

Abstract
Fermentation enables the production of reduced metabolites, such as the biofuels ethanol and butanol, from fermentable sugars. This work demonstrates a general approach for designing and constructing a production host that uses a heterologous pathway as an obligately fermentative pathway to produce reduced metabolites, specifically, the biofuel isobutanol. Elementary mode analysis was applied to design an Escherichia coli strain optimized for isobutanol production under strictly anaerobic conditions. The central metabolism of E. coli was decomposed into 38,219 functional, unique, and elementary modes (EMs). The model predictions revealed that during anaerobic growth E. coli cannot produce isobutanol as the sole fermentative product. By deleting 7 chromosomal genes, the total 38,219 EMs were constrained to 12 EMs, 6 of which can produce high yields of isobutanol in a range from 0.29 to 0.41 g isobutanol/g glucose under anaerobic conditions. The remaining 6 EMs rely primarily on the pyruvate dehydrogenase enzyme complex (PDHC) and are typically inhibited under anaerobic conditions. The redesigned E. coli strain was constrained to employ the anaerobic isobutanol pathways through deletion of 7 chromosomal genes, addition of 2 heterologous genes, and overexpression of 5 genes. Here we present the design, construction, and characterization of an isobutanol-producing E. coli strain to illustrate the approach. The model predictions are evaluated in relation to experimental data and strategies proposed to improve anaerobic isobutanol production. We also show that the endogenous alcohol/aldehyde dehydrogenase AdhE is the key enzyme responsible for the production of isobutanol and ethanol under anaerobic conditions. The glycolytic flux can be controlled to regulate the ratio of isobutanol to ethanol production.
INTRODUCTION
Society currently relies on the use of fossil fuels to meet its growing energy demands (16). Oil, natural gas, and coal contribute about 84% of the world's energy consumption (International Energy Outlook 2010 [39]). There is thus a pressing need to develop alternative technologies to reduce reliance on fossil fuels (26). One such alternative is the biological conversion of lignocellulosic biomass into biofuels, which has emerged as a promising route to sustainable energy (1, 9, 24).
Recent efforts have focused on engineering Saccharomyces cerevisiae to ferment sugars derived from lignocellulosic biomass hydrolysates to produce ethanol as a biofuel (17). Other microorganisms, such as Zymomonas mobilis (40) and Escherichia coli (20), have also been engineered to produce ethanol. However, ethanol is not an ideal biofuel, due to its miscibility in water and low energy content relative to gasoline. Recently, metabolic engineering and synthetic biology have been applied to engineer recombinant microorganisms to produce biofuels, such as butanol, isobutanol, isopentanol, fatty alcohols, biodiesel, and isoprenoid-derived biofuels, that have similar properties to gasoline or jet fuel (3, 10, 18, 25, 31). However, most of these biofuels are typically produced at low titers and with poor yields, in part because the producing organism must be cultivated under aerobic or oxygen-limited conditions to address a redox imbalance inherent in the production pathways employed.
Fermentation is the anaerobic metabolic conversion of sugars to products, involves endogeneous electron acceptors, and produces reducing equivalents, such as NADH. Fermentation is the most efficient route to synthesize biofuels that are reduced relative to the sugar source. Relevant examples are ethanol production from S. cerevisiae and butanol production from Clostridium acetobutylicum (17, 30). Under anaerobic conditions, the NADH generated from sugar metabolism is recycled to NAD+ by the formation of reduced metabolites, such as alcohols, thus helping to maintain the appropriate redox balance within the cell. However, most pathways that synthesize advanced biofuels are not obligately anaerobic and are active under aerobic conditions, even when the host is a facultative anaerobe. For example, E. coli is able to produce biodiesel and certain hydrocarbons by employing fatty acid biosynthesis pathways only under aerobic conditions (25, 31).
Under aerobic conditions, reducing equivalents in the form of NADH are most efficiently regenerated by oxidation, directly generating ATP for cell synthesis and maintenance, rather than by the production of reduced metabolites such as biofuels (37). To direct cellular metabolism toward the formation of reduced products, cell cultivation is often carried out under oxygen-limited conditions (3). Precise control of the dissolved oxygen concentration is required to partition carbon flow between the synthesis of biomass and reduced metabolites. Therefore, converting biofuel-producing pathways to obligate anaerobic pathways would result in higher yields of reduced metabolites and be advantageous for large-scale production, because oxygen supply and control would not be required.
Isobutanol formation provides an example of a nonfermentative biofuel-producing pathway. Isobutanol is produced via the valine biosynthesis pathway, which is not an obligately fermentative pathway. Saccharomyces cerevisiae was the first microorganism reported to be able to produce isobutanol from 13C-labeled valine as a substrate (14). Recently, E. coli JCL260 pSA55/pSA69 has been engineered to produce isobutanol directly from glucose by using the valine biosynthesis pathway and two additional exogenous enzymes: α-ketoacid decarboxylase (Kivd from Lactobacillus lactis) and alcohol dehydrogenase (Adh2 from Saccharomyces cerevisiae) (3). However, this recombinant isobutanol-producing E. coli was reported to grow and produce isobutanol only in the presence of oxygen, even though E. coli is a facultative anaerobe. The engineered strain could not consume glucose anaerobically because the deletion of the competing pathways resulted in a redox imbalance. The behavior of this phenotype will be clarified by the model predictions and experimental results of the present work.
In the present study, we demonstrate as a proof of concept that it is possible to “retrofit” E. coli to produce isobutanol through a nonnative obligate anaerobic pathway. Elementary mode (EM) analysis was employed to redesign E. coli metabolism. This approach deconstructs the complex metabolic network of E. coli into a complete set of EMs. Each EM contains a minimum and unique set of enzymes that cells can use to function under quasi-steady-state conditions (23, 27–29). Knowledge of the complete set of EMs allows the selection of routes that enable cell growth and synthesis of isobutanol at high yields under anaerobic conditions (32, 33, 35, 36, 38). Herein we illustrate why the former engineered E. coli JCL260 pSA55/pSA69 could not convert glucose to isobutanol anaerobically. We provide details of how the reengineered strain was designed, constructed, and characterized to employ a heterologous, obligate anaerobic isobutanol-producing pathway. Our goals were (i) to demonstrate how E. coli metabolism can be retrofitted for anaerobic isobutanol production and (ii) to identify what rate-limiting steps must be overcome to improve anaerobic isobutanol production. This approach will also guide strategies to rationally design microbes that can synthesize reduced metabolites by retrofitting them with heterologous pathways that function as obligate anaerobic pathways. This paves the way for more efficient and cost-effective routes to produce industrial biotechnological products that are reduced with respect to their more oxidized carbohydrate feedstocks.
MATERIALS AND METHODS
Organisms and plasmids.
Table 1 shows a list of strains and plasmids used in the study.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Source |
|---|---|---|
| E. coli strains | ||
| BW25113 | K-12; λ−; F−lacIqrrnBT14 ΔlacZWJ16hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | 12 |
| JW0855 | BW25113, poxB::Kanr | 6 |
| JW2294 | BW25113, pta::Kanr | 6 |
| JW1841 | BW25113, zwf::Kanr | 6 |
| JW3205 | BW25113, mdh::Kanr | 6 |
| JW1375 | BW25113, ldhA::Kanr | 6 |
| JW4115 | BW25113, frdA::Kanr | 6 |
| JW1095 | BW25113, ndh::Kanr | 6 |
| JW1228 | BW25113, adhE::Kanr | 6 |
| BFA7.001(λDE3) | BW25113, λDE3 Δzwf Δmdh ΔfrdA Δndh Δpta ΔpoxB ΔldhA::Kan | This study |
| BFA8.001(λDE3) | BW25113, λDE3 Δzwf Δmdh ΔfrdA Δndh Δpta ΔpoxB ΔldhA ΔadhE::Kan | This study |
| BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm (λDE3) | Invitrogen (catalog no. C600003) |
| Plasmids | ||
| pSA55 | ColE1 ori, plac::kivd::adh2; Ampr | 3 |
| pSA69 | ColE1 ori, plac::alsS::ilvC::ilvD; Kanr | 3 |
| pCOLADuet | ColA ori; Kanr | Novagen (catalog no. 71406-3) |
| pET24b | f1 ori; Kanr | Novagen (catalog no. 69750-3) |
| pCT01 | pCOLA pT7::alsS::ilvC::ilvD, inserted at BamHI and NotI restriction sites; Kanr | This study |
| pCT02 | pCOLA pT7::kivd::adh2, inserted at NdeI and XhoI restriction sites; Kanr | This study |
| pCT03 | pCOLA pT7::alsS::ilvC::ilvD::pT7::kivd::adh2; Kanr | This study |
| pCT06 | pCOLA pT7::alsS::ilvC::ilvD::pT7::adhE; Kanr | This study |
| pCT13 | pCOLA pT7::alsS::ilvC::ilvD::pT7::kivd::adhE; Kanr | This study |
| pET24b adhE | pET24b pT7::adhE; Kanr | This study |
| pPC20 | FLP+, λ cI857+, λ pR Rep(Ts); Ampr Cmr | 11 |
Strain construction.
All mutants with single deleted genes were obtained from the single-gene knockout library of the Keio Collection (6). These mutants were derived from BW25113, a derivative of E. coli MG1655, and constructed using the technique of one-step disruption of chromosomal genes (12). Mutants with multiple deleted genes were created by multiple steps of P1 transduction from strains with a single deleted gene (33). At each step, the recipient strain that contained one or more deleted genes had the kanamycin cassette removed by using the temperature-sensitive helper plasmid pCP20 (11). Donor strains used to prepare P1 lysate had a single deleted gene with an intact kanamycin cassette. The PCR test was designed to detect complete gene disruption by using primers located outside the undeleted portion of the structural gene (see Table S1 in the supplemental material). To construct a mutant with the T7 polymerase gene, the prophage λDE3 (Novagen λDE3 lysogenization kit, catalog number 69734-3) was used to insert the gene into the specific site of the mutant chromosome.
Plasmid construction.
To construct pCT01, the operon alsS::ilvC::ilvD was amplified from the plasmid pSA69 with the primers alsS_pCOLA_1f (5′-AAA AAA GGA TCC GTT GAC AAA AGC AAC AAA AGA ACA AAA ATC CCT TGT G-3′) and ilvD_pCOLA_1r (5′-AAA AAA GCG GCC GCT TAA CCC CCC AGT TTC GAT TTA TCG CGC ACC-3′). The operon amplification was carried out by PCR with a high-fidelity Pfu polymerase (Agilent Technologies catalog number 600674). The PCR product was doubly digested with BamHI and NotI and ligated into the backbone vector pCOLADuet (Novagen).
To construct pCT02, the operon kivd::adh2 was amplified from the plasmid pSA55 with the primers kivd_pCOLA_1f_d (5′-AAA AAA CAT ATG TAT ACA GTA GGA GAT TAC-3′) and adh2_pCOLA_1r _d (5′-AAA AAA CTC GAG TTA TTT AGA AGT GTC AAC-3′). The PCR product was doubly digested with NdeI and XhoI and ligated into the backbone vector pCOLADuet. To construct pCT03, the operon alsS::ilvC::ilvD was doubly digested with BamHI and NotI and ligated into the vector pCT01.
The operon kivd::adhE was constructed using the overlapping PCR extension technique. The gene kivd was amplified from the plasmid pSA55 by using the primers kivd_adhE_1f_1 5′-AAA AAA CAT ATG TAT ACA GTA GGA GAT TAC-3′) and kivd_adhE_1r_1 (5′-TTC AGC GAC ATT AGT AAC AGC CAT TAT ATC TCC TTT TAT GAT TTA TTT TGT TC-3′), and the gene adhE was amplified from the genomic DNA of E. coli MG1655 by using the primers kivd_adhE_1f_2 (5′-TTT GCT GAA CAA AAT AAA TCA TAA AAG GAG ATA TAA TGG CTG TTA CTA ATG TC-3′) and kivd_adhE_1r_2 (5′-AAA AAA CTC GAG TTA AGC GGA TTT TTT CGC-3′). The operon kivd::adhE was constructed by amplifying both of the amplified genes kivd and adhE with the primers kivd_adhE_1f_1 and kivd_adhE_1r_2. The operon kivd::adhE was digested with NdeI and XhoI and ligated into the backbone vector pCOLADuet to create the vector pCT13A. To construct the vector pCT13, the operon alsS::ilvC::ilvD was doubly digested with BamHI and NotI and ligated into the vector pCT13A. All constructed vectors were confirmed by gel electrophoresis after being digested with either BamHI/NotI or NdeI/XhoI and by sequencing the inserts.
To construct the plasmid pET24b with the adhE gene, the gene adhE was amplified from the genomic DNA of E. coli MG1655 by using the primers adhE_pET24b_1f (5′-AAA AAA CAT ATG GCT GTT ACT AAT GTC GCT-3′) and adhE_pET24b_1r (5′-AAA AAA CTC GAG AGC GGA TTT TTT CGC TTT-3′). The PCR product was doubly digested with NdeI and XhoI and ligated into the vector pET24b to create the vector pET24b adhE. This gene also contained an additional 18-nucleotide sequence from pET24b that encoded the His-tagged sequence at the C terminus of the enzyme AdhE.
Cell cultivation. (i) Medium.
Strains were characterized in a medium containing 6.8 g/liter Na2HPO4, 3.0 g/liter KH2PO4, 1.0 g/liter NH4Cl, 2 ml/liter 1 M MgSO4, 100 μl/liter CaCl2, 1 ml/liter stock trace metals solution, 5 g/liter yeast extract, 100 μg/ml kanamycin, and 20 g/liter glucose (unless otherwise specified). The stock trace metals solution contained 0.15 g/liter H3BO4, 0.065 g/liter CoSO4, 0.05 g/liter ZnSO4·7H2O, 0.015 g/liter MnCl2·4H2O, 0.015 g/liter NaMo4·2H2O, 0.01 g/liter NiCl2·6H2O, 0.005 g/liter CuSO4·5H2O, 3 g/liter Fe(NH4) citrate. A stock of 1 M isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a cell culture at 1 ml/liter until the optical density at 600 nm (OD600) of the cell culture reached about 0.5. Antifoam 204 (Sigma-Aldrich catalog number A6426) was added to the medium at 100 μl/liter before cell inoculation.
(ii) Fermentation.
Batch fermentations were conducted in 3-liter bioreactors (Bioengineering AG, Switzerland) with a working volume of 2 liters, under anaerobic growth conditions. The operating conditions were 200 rpm, pH 7 (unless otherwise specified), and 37°C. Anaerobic conditions were maintained by continuously sparging the bioreactor with N2 at a volumetric flow rate of 100 ml/min throughout the fermentation. Dissolved oxygen was measured by using a polarographic dissolved oxygen probe and found to be 0%, confirming that the bioreactor was anaerobic. Exponentially grown cell cultures (OD600, 0.5 to ∼1) were used for inoculation, and the OD600 of initial cultures in the bioreactors following inoculation was about 0.05. Optical density was measured at 600 nm by using a spectrophotometer (Genesys 20, Thermo Scientific). A correlation between the OD600 and dry weight of cells (DCW) was used to calculate the biomass concentration during fermentation (1 OD600 = 0.5 g DCW/liter). All batch fermentations were performed at least twice in independent experiments, with three samples taken at each time point.
Data analysis.
Product yields were calculated, and a carbon balance was determined as previously described (34, 36). Statistical analysis was performed using SigmaPlot (Systat Software, Inc.).
Protein expression and purification.
From the petri dish, a single colony of BL21(DE3) pET24b adhE was picked and grown overnight in a shake tube containing Luria-Bertani (LB) medium plus 10 g/liter glucose and 50 μg/ml kanamycin. The overnight culture was transferred into a 1-liter shake flask containing 500 ml of the identical medium at an inoculum ratio of 1:100. After the OD600 reached 0.5, IPTG was added to a final concentration of 0.5 mM to induce expression of the His-tagged AdhE. The cell pellet was collected after 4 h of induction by centrifugation at 5,000 × g for 10 min and at 4°C. Cells were lysed by using a French press cell operated at a constant pressure of 21,300 lb/in2. The lysed cells were used to purify the His-tagged AdhE by using Ni-nitrilotriacetic acid (NTA) agarose beads. The purification procedure followed protocols 9 and 12 in the QIA expressionist manual (Qiagen Inc.). The protein concentration was measured by the Bradford method (catalog number 500-0006; Bio-Rad, Hercules, CA).
Enzyme assay.
The enzyme activity of AdhE was measured based on the oxidation of NADH by different aldehydes, including acetaldehyde, isobutyraldehyde, and butyraldehyde, at the wavelength of 340 nm. A 1-ml reaction mixture in morpholinepropanesulfonic acid buffer (pH 7.0) containing 10 μg/ml of the purified protein AdhE, 5 mM aldehyde, and various concentrations of NADH (40 to 150 μM) at 37°C. The reaction was initiated by adding the aldehyde last, and the OD340 was measured and recorded every 10 s during the course of 180 to 360 s.
Analytical techniques.
Metabolites such as glucose, isobutanol, succinate, lactate, acetate, and formate were measured using the high-performance liquid chromatography Prominence system (10A; Shimadzu, Columbia, MD) as described previously (34).
Metabolic network reconstruction and analysis.
The metabolic network of E. coli central metabolism was constructed as previously reported (36). In this work, the isobutanol-producing pathway consisting of 5 enzymatic reactions that convert pyruvate into isobutanol (Fig. 1) was also added. These reactions were as follows: IBUT1, AlsS (acetolactate synthase), 2 pyruvate = acetolactate + CO2; IBUT2, IlvC (2,3-dihydroxy isovalerate oxidoreductase), acetolactate + NADPH + H+ = 2,3-dihydroxy isovalerate + NADP+; IBUT3, IlvD (2,3-dihydroxy isovalerate dehydratase), 2,3-dihydroxy isovalerate = isovalerate + H2O; IBUT4, Kivd/AlsS (ketoacid decarboxylase), isovalerate = isobutanol + CO2; IBUT5, Adh (alcohol dehydrogenase), isobutanal + NADH + H+ = isobutanol + NAD+. EM analysis was used as a metabolic pathway analysis tool to study the metabolic network structure (27) and to rationally design optimal cells with efficient minimized metabolic functionalities (32). EMs of the metabolic network were calculated using the publicly available software program METATOOL 5.0.
Fig. 1.
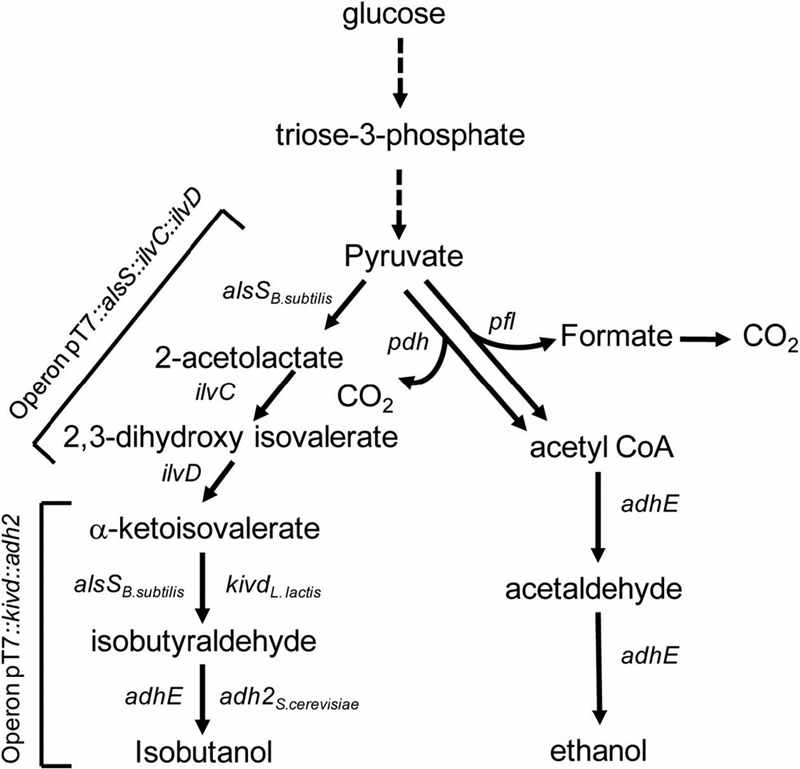
Simplified metabolic network of the designed isobutanol-producing cells, optimized for anaerobic isobutanol synthesis. Pfl (encoded by the gene pflB), pyruvate formate lyase; PDHC (pdh), PDHC enzyme; AlsSB. subtilis (alsSB. subtilis), acetolactate synthase from B. subtilis. It should be noted that E. coli has the endogenous acetolactate synthase enzyme encoded by ilvHI, ilvGM, or ilvNB (http://www.ecocyc.org). Here, AlsSB. subtilis is used to bypass the allosteric inhibition by valine (2, 21). IlvC (ilvC), 2,3-dihydroxy isovalerate oxidoreductase; IlvD (ilvD), 2,3-dihydroxy isovalerate dehydratase; KivdL. lactis (kivdL. lactis), α-ketoacid decarboxylase from L. lactis; Adh2S. cerevisiae, alcohol/aldehyde dehydrogenase from S. cerevisiae; AdhE, aldehyde/alcohol dehydrogenase from E. coli.
RESULTS
Metabolic network analysis. (i) Metabolic network structure.
EM analysis identified a total of 38,219 EMs that used glucose as a carbon source, 5,621 of which operated under anaerobic conditions. These anaerobic EMs included 1,899 isobutanol-producing EMs, only 1,458 of which could coproduce isobutanol and cell biomass (Fig. 2A). The solution space of all admissible EMs was used to study anaerobic isobutanol metabolism in E. coli and to identify the most efficient pathways for anaerobic isobutanol production.
Fig. 2.

Identification of multiple deleted genes in the central metabolism of E. coli for the design of the most efficient fermentative pathways for anaerobic isobutanol production. (A) Effects of multiple deleted genes on the remaining total EMs, biomass-producing EMs, isobutanol-producing EMs, and biomass- and isobutanol-coproducing EMs. (B) Effects of multiple deleted genes on minimal and maximal isobutanol yields under anaerobic conditions. The presented yields were determined from the model prediction.
(ii) Infeasibility of E. coli fermentation for isobutanol production as the sole product.
To examine whether fermentation for isobutanol production as the sole product is feasible in E. coli, we searched for EMs that support both cell biomass and isobutanol production under anaerobic growth conditions. By eliminating all fermentative pathways that produced succinate, lactate, acetate, and ethanol (Δfrd, ΔldhA, Δpta, ΔpoxB, and ΔadhE), we found no existing EMs that could coproduce cell biomass and isobutanol under anaerobic growth conditions (Table 2). All 14 remaining EMs produced isobutanol as the sole product at a theoretical yield of 0.41 g isobutanol/g glucose. A similar result was obtained if only the anaerobic pathways that produced succinate and ethanol, but neither lactate nor acetate, were eliminated. However, if the isobutanol-producing pathway was forced to couple with one of the anaerobic pathways that could oxidize NADH, there existed EMs that coproduced both cell biomass and isobutanol. For instance, if the isobutanol-producing pathway was coupled with the ethanol-producing pathway, then 814 out of 1150 EMs could produce isobutanol. Among these isobutanol-producing pathways, 282 EMs could coproduce cell biomass and isobutanol (Table 2). This result indicates that anaerobic production of isobutanol as the sole product is infeasible due to an imbalance of the reducing equivalents. Isobutanol must be coproduced with other reduced metabolites, such as succinate and ethanol, to balance the reducing equivalents by recycling NADH. Our strategy was thus directed toward production of biofuels by coupling the biosynthesis of isobutanol and ethanol.
Table 2.
Effects of deleting anaerobic pathways on feasibility of producing isobutanol as the sole product
| Deletionsa | No. of EMs based on: |
Range of iBuOH yield on glucose (g/g) |
||||
|---|---|---|---|---|---|---|
| Total | Biomass | iBuOHb | Biomass and iBuOH | Minimum | Maximum | |
| Δfrd ΔldhA Δpta ΔpoxB ΔadhE | 14 | 0 | 14 | 0 | 0.41 | 0.41 |
| Δfrd ΔldhA Δpta ΔpoxB | 1,150 | 900 | 814 | 282 | 0.04 | 0.41 |
| Δfrd ΔldhA ΔadhE | 14 | 0 | 14 | 0 | 0.41 | 0.41 |
| Δfrd Δpta ΔpoxB ΔadhE | 28 | 0 | 14 | 0 | 0.41 | 0.41 |
| ΔldhA Δpta ΔpoxB ΔadhE | 427 | 251 | 427 | 251 | 0.06 | 0.41 |
| Δfrd ΔadhE | 28 | 0 | 14 | 0 | 0.41 | 0.41 |
Δfrd, removal of succinate-producing pathway; ΔldhA, removal of lactate-producing pathway; Δpta ΔpoxB, removal of acetate-producing pathway; ΔadhE, removal of ethanol-producing pathway.
iBuOH, isobutanol.
Rational design of the most efficient isobutanol-producing E. coli strain. (i) Strain design strategy.
The optimal pathways for supporting cell growth and converting glucose into isobutanol can be selected and strictly enforced by removing the inefficient pathways from the metabolic network. The search algorithm to remove the inefficient pathways has been developed and reported in previous publications (15, 32, 33, 35, 36, 38). Briefly, the algorithm identifies the minimal set of reactions that should be deleted from the metabolic network in order to (i) remove as many inefficient pathways for isobutanol production as possible, (ii) retain a small subset of efficient isobutanol-producing pathways that can achieve maximal isobutanol yield, and (iii) tightly couple isobutanol and biomass synthesis during cell growth.
(ii) Strain design implementation.
In order to achieve maximum efficiency of isobutanol production, cellular metabolism must be redirected to use only the optimal anaerobic isobutanol-producing pathways. By deleting 6 reactions (ΔPPP1, ΔTCA9r, ΔTRA5, ΔOPM4r, ΔTRA2, and ΔTRA4), corresponding to knockout of 7 genes (Δzwf, Δmdh, ΔfrdA, Δndh, Δpta, ΔpoxB, and ΔldhA), the total numbers of EMs, biomass-producing EMs, isobutanol-producing EMs, and biomass- and isobutanol-coproducing EMs were reduced from 5,621, 4,500, 1,899, and 1,458 to 12, 4, 6, and 2, respectively (Fig. 2A). After removing the 6 reactions, half of the total 12 EMs produced solely isobutanol while the other half made only ethanol. Interestingly, all 6 EMs that produced solely ethanol used the pyruvate dehydrogenase complex (PDHC) enzyme (the reaction GG13) to convert pyruvate into acetyl coenzyme A (CoA). This enzyme is known to be downregulated by a high intracellular redox state (NADH/NAD+ ratio) under anaerobic conditions (13). In principle, these 6 EMs should not be functional under anaerobic conditions.
If the additional reaction GG13 was deleted by knocking out the gene aceF, the total number of EMs, biomass-producing EMs, isobutanol-producing EMs, and biomass- and isobutanol-coproducing EMs could be reduced from 12, 4, 6, and 2 to 6, 2, 6, and 2, respectively (Fig. 2A). With the deletion of the complete set of seven reactions, the total number of EMs was equal to that of the isobutanol-producing EMs, which emphasizes that the cell was designed to obligately utilize the isobutanol-producing pathway for redox balancing. In addition, the number of the biomass-producing EMs was equal to that of the biomass- and isobutanol-coproducing EMs, which indicated that cell growth and isobutanol production were coupled during the growth-associated phase. In this phase, the designed E. coli strain could produce isobutanol with a yield of 0.29 (g isobutanol/g glucose). In the no-growth phase, all 4 EMs could produce isobutanol at the maximum theoretical yield of 0.41 (g isobutanol/g glucose) (Fig. 2B).
From the analysis, the optimal designed E. coli strain that can efficiently convert glucose into isobutanol by retrofitting the heterologous isobutanol-producing pathway as an obligately anaerobic pathway has the following deletions: Δzwf, Δmdh, ΔfrdA, Δndh, Δpta, ΔpoxB, ΔldhA, and ΔaceF*. The asterisk for the deleted gene aceF implies that if the negative regulation of the high intracellular redox state NADH/NAD+ with the PDHC enzyme is strongly exerted under anaerobic conditions, the PDHC enzyme will be completely downregulated and replaced by pyruvate formate lyase (PFL) to convert pyruvate to acetyl-CoA. Thus, the gene aceF may not need to be deleted.
Phenotypic operating space of the designed isobutanol-producing E. coli strain.
From the stoichiometry reactions of the 12 remaining EMs of the designed isobutanol-producing strain (see Table S2 of the supplemental material), the yields of ethanol and isobutanol on glucose were calculated and mapped on the two-dimensional (2-D) phenotypic space (Fig. 3). This phenotypic space is shown by the bold trapezoid and constructed by connecting the four star symbols located at the edge of the trapezoid. These symbols represent four groups of EMs. The group G1 is comprised of EMs that can produce isobutanol at the theoretical yield of 0.41 (g/g) without biomass production, while the group G2 coproduces isobutanol and biomass with an isobutanol yield of 0.29 (g/g). It should be emphasized that both groups G1 and G2 contain the 6 optimal isobutanol-producing pathways that do not use PDHC. This group of EMs is also constrained to produce a small amount of ethanol, 0.0012 (g/g), to maintain the redox balance. The isobutanol-to-ethanol mass ratio in this group is 236:1.
Fig. 3.

Phenotypic operating space of the designed isobutanol-producing strain and model validation for isobutanol production by strains BFA7.001(DE3) pCT01, BFA7.001(DE3) pCT02, BFA7.001(DE3) pCT03, and BFA7.001(DE3) pCT13.
The group G3 produces only ethanol, at a theoretical yield of 0.51 (g/g), whereas group G4 coproduces ethanol and biomass with a high ethanol yield of 0.36 (g/g). The EMs of both G3 and G4 use the PDHC enzyme to convert pyruvate into ethanol, which is typically inhibited under anaerobic conditions. The designed isobutanol-producing strain can operate anywhere in the trapezoidal phenotypic space (Fig. 3). For isobutanol production, it is desirable to have the designed strain operate close to groups G1 and G2.
Strain comparisons.
Other E. coli strains have been engineered to produce isobutanol, including JCL260 pSA55/pSA69 (3). The following genes are deleted from the host JCL260: ΔadhE, Δfnr, ΔpflB, ΔfrdBC, Δpta, and ΔldhA. According to EM analysis, deletion of these genes disrupts the cell metabolism required to support cell growth, since no existing EMs can produce cell biomass (Table 3).
Table 3.
Elementary mode analysis for the effects of multiple deleted genes applied to different designed E. coli strains, including JCL260 pSA55/pSA69 and BFA7.001(DE3) pCT03 or BFA7.001(DE3) pCT13a under anaerobic conditions
| Deleted gene or summary value | Result for strain |
|
|---|---|---|
| JCL260 pSA55/pSA69 | BFA7.001(DE3) pCT03 | |
| Deleted genes | ||
| ΔadhE | + | |
| Δfnr | + | |
| ΔpflB | + | |
| ΔfrdBC | + | + |
| Δpta | + | + |
| ΔldhA | + | + |
| Δndh | + | |
| ΔpoxB | + | |
| Δzwf | + | |
| Δmdh | + | |
| ΔaceF | + | |
| Total no. of anaerobic EMs | 14 | 6 |
| No. of biomass-producing EMs | 0 | 2 |
| No. of isobutanol-producing EMs | 14 | 6 |
| Isobutanol yield (g/g) | 0.41 | 0.29–0.41 |
Information on JCL260 pSA55/pSA69 is from reference 3, and that for BFA7.001(DE3) pCT03 and BFA7.001(DE3) pCT13 is from this study.
Construction of the designed E. coli strain. (i) Host strain construction.
The designed host E. coli strain BFA7.001(DE3) was constructed with seven genes knocked out (Δzwf, ΔldhA, Δpta, ΔpoxB, Δmdh, Δndh, ΔfrdA, and ΔaceF*) according to the model prediction for anaerobic conversion of glucose to isobutanol. In this study, we did not delete the gene aceF to remove the PDHC reaction GG13, operating under the assumption that the PDHC enzyme is downregulated and the PFL enzyme is solely responsible for converting pyruvate to acetyl-CoA. In addition, we were interested in exploring how the level of downregulation of the PDHC enzyme affected the production of isobutanol and ethanol. The knockout genes in the designed strain were confirmed by PCR. The designed strain also had the T7 polymerase gene inserted into the chromosome (see Fig. S1 in the supplemental material).
(ii) Construction of the isobutanol-producing pathway.
The isobutanol-producing pathway (Fig. 1; see also Fig. S2 in the supplemental material) was constructed by using a single plasmid, pCT03, that had a low copy number, a strong T7 promoter, and a kanamycin selection. The plasmid pCT03 derived from pCOLADuet contains two operons, including a complete set of five genes for converting pyruvate to isobutanol. The first operon, pT7::alsS::ilvC::ilvD, contained 3 genes (alsSB. subtilis, ilvC, and ilvD) under the T7 promoter that were used to amplify fluxes from pyruvate to α-ketoisovalerate. It should be noted that the native E. coli has these endogeneous enzymes to generate the precursor α-ketoisovalerate for l-valine biosynthesis. The second operon (pT7::kivd::adh2) contained two genes (kivdL. lactis and adh2S. cerevisiae) under the T7 promoter that were used to convert α-ketoisovalerate to isobutanol. Native E. coli does not have α-ketoacid decarboxylase (encoded by the KivdL. lactis gene), an important enzyme for converting α-ketoisovalerate to isobutyraldehyde.
Besides pCT03, we also constructed different incomplete parts of the isobutanol-producing pathway and expressed different alcohol dehydrogenases, such as AdhE (encoded by adhEE. coli). Specifically, the plasmid pCT01 contained the first operon, pT7::alsS::ilvC::ilvD. The plasmid pCT02 contained the second operon, pT7::kivd::adh2. The plasmid pCT13 contained both the first operon, pT7::alsS::ilvC::ilvD, and the second operon, pT7::kivd::adhE. It should be noted that pCT03 had the alcohol dehydrogenase Adh2 from S. cerevisiae while pCT13 had the alcohol dehydrogenase AdhE from E. coli.
Characterization of the designed isobutanol-producing strains. (i) Anaerobic growth of isobutanol-producing E. coli strains.
The designed isobutanol-producing E. coli strain BFA7.001(DE3) pCT03 was first tested for its ability to grow anaerobically under defined cultivation conditions (pH 7, 37°C, 200 rpm, 100 ml/min nitrogen). Consistent with the model prediction, the designed strain was able to completely ferment 20 g/liter glucose after 36 h and had a specific growth rate of 0.37 ± 0.03 h−1 (mean ± standard deviation) (Fig. 4).
Fig. 4.
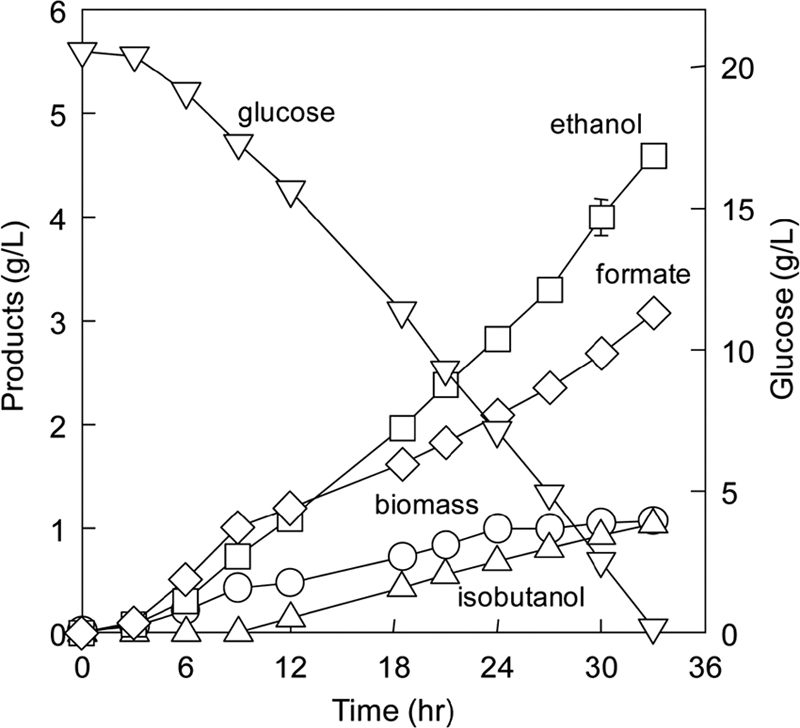
Fermentation of the designed isobutanol-producing E. coli BFA7.001(DE3) pCT03.
To confirm that the ethanol-producing pathway is required to couple with the isobutanol-producing pathway in order to support cell growth, we constructed a variant E. coli strain, BFA8.001(DE3), that was derived from BFA7.001(DE3) and had the endogenous alcohol dehydrogenase gene adhE deleted. Strains BFA8.001(DE3) pCT01, BFA8.001(DE3) pCT02, and BFA8.001(DE3) pCT03, which contained different portions of the isobutanol-producing pathway, were also constructed and tested for their fermentation capabilities. The results show that none of these strains was able to grow anaerobically (Fig. 5), presumably due to the reducing equivalent (NADH) imbalance. However, when the ethanol-producing pathway containing the endogenous enzyme AdhE was restored, the resulting E. coli strain, BFA8.001(DE3) pCT06, was able to grow even though it exhibited a long lag phase (Fig. 5). Here, the plasmid pCT06 contained two operons, pT7::alsS::ilvC::ilvD and pT7::adhE.
Fig. 5.

Cell growth of BFA8.001(DE3) containing plasmids pCT01 (○), pCT02 (▵), pCT03 (□), and pCT06 (▿) under identical anaerobic growth conditions. The medium contained 40 g/liter of glucose as the carbon source.
We also tested the fermentation capability of JCL260 pSA55 pSA69 (3). The results showed that the strain was unable to consume glucose anaerobically as a result of a genetic manipulation that led to a redox imbalance, as predicted by EM analysis. The strain was only able to grow aerobically with a supply of 100 ml/min air instead of nitrogen, with other operating conditions remaining the same (Fig. 6). In the experiment, the oxygen transfer coefficient (kLa) was set at 1.2 h−1 to test the growth sensitivity of JCL260 pSA55/pSA69 under oxygen-limited growth conditions. Within 3 h after inoculation, the dissolved oxygen in the bioreactor was reduced to zero, indicating that oxygen became limiting. The strain had an oxygen consumption rate of 8.16 mg/liter/h. Under the oxygen-limited condition, the growth of JCL260 pSA55/pSA69 was dramatically affected and exhibited a specific growth rate of 0.08 ± 0.00 h−1 (Fig. 6). This result demonstrates the challenge of precisely supplying oxygen to support cell growth and isobutanol production under oxygen-limited conditions, which becomes especially difficult for commercial production of isobutanol.
Fig. 6.
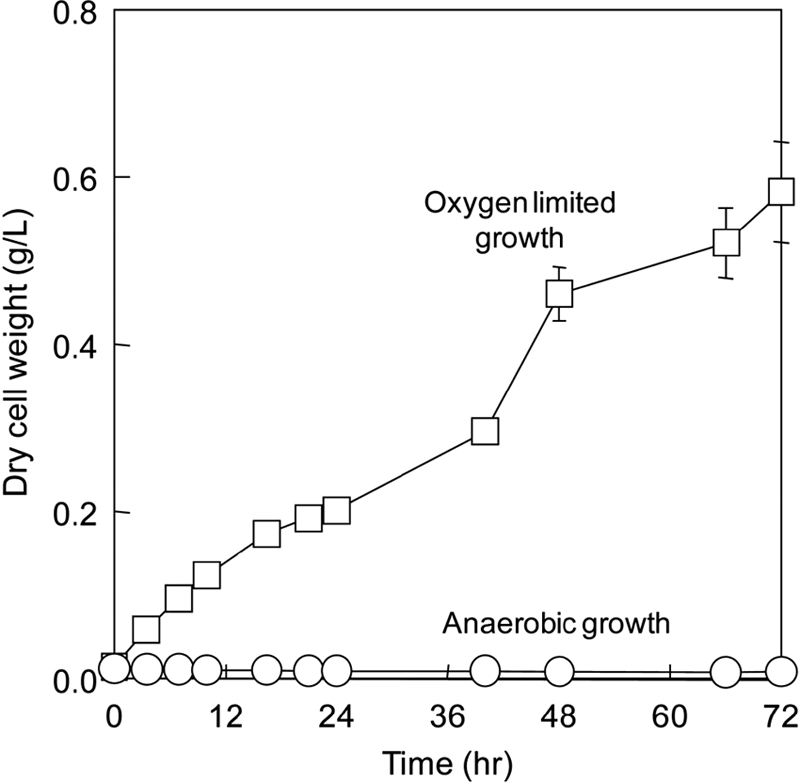
Cell growth of JCL260 pSA55 pSA69 under anaerobic and oxygen-limited conditions. The medium contained 40 g/liter of glucose as the carbon source.
Metabolite production of the designed isobutanol-producing strain.
Figure 4 shows the distribution of the fermentative products that BFA7.001(DE3) pCT03 produced during fermentation. The fraction of carbon in the products that were derived from glucose was 0.83 ± 0.02. Ethanol and isobutanol were produced as the main alcohols, with a mass ratio of (0.26 ± 0.01):1. The designed strain was able to produce isobutanol during the growth-associated (7 to 24 h) and no-growth (>24 h) phases and to recycle the reducing equivalent NADH. Ethanol was produced as the major product, indicating that the isobutanol-producing pathway was limiting and the ethanol-producing pathway contributed more in recycling NADH. The low mass ratio of isobutanol to ethanol implies that pyruvate dehydrogenase may be active in converting pyruvate to acetyl-CoA, as suggested by the EM analysis.
Formate was also produced as an unavoidable fermentative product (Fig. 4). From the stoichiometry of E. coli fermentative metabolism, for every mole of ethanol (46 g/g-mole) produced from pyruvate via the PFL enzyme, one mole of formate (46 g/g-mole) is made, together with 1 mole of acetyl-CoA, which is a precursor for cell biomass. During the first 12 h of fermentation, formate production paralleled ethanol production, indicating the major role of the PFL enzyme. After 12 h, formate production decreased as a result of either the oxidation of formate to CO2 by formate hydrogenlyase (FHL) or the dehydrogenation of pyruvate directly to CO2 and acetyl-CoA by the PDHC enzyme.
Effect of overexpression of different portions of isobutanol-producing pathways on isobutanol production and cell physiology.
Different portions of the isobutanol-producing pathways were overexpressed and characterized in the designed host strain BFA7.001(DE3) under identical anaerobic conditions. Figure 7 compares fermentations of all strains, including BFA7.001(DE3) pCT01, BFA7.001(DE3) pCT02, and BFA7.001(DE3) pCT03. Both BFA7.001(DE) pCT01 (μ = 0.45 ± 0.02 h−1) and BFA7.001(DE3) pCT02 (μ = 0.49 ± 0.01 h−1) grew faster than BFA7.001(DE) pCT03 (μ = 0.37 ± 0.03 h−1), which probably suffered from the significant metabolic burden of carrying a larger plasmid with the complete isobutanol-producing pathway. Similarly, both BFA7.001(DE) pCT01 (4.71 ± 0.20 g glucose/g DCW/h) and BFA7.001(DE3) pCT02 (4.43 ± 0.15 g glucose/g DCW/h) had higher specific glucose uptake rates than BFA7.001(DE3) pCT03 (4.20 ± 0.39 g glucose/g DCW/h) (Fig. 7A).
Fig. 7.
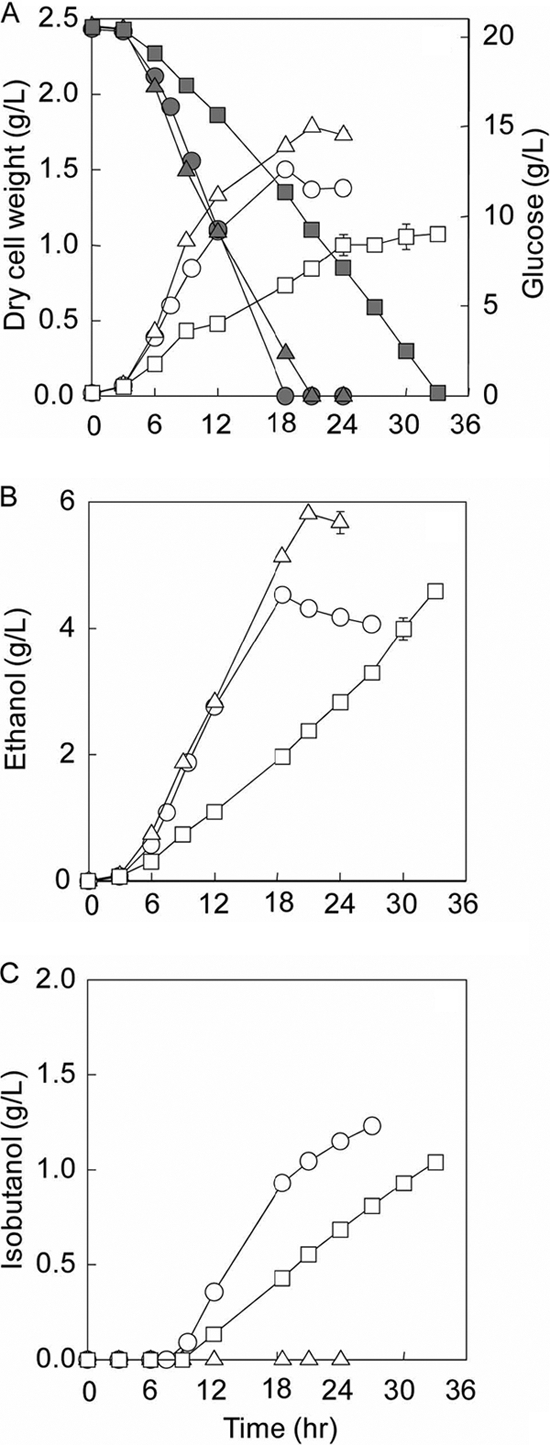
Effects of overexpressing different portions of the isobutanol-producing pathway on the designed host BFA7.001(DE3), including glucose consumption and biomass production (A), ethanol production (B), and isobutanol production (C). The characterized strains were BFA7.001(DE3) pCT01 (○), BFA7.001(DE3) pCT02 (▵), and BFA7.001(DE3) pCT03 (□). In panel A, the filled symbols refer to glucose consumption, while the open symbols represent the biomass production.
The strain BFA7.001(DE3) pCT01 overexpressed the first portion of the isobutanol-producing pathway to convert pyruvate to α-ketoisovalerate. It was not expected to produce isobutanol, because the native E. coli is not known to have an α-ketoacid decarboxylase to convert α-ketoisovalerate into isobutyraldehyde. However, the strain was unexpectedly capable of producing isobutanol under anaerobic conditions (Fig. 7C). This result implies that AlsSB. subtilis has the dual function of not only a known acetotalactate synthase, converting pyruvate into 2-acetolactate, but also an α-ketoacid decarboxylase converting α-ketoisovalerate into isobutyraldehyde. In addition, the result strongly suggests that the endogenous alcohol dehydrogenase(s) of E. coli converted isobutyraldehyde to isobutanol. These observations are consistent with the recent reports of Atsumi et al. (4, 5).
In strain BFA7.001(DE3) pCT02 the second portion of the isobutanol-producing pathway was amplified to convert α-ketoisovalerate into isobutanol. Nevertheless, BFA7.001(DE3) pCT02 could not produce isobutanol (Fig. 7C) but did secrete pyruvate, a precursor metabolite for isobutanol production that was not detected in the fermentation of either BFA7.001(DE3) pCT01 or BFA7.001(DE3) pCT03 (data not shown). This result implies that the first section of the isobutanol-producing pathway was limiting. Because BFA7.001(DE3) pCT02 could not synthesize isobutanol, it produced ethanol at a higher rate and titer than BFA7.001(DE3) pCT01 and BFA7.001(DE3) pCT03 in order to recycle the reducing equivalent of NADH (Fig. 7B).
Strain BFA7.001(DE3) pCT03 contained not only the overexpressed α-ketoacid decarboxylase from KivdL. lactis and AlsSB. subtilis but also the amplified alcohol dehydrogenase from Adh2S. cerevisiae. However, the strain produced isobutanol at almost the same titer as BFA7.001(DE3) pCT01 (Fig. 7C). This result strongly implies that Adh2S. cerevisiae had either low or no activity for isobutyraldehyde, at least under anaerobic conditions, in sharp contrast to the report by Atsumi et al. (3), and that alcohol/aldehyde dehydrogenase was a rate-limiting step. Recently, Atsumi et al. (5) demonstrated that the endogenous alcohol/aldehyde dehydrogenase YqhD was responsible for aerobic isobutanol production, not the exogenous enzyme Adh2S. cerevisiae as originally reported (3). Because the enzyme YqhD is NADPH dependent and the source of NADPH produced by the glucose-6-phosphate dehydrogenase (Zwf) from the oxidative pentose phosphate pathway was knocked out in the designed isobutanol-producing strain, it is unlikely that YqhD played a role in anaerobic isobutanol production.
Role of endogenous AdhE in the isobutanol-producing pathway.
BFA7.001(DE3) pCT01 produced isobutanol without the exogenous alcohol dehydrogenase Adh2S. cerevisiae, indicating that the endogenous alcohol dehydrogenase(s) of E. coli was responsible for anaerobic isobutanol production. BFA7.001(DE3) pCT01 also produced isobutanol at the same titer as BFA7.001(DE3) pCT03, indicating that Adh2S. cerevisiae was not important in anaerobic isobutanol production. In addition, BFA8.001(DE3), which was derived from the parent strain BFA7.001(DE3) and had the gene adhE deleted, could not consume glucose anaerobically when carrying the plasmids pCT01, pCT02, or pCT03 (Fig. 5). Together, these results suggest that the endogenous enzyme AdhE is involved in anaerobic isobutanol production. To confirm the role of AdhE in anaerobic isobutanol production, His-tagged AdhE was produced in BL21(DE3) pET24b adhE, purified using Ni-NTA agarose beads (see Fig. S3 in the supplemental material), and assayed for activity. The activity assay showed that AdhE was NADH dependent and had activity not only for acetaldehyde but also for isobutyraldehyde and butyraldehyde. It is interesting that AdhE had the highest enzymatic activity for butyraldehyde under the assay conditions employed (Table 4).
Table 4.
Kinetic parameters for the endogeneous alcohol/aldehyde dehydrogenase AdhE reacting with acetaldehyde, isobutyraldehyde, or butyraldehydea
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (103) (mM−1 s−1) |
|---|---|---|---|
| Acetaldehyde | 48.6 ± 3.5 | 5.72 ± 0.07 | 118 ± 7 |
| Isobutyraldehyde | 54.3 ± 5.1 | 2.13 ± 0.05 | 39.5 ± 2.8 |
| Butyraldehyde | 196 ± 38 | 59.2 ± 8.1 | 305 ± 17.1 |
Each value is a mean ± average deviation from two independent measurements. Nonlinear regression (SigmaPlot) was used to determine Km and kcat values with P values of <0.05. The R2 coefficients for nonlinear regression were greater than 0.99 for all cases.
Alleviating the alcohol dehydrogenase rate-limiting step.
To alleviate the alcohol dehydrogenase rate limitation, we constructed strain BFA7.001(DE3) pCT13 to overexpress the entire isobutanol-producing pathway and to amplify the endogenous alcohol dehydrogenase AdhE instead of Adh2S. cerevisiae. Even though BFA7.001(DE3) pCT13 (0.42 ± 0.01 h−1) carried a larger plasmid than BFA7.001(DE3) pCT03 (see Fig. S2 in the supplemental material), it was still able to grow at a higher rate under identical anaerobic conditions (pH 7, 200 rpm, 37°C, 100 ml/min nitrogen), further supporting the unimportant role of Adh2S. cerevisiae (Fig. 8A). BFA7.001(DE3) pCT13 produced isobutanol at a higher titer than either BFA7.001(DE3) pCT01 (66% higher) or BFA7.001(DE3) pCT03 (28% higher) (Table 5).
Fig. 8.
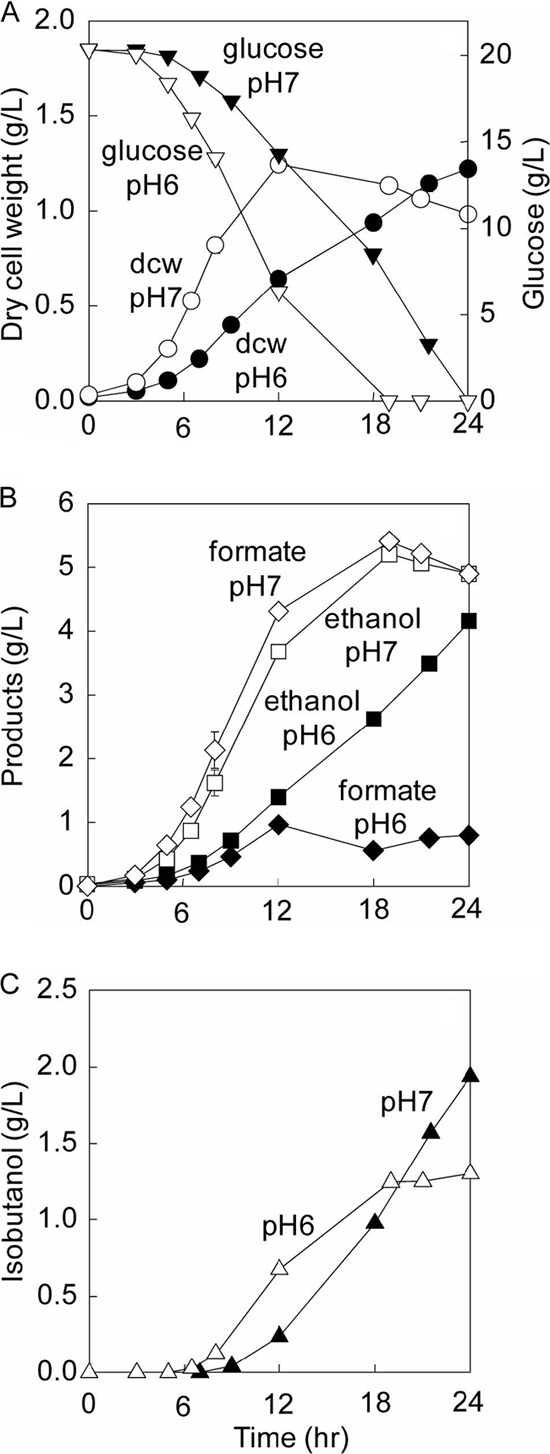
Fermentation of the designed isobutanol-producing E. coli BFA7.001(DE3) pCT13 at pH 6 and pH 7. Kinetic profiles include glucose consumption and biomass production (A), ethanol and formate production (B), and isobutanol production (C).
Table 5.
Specific growth rates, isobutanol concentrations, and mass ratios of isobutanol to ethanol of the strains BFA7.001(DE3) pCT01, BFA7.001(DE3) pCT02, BFA7.001(DE3) pCT03, and BFA7.001(DE3) pCT13, characterized in batch culture under anaerobic conditions
| Strain | pH | Specific growth rate (h−1) | Isobutanol (g/liter) | IBUT:ETOH ratio (g/g) |
|---|---|---|---|---|
| BFA7.001(DE3) pCT01 | 7 | 0.45 ± 0.02 | 0.81 ± 0.13 | 0.28 ± 0.04 |
| BFA7.001(DE3) pCT02 | 7 | 0.49 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| BFA7.001(DE3) pCT03 | 7 | 0.37 ± 0.03 | 1.11 ± 0.07 | 0.26 ± 0.01 |
| BFA7.001(DE3) pCT13 | 7 | 0.42 ± 0.01 | 1.34 ± 0.05 | 0.31 ± 0.02 |
| BFA7.001(DE3) pCT13 | 6 | 0.35 ± 0.01 | 1.72 ± 0.22 | 0.52 ± 0.08 |
Effect of glycolytic flux on isobutanol production.
Our recent study of E. coli KO11 in both batch and continuous fermentation showed that an increase in glycolytic flux led to an increasing specific glucose uptake rate. This increased uptake rate of glucose resulted in rapid cell growth and production of lactate from pyruvate as a route to balance the redox potential (34). However, since the designed strain BFA7.001(DE3) pCT13 had the lactate dehydrogenase LdhA deleted and the endogenous alcohol/aldehyde dehydrogenase AdhE had higher specific activity toward acetaldehyde than isobutyraldehyde (Table 4), we hypothesized that the reducing equivalent NADH was more quickly and efficiently recycled for faster cell growth by using the ethanol-producing pathway. This phenotype might explain the higher production of ethanol than isobutanol, with a mass ratio of isobutanol to ethanol of (0.31 ± 0.02):1 (Table 5).
To test this hypothesis, BFA7.001(DE3) pCT13 was grown at pH 6 and 7 in order to change both the cell growth and specific glucose uptake rates that regulate the glycolytic fluxes. The results in Fig. 8A show that when the pH was reduced from 7 to 6, the specific growth rate decreased by 16%, from 0.42 ± 0.01 h−1 to 0.35 ± 0.01 h−1, and the specific glucose uptake rate dropped by 21%, from 5.23 ± 0.32 to 4.13 ± 0.65 (g glucose/g DCW/h). However, the isobutanol production increased by 28%, from 1.34 ± 0.05 to 1.74 ± 0.22 g/liter, and the ethanol production decreased by 12%, from 4.71 ± 0.20 to 4.17 ± 0.05 g/liter (Fig. 8B and C). The mass ratio of isobutanol to ethanol shifted from (0.31 ± 0.02):1 to (0.53 ± 0.08):1 (Table 5). It should be noted that each value represents the mean ± the standard deviation (n ≥ 6). The t test for statistical analysis in SigmaPlot gave P values of <0.05 for comparing the respective concentrations, indicating that these results are statistically significant with greater than 95% confidence.
One interesting observation during fermentation at low pH is that formate production was significantly reduced and was lower than that of ethanol (Fig. 8B). The lower production was due to the conversion of formate into CO2 and H2 via formate hydrogenlyase, which is known to be overexpressed in acidic environments (8, 22, 41). This phenotype is useful for the cell because the accumulation of formate is toxic to cell growth and hence benefits cell maintenance.
Model validation.
Experimental data on the physiological states of all isobutanol-producing strains, including BFA7.001(DE3) pCT01, BFA7.001(DE3) pCT02, BFA7.001(DE3) pCT03, and BFA7.001(DE3) pCT13, were projected on the 2-D phenotypic space shown in Fig. 3. The physiological states of BFA7.001(DE3) pCT13 fit very well on the predicted phenotypic space when operated at both pH 6 and pH 7. As the pH was reduced from 7 to 6, the designed strain was able to shift from a lower isobutanol-producing and higher ethanol-producing phenotype to a higher isobutanol-producing and lower ethanol-producing phenotype. Strain optimization can be employed to improve the isobutanol-to-ethanol selectivity, but this was not an objective of the present work.
DISCUSSION
In this proof-of-concept study, the central metabolism of E. coli was rewired to convert the heterologous isobutanol-producing pathway to an obligately anaerobic pathway. The engineered E. coli strain was rationally designed based on EM analysis in order to identify the optimal pathways for the strain to grow and produce isobutanol anaerobically. The designed strain was then constructed and its performance characterized under controlled anaerobic conditions.
It is noteworthy that the designed isobutanol-producing strain BFA7.001(DE3) pCT13 could operate in the predicted phenotypic space (Fig. 3). The designed strain was able to grow and produce isobutanol anaerobically. Due to the infeasibility of solely producing isobutanol, ethanol must be coproduced with isobutanol to balance the high intracellular redox state under anaerobic conditions. Thus, the designed strain BFA7.001(DE3) pCT13 produced mixed alcohols, with isobutanol and ethanol as the major fermentative products. The adhE-deficient strain derived from the host BFA7.001(DE3) could not grow anaerobically unless complemented with the gene adhE.
In a previous study, JCL260 pSA55/pSA69 was reported to produce isobutanol under oxygen-limited conditions (3). The strain could not consume glucose anaerobically because multiple genes were inactivated to remove competitive pathways, resulting in a redox imbalance. This was predicted by EM analysis and experimentally verified in the controlled bioreactor experiments performed in the present study. In particular, EM analysis showed that deleting only fumarate reductase (frdABC) and alcohol/aldehyde dehydrogenase (adhE) was sufficient to inactivate anaerobic growth in JCL260 pSA55/pSA69 (Table 2). Even though JCL260 pSA55/pSA69 could convert glucose to isobutanol microaerobically, the precise control of oxygen to support cell growth and enable isobutanol production is a difficult challenge.
Both the isobutanol yield (∼20% of the theoretical yield) and mass ratio of isobutanol to ethanol (0.52) were lower than the predicted values derived from the 6 most efficient isobutanol-producing pathways (Fig. 2; see also Table S2 in the supplemental material), which are located on the line connecting groups G1 and G2 in Fig. 3. The key reason is that the PDHC enzyme that was assumed to be strongly inhibited under anaerobic conditions could be unexpectedly employed for anaerobic isobutanol production. The PDHC enzyme is typically present at low levels under anaerobic conditions due to the inhibition of NADH on the E2 subunit of the enzyme, reducing the production of NADH. Its anaerobic function is replaced by the PFL enzyme (13), but this does not imply that the PDHC enzyme is not expressed and produced (19). In the designed isobutanol-producing strains, the PDHC enzyme was likely activated, because overexpression of the endogenous alcohol/aldehyde dehydrogenase AdhE could help the cell to quickly recycle NADH and alleviate NADH inhibition of the PDHC enzyme. In addition, the low or nonspecific activity of the isobutyraldehyde dehydrogenase Adh2S. cerevisiae contributed significantly to the low isobutanol and high ethanol production. Thus, the isobutanol-producing pathway is limiting at the final conversion step. The observed phenotype provides valuable insights into the metabolism of the designed isobutanol-producing strain and suggests the next step of strain optimization. Anaerobic isobutanol production can be improved by (i) using a more specific isobutyraldehyde dehydrogenase in place of either AdhEE. coli or Adh2S. cerevisiae (potential candidates are AdhE2 from C. acetobutylicum and engineered AdhA from L. lactis [7]) and (ii) completely inactivating the PDHC enzyme in BFA7.001(DE3) by deleting aceF.
An important finding of this study is that the endogenous NADH-dependent alcohol/aldehyde dehydrogenase AdhE plays a key role in anaerobic isobutanol production by converting isobutyraldehyde to isobutanol. Its unique role is supported by several lines of evidence. First, the designed strains BFA8.001(DE3) pCT01, BFA8.001(DE3) pCT02, and BFA8.001(DE3) pCT03, in which the gene adhE was deleted, were unable to grow. However, the complemented strain BFA8.001(DE3) pCT06 was able to grow when AdhE function was restored. In addition, overexpression of the gene adhE in the designed isobutanol-producing strain BFA7.001(DE3) pCT13 increased isobutanol production. Finally, the role of AdhE role was confirmed by the in vitro enzyme assay with the purified His-tagged AdhE. Interestingly, the enzyme AdhE had the highest activity on butyraldehyde, and its activity on acetaldehyde was about 3-fold higher than on isobutyraldehyde. This result explains why overexpression of adhE in the designed strain did not significantly improve isobutanol production.
Control of glycolytic flux plays a significant role in improving isobutanol production and changing the isobutanol-to-ethanol mass ratio in the designed isobutanol-producing strain. The high glycolytic flux causes a high intracellular redox state (ratio of NADH to NAD+) that requires the strain to recycle NADH as quickly as possible from the ethanol-producing pathway. Thus, the strain will make more ethanol and less isobutanol. However, if the glycolytic flux is adjusted, the isobutanol-to-ethanol mass ratio can be controlled. We demonstrated this by carrying out controlled batch fermentations at pH 6 and 7. Lowering the pH decreased the glycolytic flux, reduced the high intracellular redox state, and hence increased the isobutanol-to-ethanol mass ratio. Other cultivation techniques, such as chemostat operation, can also be applied to demonstrate the observed phenotype.
In summary, we have demonstrated a powerful approach to rationally design an E. coli strain that employs the heterologous isobutanol-producing pathway as an obligately anaerobic pathway. Metabolic pathway analysis was used to dissect E. coli cellular metabolism and identify the most efficient pathways for anaerobic isobutanol production. The designed strain functioned in accordance with these selected optimal pathways, and its metabolism closely matched the model predictions. With the anaerobic isobutanol metabolism of the designed strain characterized, strain optimization to improve the isobutanol production and control the isobutanol-to-ethanol mass ratio can be addressed. The present approach has thus proven useful in rational strain development for conversion of renewable and sustainable biomass feedstocks into biofuels and biochemicals.
Supplementary Material
ACKNOWLEDGMENT
We thank James C. Liao at UCLA for kindly providing the strain JCL260 and the plasmids pSA55 and pSA69.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 3 June 2011.
REFERENCES
- 1. Alper H., Stephanopoulos G. 2009. Engineering for biofuels: exploiting innate microbial capacity or importing biosynthetic potential? Nat. Rev. Microbiol. 7:715–723 [DOI] [PubMed] [Google Scholar]
- 2. Atsumi S., et al. 2008. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 10:305–311 [DOI] [PubMed] [Google Scholar]
- 3. Atsumi S., Hanai T., Liao J. C. 2008. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–89 [DOI] [PubMed] [Google Scholar]
- 4. Atsumi S., Li Z., Liao J. C. 2009. Acetolactate synthase from Bacillus subtilis serves as a 2-ketoisovalerate decarboxylase for isobutanol biosynthesis in Escherichia coli. Appl. Environ. Microbiol. 75:6306–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atsumi S., et al. 2010. Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl. Microbiol. Biotechnol. 85:651–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baba T., et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio Collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bastian S., et al. 2011. Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab. Eng. 13:345–352 [DOI] [PubMed] [Google Scholar]
- 8. Birkmann A., Zinoni F., Sawers G., Böck A. 1987. Factors affecting transcriptional regulation of the formate-hydrogen-lyase pathway of Escherichia coli. Arch. Microbiol. 148:44–51 [DOI] [PubMed] [Google Scholar]
- 9. Blanch H. W., et al. 2008. Addressing the need for alternative transportation fuels: the Joint BioEnergy Institute. ACS Chem. Biol. 3:17–20 [DOI] [PubMed] [Google Scholar]
- 10. Cann A., Liao J. 2010. Pentanol isomer synthesis in engineered microorganisms. Appl. Microbiol. Biotechnol. 85:893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cherepanov P. P., Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 12. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Graef M. R., Alexeeva S., Snoep J. L., Teixeira de Mattos M. J. 1999. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 181:2351–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dickinson J. R., Harrison S. J., Hewlins M. J. E. 1998. An investigation of the metabolism of valine to isobutyl alcohol in Saccharomyces cerevisiae. J. Biol. Chem. 273:25751–25756 [DOI] [PubMed] [Google Scholar]
- 15. Hädicke O., Klamt S. 2010. CASOP: a computational approach for strain optimization aiming at high productivity. J. Biotechnol. 147:88–101 [DOI] [PubMed] [Google Scholar]
- 16. Herrera S. 2006. Bonkers about biofuels. Nat. Biotechnol. 24:755–760 [DOI] [PubMed] [Google Scholar]
- 17. Ho N. W., Chen Z., Brainard A. P. 1998. Genetically engineered Saccharomyces yeast capable of effective cofermentation of glucose and xylose. Appl. Environ. Microbiol. 64:1852–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Inui M. 2008. Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl. Microbiol. Biotechnol. 77:1305–1316 [DOI] [PubMed] [Google Scholar]
- 19. Murarka A., Clomburg J. M., Moran S., Shanks J. V., Gonzalez R. 2010. Metabolic analysis of wild-type Escherichia coli and a pyruvate dehydrogenase complex (PDHC)-deficient derivative reveals the role of PDHC in the fermentative metabolism of glucose. J. Biol. Chem. 285:31548–31558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohta K., Beall D. S., Mejia J. P., Shanmugam K. T., Ingram L. O. 1991. Genetic improvement of Escherichia coli for ethanol production: chromosomal integration of Zymomonas mobilis genes encoding pyruvate decarboxylase and alcohol dehydrogenase II. Appl. Environ. Microbiol. 57:893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park J. H., Lee K. H., Kim T. Y., Lee S. Y. 2007. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. U. S. A. 104:7797–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pecher A., et al. 1983. On the redox control of synthesis of anaerobically induced enzymes in enterobacteriaceae. Arch. Microbiol. 136:131–136 [DOI] [PubMed] [Google Scholar]
- 23. Pfeiffer T., Sanchez-Valdenebro I., Nuno J. C., Montero F., Schuster S. 1999. METATOOL: for studying metabolic networks. Bioinformatics 15:251–257 [DOI] [PubMed] [Google Scholar]
- 24. Ragauskas A. J., et al. 2006. The path forward for biofuels and biomaterials. Science 311:484–489 [DOI] [PubMed] [Google Scholar]
- 25. Schirmer A., Rude M. A., Li X., Popova E., del Cardayre S. B. 2010. Microbial biosynthesis of alkanes. Science 329:559–562 [DOI] [PubMed] [Google Scholar]
- 26. Schubert C. 2006. Can biofuels finally take center stage? Nat. Biotechnol. 24:777–784 [DOI] [PubMed] [Google Scholar]
- 27. Schuster S., Fell D. A., Dandekar T. 2000. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 18:326–332 [DOI] [PubMed] [Google Scholar]
- 28. Schuster S., Hilgetag C., Woods J. H., Fell D. A. 1994. Elementary modes of functioning in biochemical networks, p. 151–165 In Cuthbertson R., Holcombe M., Paton R. (ed.), Computation in cellular and molecular biological systems. World Scientific, Singapore [Google Scholar]
- 29. Schuster S., Hilgetag C., Woods J. H., Fell D. A. 2002. Reaction routes in biochemical reaction systems: algebraic properties, validated calculation procedure and example from nucleotide metabolism. J. Math. Biol. 45:153–181 [DOI] [PubMed] [Google Scholar]
- 30. Sillers R., Chow A., Tracy B., Papoutsakis E. T. 2008. Metabolic engineering of the non-sporulating, non-solventogenic Clostridium acetobutylicum strain M5 to produce butanol without acetone demonstrate the robustness of the acid-formation pathways and the importance of the electron balance. Metab. Eng. 10:321–332 [DOI] [PubMed] [Google Scholar]
- 31. Steen E. J., et al. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559–562 [DOI] [PubMed] [Google Scholar]
- 32. Trinh C., Wlaschin A., Srienc F. 2009. Elementary mode analysis: a useful metabolic pathway analysis tool for characterizing cellular metabolism. Appl. Microbiol. Biotechnol. 81:813–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trinh C. T., Carlson R., Wlaschin A., Srienc F. 2006. Design, construction and performance of the most efficient biomass producing E. coli bacterium. Metab. Eng. 8:628–638 [DOI] [PubMed] [Google Scholar]
- 34. Trinh C. T., Huffer S., Clark M. E., Blanch H. W., Clark D. S. 2010. Elucidating mechanisms of solvent toxicity in ethanologenic Escherichia coli. Biotechnol. Bioeng. 106:721–730 [DOI] [PubMed] [Google Scholar]
- 35. Trinh C. T., Srienc F. 2009. Metabolic engineering of Escherichia coli for efficient conversion of glycerol to ethanol. Appl. Environ. Microbiol. 75:6696–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trinh C. T., Unrean P., Srienc F. 2008. Minimal Escherichia coli cell for the most efficient production of ethanol from hexoses and pentoses. Appl. Environ. Microbiol. 74:3634–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Unden G., Bongaerts J. 1997. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320:217–234 [DOI] [PubMed] [Google Scholar]
- 38. Unrean P., Trinh C. T., Srienc F. 2010. Rational design and construction of an efficient E. coli for production of diapolycopendioic acid. Metab. Eng. 12:112–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. U.S. Energy Information Administration 2010. International energy outlook 2010. U.S. Energy Information Administration, U.S. Department of Energy, Washington, DC [Google Scholar]
- 40. Zhang M., Eddy C., Deanda K., Finkelstein M., Picataggio S. 1995. Metabolic engineering of a pentose metabolism pathway in ethanologenic Zymomonas mobilis. Science 267:240–243 [DOI] [PubMed] [Google Scholar]
- 41. Zinoni F., Beier A., Pecher A., Wirth R., Böck A. 1984. Regulation of the synthesis of hydrogenase (formate hydrogen-lyase linked) of E. coli. Arch. Microbiol. 139:299–304 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.