

Abstract
Convincing correlates of protective immunity against tuberculosis have been elusive. In BALB/c mice, intranasal immunization with a replication-deficient recombinant adenovirus expressing Mycobacterium tuberculosis antigen 85A (adenovirus-85A) induces protective lower respiratory tract immunity against pulmonary challenge with Mycobacterium tuberculosis, while intradermal immunization with adenovirus-85A does not. Here we report that intranasal immunization with adenovirus-85A induces expression of the chemokine receptor CXCR6 on lung CD8 T lymphocytes, which is maintained for at least 3 months. CXCR6-positive antigen-specific T cell numbers are increased among bronchoalveolar lavage-recoverable cells. Similarly, intranasal immunization with recombinant antigen 85A with adjuvant induces CXCR6 expression on lung CD4 cells in BALB/c and C57BL/6 mice, while a synthetic ESAT61–20 peptide with adjuvant induces CXCR6 expression in C57BL/6 mice. Parenteral immunization fails to do so. Upregulation of CXCR6 is accompanied by a transient elevation of serum CXCL16 after intranasal immunization, and lung cells cultured ex vivo from mice immunized intranasally show increased production of CXCL16. Administration of CXCL16 and cognate antigen intranasally to mice previously immunized parenterally increases the number of antigen-specific T lymphocytes in the bronchoalveolar lavage-recoverable population, which mediates inhibition of the early growth of Mycobacterium tuberculosis after challenge. We conclude that expression of CXCR6 on lung T lymphocytes is a correlate of local protective immunity against Mycobacterium tuberculosis after intranasal immunization and that CXCR6 and CXCL16 play an important role in the localization of T cells within lung tissue and the bronchoalveolar lavage-recoverable compartment.
INTRODUCTION
When T cells are responsible for protective immunity against an organism, it has so far proved difficult to generate complete protection against disease by immunization, and there is increasing evidence that systemic T cell immune responses are insufficient to prevent infection or disease. However, targeting appropriate immune responses to the portal of entry of the pathogen can provide increased protection (18, 45, 50). For tuberculosis, an attenuated vaccine, Mycobacterium bovis bacillus Calmette-Guérin (BCG), administered parenterally, provides protection against disseminated disease in infants, but protection against pulmonary disease in adults is poor. One reason for this is the delay in initiation of the lung adaptive immune response to M. tuberculosis. Activation of T cells in the draining lymph nodes is not seen until 10 days postinfection, and effector T cells do not enter the lungs until 12 days postinfection or later (7, 39). Even in BCG-immunized animals, inhibition of mycobacterial growth is not seen during the first 7 days (32, 33, 53). This allows M. tuberculosis to grow unchecked in the lungs during the early phase of infection, strongly implying that harnessing the local immune response might provide an advantage in immunization against M. tuberculosis.
We and others have shown that this is the case (13, 40, 52). When BALB/c mice are immunized intranasally (i.n.) with a replication-deficient recombinant adenovirus expressing antigen 85A of M. tuberculosis (Ad85A), a large population of lung-resident antigen-specific CD8 T cells is established, and when these mice are challenged with M. tuberculosis, mycobacterial growth is inhibited during the first 7 days after infection (40). The lung-resident antigen-specific cells are highly activated, and many are within the bronchoalveolar lavage (BAL)-recoverable compartment (20). In contrast, immunization with Ad85A intradermally (i.d.) or intramuscularly (i.m.) does not protect against pulmonary challenge with M. tuberculosis, despite inducing a large population of splenic antigen-specific CD8 T cells, as well as the entry of T cells into lung tissues, but not the BAL-recoverable population (40, 42). To identify differences between lung CD8 T cells induced by protective (i.n.) and nonprotective (i.d.) regimens, gene expression patterns were compared by microarray analysis. Strikingly, lung CD8 T cells from i.n. immunized mice showed enhanced expression of several tissue-targeting chemokines and a single chemokine receptor, CXCR6, compared to lung CD8 T cells from i.d. immunized mice (26).
Chemokines control trafficking of cells into tissues, and chemokine receptor expression on the cell surface allows cells to be directed to relevant tissues by responding to a chemokine gradient. CXCR6 was initially described as a coreceptor for HIV (2) but was subsequently found to promote T cell migration into tissues (24). Expression of CXCR6 has been detected on NK cells (47), NKT cells (35), dendritic cells (DC), and activated T cells (47) and is a marker for effector T cells in both humans and mice (6, 47, 51). CXCR6 promotes homing of lymphocytes to extralymphoid tissue such that, for example, CXCR6+ T cells have been found in the liver during hepatitis C virus infection (4, 5, 37) and in tumor sites, where they may help to limit metastases (8, 31). CXCR6+ cells have also been implicated in a number of T cell-mediated pathologies, such as atherosclerosis and arthritis (17, 36, 48).
In humans, CXCL16, the only known ligand for CXCR6, is expressed constitutively in lung epithelia, may be responsible for the long-term retention of CD3+ T cells in lungs, and may also play a role in chronic obstructive pulmonary disease (9, 10, 15, 34). Because CXCR6-CXCL16 was the only chemokine receptor-chemokine pair shown by our microarray analysis to be upregulated in the lungs of protected, i.n. Ad85A-immunized mice compared to unprotected, i.d. Ad85A-immunized mice, we investigated the role of this receptor-ligand pair in protective immunity following immunization against M. tuberculosis.
MATERIALS AND METHODS
Mice and immunization.
All experiments were performed with 6- to 8-week-old female BALB/c or C57BL/6 mice (Harlan Orlac, Blackthorn, United Kingdom), were approved by the animal use ethical committee of Oxford University, and fully complied with the relevant Home Office guidelines. Mice were immunized with a recombinant replication-deficient adenovirus of serotype 5 containing the 85A antigen of M. tuberculosis (Ad85A) (40). For i.d. immunization, mice were anesthetized and injected in each ear with 25 μl of preparation containing a total of 2 × 109 virus particles (v.p.) of Ad85A per mouse, and for i.n. immunization, mice were allowed to slowly inhale 5 or 50 μl of phosphate-buffered saline (PBS) containing 2 × 109 v.p. of Ad85A. Mice were also immunized with recombinant antigen 85A protein (rec85A) or with a peptide carrying the first 20 amino acids of ESAT6 (6-kDa early secreted antigenic target; ESAT61–20). Intranasal protein immunization was conducted as described above, delivering 2 μg of rec85A protein or 20 μg of ESAT61–20 mixed with 2 μg of cholera toxin (CT; Sigma-Aldrich, Dorset, United Kingdom) into the nostrils in a 50-μl volume. Subcutaneous (s.c.) immunization was performed by administering 2 μg of rec85A protein or 20 μg of ESAT61–20 peptide in 200 μl of Sigma's monophosphoryl lipid A adjuvant system (Sigma-Aldrich) according to the manufacturer's instructions. Proteins were administered 3 times at twice-weekly intervals.
In some experiments, mice were immunized with Ad85A i.d. as described above. At 3 weeks postimmunization, 2 μg recombinant CXCL16 (R&D Systems), 2.5 μg rec85A, or a mixture of the two in 50 μl PBS plus 0.5% bovine serum albumin (BSA) was delivered i.n. to groups of mice. Four days later, the mice were sacrificed and the BAL fluid and lung tissue collected for analysis, or the mice were challenged with M. tuberculosis as described below.
Production of rec85A.
rec85A was produced as previously described (14). Briefly, the M. tuberculosis Rv3804c gene was amplified by PCR from genomic H37Rv DNA and cloned by Gateway technology (Invitrogen, Carlsbad, CA) into a bacterial expression vector containing a histidine tag at the N terminus. The protein was overexpressed in Escherichia coli BL21(DE3) and purified. The size and purity of rec85A were analyzed by gel electrophoresis and Western blotting with an anti-His antibody (Invitrogen, Carlsbad, CA) and an anti-E. coli polyclonal antibody (Abcam, Cambridge, United Kingdom). The endotoxin content was below 50 IU/mg recombinant protein, as tested using a Limulus amebocyte lysate (LAL) assay (Cambrex, East Rutherford, NJ). Subsequently, the protein was tested in a lymphocyte stimulation assay to exclude antigen-nonspecific T cell stimulation and cellular toxicity, using peripheral blood mononuclear cells (PBMC) from in vitro purified protein derivative (PPD)-negative healthy Dutch donors (28).
Isolation of lymphocytes from BAL fluid, blood, lungs, liver, facial lymph nodes, spleen, and NALT.
Blood was collected from the jugular vein or by cardiac puncture into heparin tubes and diluted in PBS, and lymphocytes were isolated by density centrifugation over Lymphoprep (Axis-Shield, Oslo, Norway). Cells from the interface were collected and washed. BAL fluid was collected from lungs of mice by lung washes, and samples were pooled for each group. The collected cells were centrifuged and resuspended in medium. Lungs were perfused with PBS, cut into small pieces, and digested with 0.7 mg/ml collagenase type I (Sigma-Aldrich) and 30 μg/ml DNase I (Sigma-Aldrich) for 45 min at 37°C. Lung fragments were then crushed through a cell strainer by use of a 5-ml syringe plunger, washed, layered over Lympholyte (Cedarlane, Ontario, Canada), and centrifuged at 1,000 × g for 25 min. Interface cells were collected and washed. Spleens were passed through a cell strainer by use of a 5-ml syringe plunger, red blood cells (RBC) were lysed using RBC lysis buffer (Qiagen, Crawley, United Kingdom), and the cells were washed. Livers were perfused with PBS, cut into small pieces, and passed through a cell sieve. The single-cell suspension was resuspended in 30% Percoll (GE Healthcare, Little Chalfont, United Kingdom) and layered over 70% Percoll. The gradient was centrifuged at 800 × g for 25 min, and the interface cells were collected and washed. Pooled parotid, mandibular, and superficial cervical lymph nodes were collected and pooled for 3 mice per group. Single-cell suspensions were made by crushing nodes between microscope slides, and the cells were washed once. Cells from the organized nasal mucosa-associated lymphoid tissues (NALT) (the paired lymphoid structures at the posterior end of the hard palate) were isolated as described previously (41). Isolated washed cells from all tissues/organs were resuspended in RPMI (Invitrogen) plus 10% fetal calf serum (FCS).
Purification and depletion of lymphocyte populations and cell transfer.
CD8+ lung lymphocytes from mice immunized 4 weeks previously with Ad85A i.n. were enriched by first incubating with a cocktail of biotinylated monoclonal antibodies (MAbs) against different cell types (CD8 T cell isolation kit; Miltenyi Biotec, Surrey, United Kingdom) and then being washed and labeled with anti-biotin microbeads before separation on an LS magnetic column according to the manufacturer's instructions (Miltenyi Biotec). The flowthrough enriched CD8 T cells were divided in two. Half of the cells were labeled with CXCR6-phycoerythrin (CXCR6-PE) monoclonal antibody (clone 221002; R&D Systems, Abingdon, United Kingdom), and the remainder were not. Both aliquots of cells were passed separately through LD magnetic columns. Enriched CD8 cells contained ∼70% T cells, and 3.2% of these were CXCR6+, while CXCR6 depletion reduced this value to 1.3%. A total of 5 × 105 cells were administered i.n. in 50 μl PBS to each recipient mouse. The mice were challenged with M. tuberculosis the following day, and organs were harvested for enumeration of M. tuberculosis CFU 7 days later.
Flow cytometry.
Cells were cultured in RPMI supplemented with 10% heat-inactivated FCS, l-glutamine, penicillin, and streptomycin for 6 h prior to staining with anti-CXCR6 antibody. In some experiments, cells were stimulated with a mix of 3 peptides (Peptide Protein Research Ltd., Fareham, United Kingdom) carrying the dominant CD4 (Ag85A99-118 [TFLTSELPGWLQANRHVKPT]) and CD8 (Ag85A70–78 [MPVGGQSSF] and Ag85A145–152 [YAGAMSGL]) peptide epitopes (40). Each peptide was used at a final concentration of 2 μg/ml during the stimulation. After 2 h at 37°C, Golgi Plug (BD Biosciences, Oxford, United Kingdom) was added according to the manufacturer's instructions, and cells were incubated for an additional 4 h before intracellular cytokine staining.
Cells were washed and incubated with CD16/CD32 MAb to block Fc binding. Subsequently, the cells were stained for CD27 (LG.7F9), CD19 (1D3), CD4 (RM4-5), gamma interferon (IFN-γ) (XMG1.2), tumor necrosis factor (TNF) (MP6-XT22; eBioscience, Hatfield, United Kingdom), CXCR6 (221002; R&D Systems), and CD8 (53-6.7) (BD Bioscience), using a BD Cytofix/Cytoperm kit according to the manufacturer's instructions. In some experiments, cells were also stained with H-2Ld 85A70–78 peptide (MPVGGQSSF) tetramer (kindly provided by the NIH Tetramer Facility, Bethesda, MD). Cells were fixed with PBS plus 1% paraformaldehyde, run on an LSRII flow cytometer (BD Biosciences), and analyzed using FlowJo software (Tree Star Inc., Ashland, OR). To obtain absolute cell numbers, 50 or 20 μl of CountBright absolute counting bead suspension (Invitrogen) was added to each sample prior to analysis.
CXCL16 ELISA.
Serum samples were collected by cardiac puncture. Lung lymphocytes isolated as described above were cultured in RPMI plus 10% FCS for 6 h at 37°C without stimulation, and the culture supernatant was collected. The CXCL16 protein concentration was measured by enzyme-linked immunosorbent assay (ELISA) (R&D Systems).
Infection with M. tuberculosis and determination of mycobacterial load.
Mice were anesthetized with isoflurane and infected i.n. with M. tuberculosis (strain Erdman; kindly provided by Amy Yang, CBER/FDA) in 50 μl PBS. Deposition in the lungs was measured 24 h after challenge to determine the number of organisms deposited, which was on the order of 200 CFU. Mice were sacrificed at the indicated times, the lungs were homogenized, and bacterial loads were determined by plating 10-fold serial dilutions of tissue homogenates on Middlebrook 7H11 agar plates (E&O Laboratories Ltd., Bonnybridge, United Kingdom). Colonies were counted after 3 to 4 weeks of incubation at 37°C in 5% CO2.
RESULTS
CXCR6 is upregulated on lung CD8 T cells after i.n. immunization with Ad85A.
BALB/c mice immunized with Ad85A i.n., but not i.d., showed a statistically significant reduction in mycobacterial load following pulmonary M. tuberculosis challenge (Table 1) (26, 40). Since microarray analysis of CD8 T cells isolated from the lungs of mice immunized with Ad85A i.n. showed increased expression of CXCR6 compared to that for immunization with Ad85A i.d. (26), we analyzed CXCR6 expression in lymphoid and nonlymphoid organs of mice immunized by the two routes. Figure 1 shows that there were few CXCR6+ CD8+ T cells in the blood, spleen, and pooled facial lymph nodes after Ad85A immunization i.n. or i.d., with some upregulation on liver CD8 T cells. However, in the lungs, only lymphocytes from mice immunized with Ad85A i.n. upregulated CXCR6.
Table 1.
Lung mycobacterial loads 6 weeks after aerosol challenge with 200 CFU of M. tuberculosis
| Ad85A immunization route | Mycobacterial load (mean log CFU per lung ± SEM)a |
|---|---|
| Not immunized | 6.38 ± 0.097 |
| i.d. | 6.40 ± 0.077 |
| i.n. | 5.76 ± 0.056* |
Data are mean CFU for 5 or 6 mice per group challenged 4 weeks after immunization with Ad85A via the indicated routes (26). The data were analyzed by the Kruskal-Wallis test (overall P value, 0.007) followed by Dunn's multiple comparison test. *, P < 0.05 compared to naïve or i.d. group.
Fig. 1.

Expression of CXCR6 is upregulated on CD8 T cells in the lung after i.n. Ad85A immunization. BALB/c mice were immunized with Ad85A i.n. or i.d. Five weeks later, CD8+ T cells from several tissues were analyzed for expression of CXCR6. The results shown are the mean values for 3 mice per group for lung, spleen, and liver samples ± standard deviations (SD), while the facial lymph nodes and blood were pooled from the same mice for analysis, and their results are representative of results obtained in two independent experiments. *, P = 0.03 by Mann-Whitney testing of the data from two experiments. (B) The absolute numbers of CD8 CXCR6+ cells in the lungs and spleen were also determined. The results shown are representative of two independent experiments.
We have shown previously that in order to obtain protection against pulmonary M. tuberculosis challenge, it is essential to use a 50-μl i.n. inoculum to induce lower respiratory tract responses, while a 5-μl inoculum induces a NALT response but no protection (41). Here we analyzed CXCR6 expression after i.n. immunization with 5 or 50 μl of Ad85A. A 5-μl nonprotective inoculum did not induce CXCR6 expression on lung T cells (data not shown), confirming that CXCR6 expression is a correlate of protective lower respiratory tract immunity.
Effector CD8 T cells are typically short-lived and present during the course of an infection, but after immunization with Ad85A i.n., antigen-specific lung T cells and protection against aerosol M. tuberculosis infection are sustained for several months (40). In accordance with this, although the number and percentage of lung antigen-specific CD8 T cells declined after a peak at 4 to 6 weeks postinfection (13), we consistently demonstrated CD8+ CXCR6+ antigen-specific cells in the lungs for up to 3 months postimmunization (see Fig. 3), indicating that CXCR6 expression is long-lived after i.n. immunization with Ad85A.
Fig. 3.

Properties of CXCR6+ cells in lungs after immunization. (A) Dot plots showing percentages of CXCR6+ Tet+ cells in the CD8-gated T cell population isolated from lungs of BALB/c mice immunized with Ad85A i.n. or i.d. 7 weeks previously. (B) Histogram showing means ± SD for 2 experiments with 3 mice in each. (C) CD8+ CXCR6+ lung cells from mice immunized 11 weeks previously with Ad85A i.n. were stained for CD27. The histogram shows means ± SD for 3 mice and is representative of 2 experiments. (D) Lymphocytes from the BAL fluid of mice immunized 13 weeks previously with Ad85A i.n. were stimulated with the dominant CD4 and dominant and subdominant CD8 peptides and stained for CD8, CXCR6, and intracellular IFN-γ. The histogram shows means ± SD for 3 mice and is representative of 4 independent experiments. *, P < 0.05 by Mann-Whitney test.
CD8+ CXCR6+ cells are detected in the BAL fluid after i.n. immunization with Ad85A.
In mice infected with M. tuberculosis, the localization of effector T cells in BAL fluid has been shown to correlate with protection induced by i.n. Ad85A immunization (20, 42). To investigate if CXCR6 is expressed on these cells, BAL fluid was collected from i.n. and i.d. Ad85A-immunized mice at 3 weeks postimmunization, and the cells were stained for CXCR6. Lymphocytes were also isolated from lung tissue after BAL fluid collection to compare the numbers of CD8+ CXCR6+ cells in these two compartments (Fig. 2A). Very few CD8+ CXCR6+ cells were observed in the BAL fluid of i.d. Ad85A-immunized mice, while they were abundant in BAL fluid of i.n. Ad85A-immunized mice. Furthermore, in i.n. Ad85A-immunized mice, a large proportion (∼30%) of lung CD8+ CXCR6+ cells were in BAL fluid. Double labeling of CD8 cells with CXCR6 and a tetramer for the dominant CD8 H2d-restricted epitope of antigen 85A also showed that many of the CXCR6+ cells were antigen specific (Fig. 2B and 3), suggesting that CXCR6 may be involved in targeting antigen-specific cells to the BAL-recoverable compartment.
Fig. 2.

Numbers of CXCR6+ cells in BAL fluid versus residual lung tissue. BALB/c mice were immunized i.n. or i.d. with Ad85A. Lymphocytes from BAL fluid and residual lung tissue (processed after collection of BAL fluid) collected from mice at 3 weeks postimmunization were stained for CD8, CXCR6, and a tetramer containing the dominant CD8 epitope of antigen 85A. Numbers of cells from BAL fluid or residual lung of i.d. (black bars) or i.n. (white bars) Ad85A-immunized mice are shown. (A) CD8+ CXCR6+ cells; (B) CD8+ Tet+ CXCR6+ cells. The results are the means ± SD for three mice from one of two independent experiments. Data from all repeats were analyzed by the Kruskal-Wallis test followed by Dunn's multiple comparison test. *, P < 0.05 (similar results were obtained with the Mann-Whitney test).
Properties of lung CD8+ CXCR6+ T cells.
In order to analyze the properties of antigen-specific CXCR6+ cells, they were costained with a tetramer for the dominant CD8 T cell epitope of 85A and with antibodies to other functionally important molecules. Figure 3A and B show that at 11 or 13 weeks postimmunization, a large proportion of the CXCR6+ cells were tetramer positive (Tet+) (56%), and similarly, many Tet+ cells were CXCR6+ (27%). It is likely that some of the remaining CXCR6+ cells were adenovirus specific, since lung cells from i.n. Ad85A-immunized mice responded vigorously to a control adenovirus lacking 85A by cytokine production (data not shown). The CXCR6+ population was also stained with CD27, and expression of CD27 was downregulated, indicating activation and recent exposure to antigen in vivo (Fig. 3C) (40).
BAL fluid lymphocytes from i.n. Ad85A-immunized mice were stimulated with peptides representing the dominant CD4 and CD8 antigen 85A T cell epitopes in the presence of brefeldin and were costained for IFN-γ and CXCR6 to determine if CXCR6+ cells produce IFN-γ ex vivo. IFN-γ production was detected in a subset of CXCR6+ cells at various times postimmunization (Fig. 3D), with the percentage of CD8+ CXCR6+ IFN-γ+ cells varying from 14 to 36% of CXCR6+ cells. Stimulated CD8+ CXCR6+ BAL fluid cells also produced TNF that was detectable by intracytoplasmic staining (data not shown).
In summary, CXCR6+ cells induced after i.n. Ad85A immunization are highly activated, antigen specific, and capable of secreting IFN-γ and TNF. The cells appear to be fully functional and may contribute to the early control of M. tuberculosis that we have described previously for mice immunized with Ad85A i.n. (40).
CXCR6 is expressed after protein or peptide i.n. immunization in BALB/c and C57BL/6 mice.
We next investigated if sustained upregulation of CXCR6 in lung CD8 T cells was a response only to i.n. Ad85A. BALB/c or C57BL/6 mice were immunized with rec85A or ESAT61–20 peptide with adjuvant either i.n. or s.c. Four weeks after the last booster, the proportion of CXCR6+ cells was assessed. i.n. administration of rec85A or ESAT6 peptide induced expression of CXCR6 on lung T cells, and in contrast to the case for i.n. Ad85A immunization, protein or peptide immunization upregulated CXCR6 predominantly on CD4+ T cells (Fig. 4). CXCR6 is therefore a hallmark of successful i.n. immunization for both CD4 and CD8 cells, irrespective of the nature of the immunogen.
Fig. 4.

Expression of CXCR6 on lung T cells after immunization with rec85A by different routes. (A and B) BALB/c or C57BL/6 mice were immunized with rec85A s.c. or i.n. Four weeks after the final booster, spleen and lung lymphocytes were isolated, and CD4 (A) or CD8 (B) T cells were stained for CXCR6. The results shown are the means ± SD for 4 mice and are representative of results obtained from 2 independent experiments. (C and D) Expression of CXCR6 on lung T cells after immunization with ESAT61–20 peptide by different routes. C57BL/6 mice were immunized with ESAT61–20 peptide s.c. or i.n. Four weeks after the second immunization, spleen and lung cells were stained for CD4, CD8, and CXCR6 and analyzed by gating on CD4 (C) and CD8 (D) T cells. The results shown are means ± standard errors of the means (SEM) for 4 mice and are representative of results obtained from 2 independent experiments. *, P < 0.05 by Mann-Whitney test.
CXCL16 production after i.n. Ad85A immunization.
Trafficking of cells into tissues occurs in response to a gradient of a chemokine ligand. CXCL16 is the only known ligand for CXCR6 (30) and is usually expressed as a membrane-bound molecule, but an alternatively spliced, secreted form of the chemokine can be produced (49). Serum levels of CXCL16 were measured to determine if i.n. immunization elicited a CXCL16 gradient to recruit T cells from the circulation. Measurements were made at 6 days and 2, 3, and 12 weeks postimmunization. At day 6, serum CXCL16 was significantly elevated in i.n. compared to i.d. immunized mice (Fig. 5A), but not at later times (data not shown). Since CXCL16 may be expressed constitutively in lung epithelium (9), we investigated whether lung lymphocytes isolated from naïve or i.n. or i.d. immunized mice differed in the ability to secrete CXCL16. Lung mononuclear cells isolated from i.n. immunized mice secreted CXCL16 protein for up to 3 weeks after immunization (Fig. 5B). Stimulation with the dominant CD4 and dominant and subdominant CD8 peptides for 6 h did not alter the levels of CXCL16 produced by i.n. immunized lung lymphocytes, nor did it induce CXCL16 production in lung lymphocytes isolated from i.d. immunized mice (data not shown). Therefore, i.n. immunization increases serum CXCL16 shortly after immunization, possibly reflecting formation of a chemokine gradient to recruit activated CXCR6-expressing T cells into the lung, where they may be retained partly as a result of sustained expression of CXCL16 by lung lymphocytes.
Fig. 5.

CXCL16 protein in serum (A) and secreted by lung lymphocytes ex vivo (B) after immunization. Sera were collected from mice 6 days after immunization with Ad85A i.d. or i.n. Lung lymphocytes were isolated at 2 weeks postimmunization and incubated for 6 h at 37°C in RPMI plus 10% FCS without stimulation. CXCL16 protein in sera and cell culture supernatants was measured by ELISA. The data shown are mean concentrations ± SEM for 5 or 6 mice per group (A) or 4 to 7 mice per group (B) from 2 independent experiments. *, P < 0.05 by Mann-Whitney test.
CXCL16 and antigen increase numbers of BAL-recoverable lymphocytes.
In order to determine if CXCL16 on its own is able to increase the number of effector T cells in the BAL-recoverable compartment, BALB/c mice were immunized with Ad85A i.d. to induce a systemic CD8 T cell immune response. Three weeks later, CXCL16 protein, rec85A, or both were administered i.n. to the mice, and 4 days later, BAL fluid cells were recovered and analyzed. There was no difference in the number of CD4+, CD8+, or Tet+ T cells in the BAL fluid for mice given CXCL16 or control PBS (Fig. 6A). In contrast, delivery of rec85A protein i.n. increased the number of cells in the BAL fluid, with a preferential increase in the CD8+ and Tet+ T cell populations, but codelivery of CXCL16 and rec85A caused a much larger increase in the number of lymphocytes in the BAL fluid, indicating that CXCL16 and rec85A synergize. When identically treated groups of mice were infected with M. tuberculosis, only i.d. Ad85A-immunized mice which received both CXCL16 and antigen 85A i.n. 4 days prior to challenge were able to significantly reduce the lung mycobacterial load 7 days after challenge with M. tuberculosis (Fig. 6B). Therefore, exogenous CXCL16 in conjunction with cognate antigen leads to the presence of large numbers of CD8 and CD4 T cells in the BAL-recoverable compartment, where they can mediate protection against M. tuberculosis infection.
Fig. 6.

Numbers of CD4, CD8, and CD8 Tet+ cells isolated from the BAL fluid of i.d. Ad85A-immunized mice after i.n. delivery of CXCL16 or rec85A protein. (A) Mice were immunized with Ad85A i.d. At 3 weeks postimmunization, rec85A, CXCL16 protein, or both were administered i.n. Four days later, BAL fluid was collected from individual mice, and the numbers of CD4+, CD8+, and CD8 Tet+ cells were determined by flow cytometry. The figures show the mean number of each cell population recovered ± SD for 3 mice in one of two independent experiments. (B) Mean mycobacterial burdens in the lungs of i.d. Ad85A-immunized mice given CXCL16 or rec85A protein i.n. and subsequently challenged i.n. with a low dose of M. tuberculosis. Mice were immunized, and rec85A, CXCL16 protein, or both were administered i.n. as described above. Four days later, mice were challenged i.n. with 200 CFU of M. tuberculosis. At 7 days postchallenge, the lungs were removed, homogenized, and plated on Middlebrook agar plates to determine the mycobacterial load. The figure shows the mean log CFU per lung for 3 to 5 mice per group from a single experiment. The data were analyzed using the Kruskal-Wallis test followed by Dunn's multiple comparison test. * and **, P < 0.05 compared to PBS and CXCL16, respectively.
Depletion of CXCR6+ cells decreases the protective efficacy of CD8+ T cells.
BALB/c mice were immunized with Ad85A i.n., and 4 weeks later, T cells were isolated from the lungs. After enrichment of CD8 T cells by magnetic bead depletion of other cell types, the enriched T cells were labeled with CXCR6 antibody and depleted by passage over a second magnetic column. Equal numbers (5 × 105) of CD8+ CXCR6+ and CD8+ CXCR6-depleted cells were transferred i.n. into naïve mice, and the following day the mice were challenged with M. tuberculosis. Although both populations reduced M. tuberculosis CFU compared to those for naïve mice given no cells, CD8+ CXCR6+ cells had a significantly greater effect than CD8+ CXCR6-depleted cells (Fig. 7).
Fig. 7.
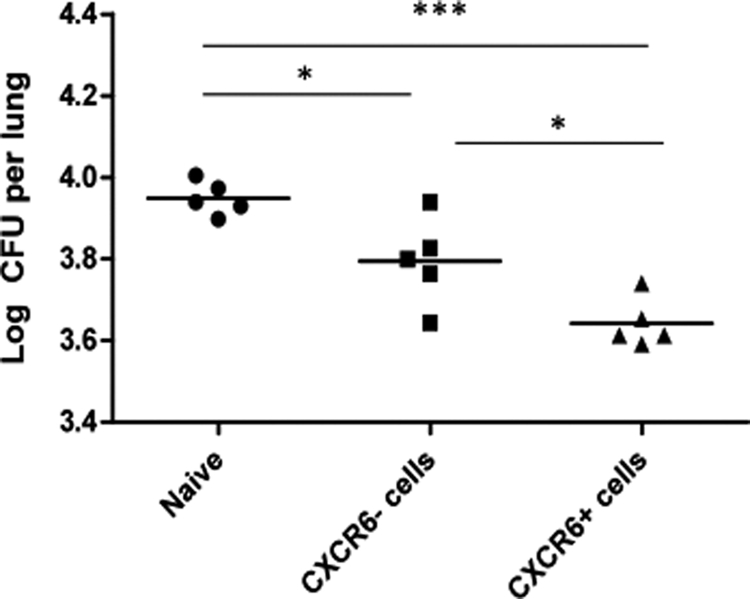
M. tuberculosis CFU in the lungs of naïve mice receiving CXCR6+ or CXCR6-depleted lymphocytes i.n. BALB/c mice were immunized with Ad85A i.n. Four weeks later, lung lymphocytes were isolated and enriched for CD8+ T cells. These were then depleted of CXCR6+ cells by use of magnetic beads. A total of 5 × 105 enriched CD8+ cells or CD8+ cells depleted of CXCR6+ cells were administered i.n. to naïve mice, which were challenged with M. tuberculosis 1 day later. Lungs were collected for M. tuberculosis CFU enumeration 7 days later. *, P < 0.05; ***, P < 0.001 by one-way analysis of variance with Tukey's posttest.
DISCUSSION
We have previously shown that a striking effect of immunization with Ad85A i.n. is an early inhibition of lung mycobacterial growth following pulmonary challenge with M. tuberculosis (40), in contrast to the late inhibitory effects of BCG and other parenterally administered vaccines (22, 23, 32, 33, 53). We propose that i.n. boosting after BCG vaccination is effective because the growth of M. tuberculosis is inhibited both early and later after challenge and that early inhibition of M. tuberculosis growth is dependent on the preferential localization of highly activated, antigen-specific effector cells secreting IFN-γ in the BAL-recoverable compartment (3, 19). Here we have shown that i.n. immunization with either a recombinant adenoviral vector or proteins/peptides with CT as an adjuvant upregulates CXCR6 on lung T cells and that CXCR6 expression persists on CD8 T cells for at least 3 months postimmunization. Immunization with recombinant adenovirus induces predominantly CD8, while protein/peptide immunization induces mainly CD4 antigen-specific T cells expressing CXCR6. CXCL16, the only known ligand for CXCR6, is also produced by lung mononuclear cells after i.n. immunization, perhaps as a consequence of IFN-γ and TNF production by effector T cells (29, 54).
Expression of CXCR6 on lung CD8 T cells 7 days after i.n. immunization with rotavirus has been reported (21), and several studies suggest that CXCR6+ cells are attracted toward and maintained in situ by the presence of antigen. For example, CXCR6+ cells migrate toward sites of acute bacterial infections (16, 43) and are found at sites of chronic inflammation (4, 27, 36), including the lung in patients with chronic obstructive pulmonary disease (15). Prolonged production of CXCL16 may aid in retention of CXCR6+ cells (48). It is thus likely that a combination of the long-term presence of antigen (40), the expression of CXCR6, and the production of CXCL16 by lung mononuclear cells contributes toward long-term maintenance of an effector population following i.n. immunization. Furthermore, CXCR6+ cells are enriched in the BAL fluid of i.n. versus i.d. Ad85A-immunized mice, suggesting that CXCL16 may play a role in their localization in the BAL-recoverable compartment, a suggestion supported by the synergistic effect of i.n. delivery of rec85A antigen and CXCL16. Since it is known that BAL fluid lymphocytes play an important role in protection against pulmonary growth of M. tuberculosis following challenge (19, 20), the presence of a lung population of antigen-specific CXCR6+ cells is effectively a signature of local protective immunity after i.n. immunization. Expression of CXCR6 elsewhere does not correlate with protection, since i.n. and i.d. Ad85A-immunized mice have equal numbers of CXCR6+ cells in the spleen, but the former are well protected and the latter are not (Fig. 1).
We previously investigated the role of the NALT in immunity to M. tuberculosis in mice (41). Here we targeted the NALT with Ad85A and CT in a small-volume inoculum (5 μl) with the aim of ensuring a powerful NALT response (12). Even with the addition of CT, no CXCR6+ cells were detected in the lung, although some were present in the NALT (data not shown). In contrast, a large-volume inoculum of Ad85A (50 μl) with no adjuvant induced CXCR6+ cells in the lung (data not shown) and protected mice from M. tuberculosis challenge (41). These data confirm the independence of NALT and lower respiratory tract immune responses in mice and support an association between the presence of CXCR6+ cells in the lung and local protective immunity following immunization.
CXCR6 expression is associated with the development of protective lower respiratory tract immunity and with enrichment of CXCR6+ antigen-specific cells in BAL fluid after i.n. immunization, but CXCL16 has been reported to be poorly chemotactic (25). Therefore, we tested the effect of administering recombinant CXCL16 i.n. to the airways of mice previously immunized i.d. We found that CXCL16 alone had little chemotactic effect (Fig. 6). However, CXCL16 in combination with cognate antigen increased the number of T cells in the BAL fluid, and many of these were antigen specific. CXCL16 is expressed constitutively by lung epithelial cells and is therefore already present in the lungs, but it is normally expressed as a membrane-bound molecule. Our results suggest that introducing only soluble (and thus potentially gradient-forming) CXCL16 into the airways has little effect but that addition of a cognate antigen has a synergistic effect with CXCL16. These data suggest two possibilities. First, presentation of antigen to T cells may make the cells more responsive to CXCL16 and thus promote migration into the BAL-recoverable compartment from the surrounding lung tissue or blood. Alternatively, CXCL16 plus antigen may induce local proliferation in the BAL-recoverable compartment. Preliminary data indicate a similar increase in the number of BAL-recoverable T cells after i.n. administration of CXCL16 with cognate antigen to that for mice previously immunized s.c. or i.m. with protein antigen plus adjuvant (data not shown).
In order to confirm that CXCR6+ cells in BAL fluid are important for protection, we isolated lymphocytes from the lungs of i.n. Ad85A-immunized mice, transferred CD8+ T cells or CD8+ T cells depleted of CXCR6+ cells into the lungs of naïve mice, and then challenged the mice with M. tuberculosis. The CXCR6-depleted cells were significantly less protective than total CD8+ cells. Clearly, and not surprisingly, since not all antigen-specific lung T cells express CXCR6, the depleted cells could also decrease the M. tuberculosis load. Nevertheless, the data support the idea that CD8+ CXCR6+ cells are important for local protection against pulmonary M. tuberculosis challenge. Both CXCR6+ and CXCR6− cells may recruit innate effector cells, and the exact mechanisms of early inhibition of M. tuberculosis growth in the lungs remain to be determined.
Successful prophylactic immunization against organisms that are normally combated by T cell immune responses has proved extremely difficult, but it was recognized recently that this may be because it is important to harness both systemic and local immunity at the portal of entry (55). In the case of M. tuberculosis, it has become clear in experimental models that it is surprisingly difficult to improve the level of protection by boosting with parenterally administered subunit vaccines after BCG priming (13, 38, 44, 46), although there are some exceptions (11). In contrast, i.n. immunization is highly effective, most likely because lung-resident activated T cells can inhibit growth of mycobacteria early after infectious challenge, in contrast to parenteral immunization, which inhibits growth much later (40). It has also been shown that the location of cells in the lung is critical and that BAL-recoverable cells are highly effective in mediating protection against M. tuberculosis (42). These data strongly support the idea that the geography of the immune response is important (19) and that an optimal strategy for development of an M. tuberculosis vaccine should take into account the properties of three populations of antigen-specific cells: those in the BAL-recoverable compartment, those in the remaining lung tissues, and those outside the lung in the immune system.
Our data suggest that expression of CXCR6 on lung T cells after i.n. immunization is a marker for local protective immunity to M. tuberculosis and that CXCR6 and CXCL16 play a role in promoting localization of T cells within the BAL-recoverable compartment. Manipulation of CXCR6-CXCL16 by i.n. immunization and codelivery of CXCL16 and antigen after parenteral immunization are strategies to recruit and establish both CD4 and CD8 antigen-specific effectors at the site of pulmonary pathogen entry, leading to quick and effective clearance upon infection.
ACKNOWLEDGMENTS
This study was supported by UK Medical Research Council grant G0701235, by the European Commission (FP7), and by the Netherlands Organization of Scientific Research.
Footnotes
Published ahead of print on 31 May 2011.
REFERENCES
- 1. Aagaard C. S., Hoang T. T., Vingsbo-Lundberg C., Dietrich J., Andersen P. 2009. Quality and vaccine efficacy of CD4+ T cell responses directed to dominant and subdominant epitopes in ESAT-6 from Mycobacterium tuberculosis. J. Immunol. 183:2659–2668 [DOI] [PubMed] [Google Scholar]
- 2. Alkhatib G., Liao F., Berger E. A., Farber J. M., Peden K. W. 1997. A new SIV co-receptor, STRL33. Nature 388:238. [DOI] [PubMed] [Google Scholar]
- 3. Beverley P. C., Tchilian E. Z. 2008. Lessons for tuberculosis vaccines from respiratory virus infection. Expert Rev. Vaccines 7:1165–1172 [DOI] [PubMed] [Google Scholar]
- 4. Billerbeck E., et al. 2010. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc. Natl. Acad.Sci. U. S. A. 107:3006–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boisvert J., et al. 2003. Liver-infiltrating lymphocytes in end-stage hepatitis C virus: subsets, activation status, and chemokine receptor phenotypes. J. Hepatol. 38:67–75 [DOI] [PubMed] [Google Scholar]
- 6. Calabresi P. A., Yun S. H., Allie R., Whartenby K. A. 2002. Chemokine receptor expression on MBP-reactive T cells: CXCR6 is a marker of IFNgamma-producing effector cells. J. Neuroimmunol. 127:96–105 [DOI] [PubMed] [Google Scholar]
- 7. Chackerian A. A., Alt J. M., Perera T. V., Dascher C. C., Behar S. M. 2002. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect. Immun. 70:4501–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cullen R., Germanov E., Shimaoka T., Johnston B. 2009. Enhanced tumor metastasis in response to blockade of the chemokine receptor CXCR6 is overcome by NKT cell activation. J. Immunol. 183:5807–5815 [DOI] [PubMed] [Google Scholar]
- 9. Day C., Patel R., Guillen C., Wardlaw A. J. 2009. The chemokine CXCL16 is highly and constitutively expressed by human bronchial epithelial cells. Exp. Lung Res. 35:272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Day C. E., et al. 2009. A novel method for isolation of human lung T cells from lung resection tissue reveals increased expression of GAPDH and CXCR6. J. Immunol. Methods 342:91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dietrich J., Billeskov R., Doherty T. M., Andersen P. 2007. Synergistic effect of bacillus Calmette Guerin and a tuberculosis subunit vaccine in cationic liposomes: increased immunogenicity and protection. J. Immunol. 178:3721–3730 [DOI] [PubMed] [Google Scholar]
- 12. Etchart N., et al. 2006. Intranasal immunisation with inactivated RSV and bacterial adjuvants induces mucosal protection and abrogates eosinophilia upon challenge. Eur. J. Immunol. 36:1136–1144 [DOI] [PubMed] [Google Scholar]
- 13. Forbes E. K., et al. 2008. Multifunctional, high-level cytokine-producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J. Immunol. 181:4955–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franken K. L., et al. 2000. Purification of His-tagged proteins by immobilized chelate affinity chromatography: the benefits from the use of organic solvent. Protein Expr. Purif. 18:95–99 [DOI] [PubMed] [Google Scholar]
- 15. Freeman C. M., Curtis J. L., Chensue S. W. 2007. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am. J. Pathol. 171:767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaida M. M., et al. 2008. Expression of the CXCR6 on polymorphonuclear neutrophils in pancreatic carcinoma and in acute, localized bacterial infections. Clin. Exp. Immunol. 154:216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galkina E., et al. 2007. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation 116:1801–1811 [DOI] [PubMed] [Google Scholar]
- 18. Haase A. T. 2010. Targeting early infection to prevent HIV-1 mucosal transmission. Nature 464:217–223 [DOI] [PubMed] [Google Scholar]
- 19. Jeyanathan M., Heriazon A., Xing Z. 2010. Airway luminal T cells: a newcomer on the stage of TB vaccination strategies. Trends Immunol. 31:247–252 [DOI] [PubMed] [Google Scholar]
- 20. Jeyanathan M., et al. 2010. Murine airway luminal antituberculosis memory CD8 T cells by mucosal immunization are maintained via antigen-driven in situ proliferation, independent of peripheral T cell recruitment. Am. J. Respir. Crit. Care Med. 181:862–872 [DOI] [PubMed] [Google Scholar]
- 21. Jiang J. Q., He X. S., Feng N., Greenberg H. B. 2008. Qualitative and quantitative characteristics of rotavirus-specific CD8 T cells vary depending on the route of infection. J. Virol. 82:6812–6819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jung Y. J., Ryan L., LaCourse R., North R. J. 2005. Properties and protective value of the secondary versus primary T helper type 1 response to airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 201:1915–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khader S. A., et al. 2007. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8:369–377 [DOI] [PubMed] [Google Scholar]
- 24. Kim C. H., et al. 2001. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J. Clin. Invest. 107:595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Latta M., Mohan K., Issekutz T. B. 2007. CXCR6 is expressed on T cells in both T helper type 1 (Th1) inflammation and allergen-induced Th2 lung inflammation but is only a weak mediator of chemotaxis. Immunology 121:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee L. N., et al. 2010. Chemokine gene expression in lung CD8 T cells correlates with protective immunity in mice immunized intranasally with adenovirus-85A. BMC Med. Genomics 3:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lehrke M., et al. 2007. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J. Am. Coll. Cardiol. 49:442–449 [DOI] [PubMed] [Google Scholar]
- 28. Leyten E. M., et al. 2006. Human T-cell responses to 25 novel antigens encoded by genes of the dormancy regulon of Mycobacterium tuberculosis. Microbes Infect. 8:2052–2060 [DOI] [PubMed] [Google Scholar]
- 29. Ludwig A., et al. 2005. Enhanced expression and shedding of the transmembrane chemokine CXCL16 by reactive astrocytes and glioma cells. J. Neurochem. 93:1293–1303 [DOI] [PubMed] [Google Scholar]
- 30. Matloubian M., David A., Engel S., Ryan J. E., Cyster J. G. 2000. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat. Immunol. 1:298–304 [DOI] [PubMed] [Google Scholar]
- 31. Matsumura S., et al. 2008. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 181:3099–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mittrucker H. W., et al. 2007. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 104:12434–12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mollenkopf H. J., Kursar M., Kaufmann S. H. 2004. Immune response to postprimary tuberculosis in mice: Mycobacterium tuberculosis and Mycobacterium bovis bacille Calmette-Guerin induce equal protection. J. Infect. Dis. 190:588–597 [DOI] [PubMed] [Google Scholar]
- 34. Morgan A. J., et al. 2008. CXCR6 identifies a putative population of retained human lung T cells characterised by co-expression of activation markers. Immunobiology 213:599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Motsinger A., et al. 2002. CD1d-restricted human natural killer T cells are highly susceptible to human immunodeficiency virus 1 infection. J. Exp. Med. 195:869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nanki T., et al. 2005. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. 52:3004–3014 [DOI] [PubMed] [Google Scholar]
- 37. Northfield J. W., et al. 2008. CD161 expression on hepatitis C virus-specific CD8+ T cells suggests a distinct pathway of T cell differentiation. Hepatology 47:396–406 [DOI] [PubMed] [Google Scholar]
- 38. Reed S. G., et al. 2009. Defined tuberculosis vaccine, Mtb72F/AS02A, evidence of protection in cynomolgus monkeys. Proc. Natl. Acad. Sci. U. S. A. 106:2301–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reiley W. W., et al. 2008. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc. Natl. Acad. Sci. U. S. A. 105:10961–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ronan E. O., Lee L. N., Beverley P. C., Tchilian E. Z. 2009. Immunization of mice with a recombinant adenovirus vaccine inhibits the early growth of Mycobacterium tuberculosis after infection. PloS One 4:e8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ronan E. O., Lee L. N., Tchilian E. Z., Beverley P. C. 2010. Nasal associated lymphoid tissue (NALT) contributes little to protection against aerosol challenge with Mycobacterium tuberculosis after immunisation with a recombinant adenoviral vaccine. Vaccine 28:5179–5184 [DOI] [PubMed] [Google Scholar]
- 42. Santosuosso M., et al. 2007. Mucosal luminal manipulation of T cell geography switches on protective efficacy by otherwise ineffective parenteral genetic immunization. J. Immunol. 178:2387–2395 [DOI] [PubMed] [Google Scholar]
- 43. Sawada S., et al. 2007. Involvement of Escherichia coli in pathogenesis of xanthogranulomatous cholecystitis with scavenger receptor class A and CXCL16-CXCR6 interaction. Pathol. Int. 57:652–663 [DOI] [PubMed] [Google Scholar]
- 44. Sharpe S. A., et al. 2010. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clin. Vaccine Immunol. 17:1170–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sui Y., et al. 2010. Innate and adaptive immune correlates of vaccine and adjuvant-induced control of mucosal transmission of SIV in macaques. Proc. Natl. Acad. Sci. U. S. A. 107:9843–9848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tchilian E. Z., et al. 2009. Immunogenicity and protective efficacy of prime-boost regimens with recombinant ΔureC hly+ Mycobacterium bovis BCG and modified vaccinia virus Ankara expressing M. tuberculosis antigen 85A against murine tuberculosis. Infect. Immun. 77:622–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Unutmaz D., et al. 2000. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J. Immunol. 165:3284–3292 [DOI] [PubMed] [Google Scholar]
- 48. van der Voort R., et al. 2005. Elevated CXCL16 expression by synovial macrophages recruits memory T cells into rheumatoid joints. Arthritis Rheum. 52:1381–1391 [DOI] [PubMed] [Google Scholar]
- 49. van der Voort R., et al. 2010. An alternatively spliced CXCL16 isoform expressed by dendritic cells is a secreted chemoattractant for CXCR6+ cells. J. Leukoc. Biol. 87:1029–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Virgin H. W., Walker B. D. 2010. Immunology and the elusive AIDS vaccine. Nature 464:224–231 [DOI] [PubMed] [Google Scholar]
- 51. Wagner C., et al. 2008. T lymphocytes in acute bacterial infection: increased prevalence of CD11b(+) cells in the peripheral blood and recruitment to the infected site. Immunology 125:503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang J., et al. 2004. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 173:6357–6365 [DOI] [PubMed] [Google Scholar]
- 53. Winslow G. M., Cooper A., Reiley W., Chatterjee M., Woodland D. L. 2008. Early T-cell responses in tuberculosis immunity. Immunol. Rev. 225:284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wuttge D. M., et al. 2004. CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 24:750–755 [DOI] [PubMed] [Google Scholar]
- 55. Xing Z. 2009. Importance of T-cell location rekindled: implication for tuberculosis vaccination strategies. Expert Rev. Vaccines 8:1465–1468 [DOI] [PubMed] [Google Scholar]