis vaccines: a review of field efficacy trials. Vaccine 26: 6759– 6767 [DOI] [PubMed] [Google Scholar]
- 33. Palatnik-de-Sousa C. B. 2008. Vaccines for leishmaniasis in the fore coming 25 years. Vaccine 26: 1709– 1724 [DOI] [PubMed] [Google Scholar]
- 34. Rafati S., et al. 2005. Protective vaccination against experimental canine visceral leishmaniasis using a combination of DNA and protein immunization with cysteine proteinases type I and II of L. infantum. Vaccine 23: 3716– 3725 [DOI] [PubMed] [Google Scholar]
- 35. Rafati S., Salmanian A. H., Taheri T., Vafa M., Fasel N. 2001. A protective cocktail vaccine against murine cutaneous leishmaniasis with DNA encoding cysteine proteinases of Leishmania major. Vaccine 19: 3369– 3375 [DOI] [PubMed] [Google Scholar]
- 36. Rafati S., Zahedifard F., Nazgouee F. 2006. Prime-boost vaccination using cysteine proteinases type I and II of Leishmania infantum confers protective immunity in murine visceral leishmaniasis. Vaccine 24: 2169– 2175 [DOI] [PubMed] [Google Scholar]
- 37. Russell D. G., Alexander J. 1988. Effective immunization against cutaneous leishmaniasis with defined membrane antigens reconstituted into liposomes. J. Immunol. 140: 1274– 1279 [PubMed] [Google Scholar]
- 38. Sacks D. L., Melby P. C. 2001. Animal models for the analysis of immune responses to leishmaniasis. Curr. Protoc. Immunol. 2001: Chapter 19, Unit 19.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sadick M. D., et al. 1990. Cure of murine leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell-dependent, interferon gamma-independent mechanism. J. Exp. Med. 171: 115– 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharifi I., et al. 1998. Randomised vaccine trial of single dose of killed Leishmania major plus BCG against anthroponotic cutaneous leishmaniasis in Bam, Iran. Lancet 351: 1540– 1543 [DOI] [PubMed] [Google Scholar]
- 41. Skeiky Y. A., et al. 1995. A recombinant Leishmania antigen that stimulates human peripheral blood mononuclear cells to express a Th1-type cytokine profile and to produce interleukin 12. J. Exp. Med. 181: 1527– 1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skeiky Y. A., et al. 1998. LeIF: a recombinant Leishmania protein that induces an IL-12-mediated Th1 cytokine profile. J. Immunol. 161: 6171– 6179 [PubMed] [Google Scholar]
- 43. Squires K. E., et al. 1989. Experimental visceral leishmaniasis: role of endogenous IFN-gamma in host defense and tissue granulomatous response. J. Immunol. 143: 4244– 4249 [PubMed] [Google Scholar]
- 44. Stager S., Smith D. F., Kaye P. M. 2000. Immunization with a recombinant stage-regulated surface protein from Leishmania donovani induces protection against visceral leishmaniasis. J. Immunol. 165: 7064– 7071 [DOI] [PubMed] [Google Scholar]
- 45. Trigo J., et al. 2010. Treatment of canine visceral leishmaniasis by the vaccine Leish-111f+MPL-SE. Vaccine 28: 3333– 3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Webb J. R., et al. 1998. Human and murine immune responses to a novel Leishmania major recombinant protein encoded by members of a multicopy gene family. Infect. Immun. 66: 3279– 3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Webb J. R., Kaufmann D., Campos-Neto A., Reed S. G. 1996. Molecular cloning of a novel protein antigen of Leishmania major that elicits a potent immune response in experimental murine leishmaniasis. J. Immunol. 157: 5034– 5041 [PubMed] [Google Scholar]