

Abstract
Egress, which describes the mechanism that some intracellular parasites use to exit from parasitophorous vacuoles and host cells, plays a very important role in the parasite life cycle and is central to Eimeria propagation and pathogenesis. Despite the importance of egress in the intracellular parasite's life cycle, very little information is known on this process compared to other steps, e.g., invasion. The present study was conducted to investigate the interplay between the host adaptive immune system and Eimeria egression. Splenic lymphocytes or soluble immune factors were incubated with parasite-infected host cells for 3 or 5 h, and the percentage of egress was calculated according to an established formula. Viability of egressed parasites and host cells was tested using trypan blue exclusion and annexin V and propidium iodide staining, respectively. We found that premature egression of sporozoites from Eimeria tenella-infected primary chicken kidney cells or from chicken peripheral blood mononuclear cells occurred when the cells were cocultured in vitro with spleen lymphocytes from E. tenella-infected chickens but not when they were cocultured with splenocytes from uninfected chickens. Eimeria-specific antibodies and cytokines (gamma interferon [IFN-γ], interleukin-2 [IL-2], and IL-15), derived from E. tenella-primed B and T lymphocytes, respectively, were capable of promoting premature egress of sporozoites from infected host cells. Both egressed parasites and host cells were viable, although the latter showed reduced reinvasion ability. These results suggest a novel, immune-mediated mechanism that the host exploits to interrupt the normal Eimeria life cycle in vivo and thereby block the release of mature parasites into the environment.
INTRODUCTION
Apicomplexan parasites, such as Plasmodium, Toxoplasma, Cryptosporidium, and Eimeria, comprise a large group of human and/or animal pathogens, most of which possess an apical complex containing characteristic structures such as the conoid, rhoptries, and micronemes. Apicomplexan parasites possess a complex life cycle consisting of cell invasion, intracellular development and propagation, morphogenesis, and egression. Normally, infectious sporozoites rapidly enter host cells aided by the glideosome, a molecular machine that powers parasite motility, migration, cell invasion, and egression (14). Intracellular Eimeria sporozoites have to differentiate into schizonts and subsequently into merozoites, which eventually cause lethal lysis of the host cells in a mechanism termed egress. During egression, the contents of micronemes are discharged, the conoid becomes extended, and the microorganisms acquire motility (31).
Postinfection parasite egression has been studied with the goal of identifying potential therapeutic approaches to interrupt cell exit and thereby disrupt the parasite's life cycle (29). For example, several proteases have been described which are essential for egression of the malaria parasite, Plasmodium, at different life cycle stages (5). Toxoplasma gondii egression is dependent on K+ ion efflux and can be mimicked experimentally using the ionophore nigericin (10). Furthermore, increasing intracellular Ca2+ levels can also induce T. gondii egression (6, 9, 35), which can be inhibited using Ca2+ chelators (4). In this report, we describe a premature egression of sporozoites from Eimeria tenella-infected host cells which occurred when the infected host cells were cocultured in vitro with spleen lymphocytes from E. tenella-infected chickens.
MATERIALS AND METHODS
Animals.
Outbred, specific-pathogen-free White Leghorn chickens from a local hatchery (Beijing Meiliyaweitong Experimental Animal Technology Co., Ltd., Beijing, China) and broiler chickens from Longenecker's Hatchery (Ross/Ross, Elizabethtown, PA) were used. All animals were housed under Eimeria-free conditions, with food and water provided ad libitum. All experiments were performed following Institutional Animal Care and Use Committee guidelines.
Parasites.
Oocysts of E. tenella strains BJ and Beltsville WLR-1 were propagated, isolated, and sporulated as described previously (24). Sporozoites were purified using DE-52 anion-exchange chromatography (32).
Preparation of chicken cells for E. tenella infection.
Primary chicken kidney (PCK) cells were prepared as previously described (27), with modifications. Kidneys were aseptically removed from 7- to 14-day-old chickens, placed in Ca2+- and Mg2+-free Hanks' balanced salt solution (CMF-HBSS) containing 100 U/ml penicillin and 100 μg/ml streptomycin, and cut into small pieces. Kidney pieces were incubated with 0.25% trypsin (Sigma, St. Louis, MO) for 5 min at 37°C, trypsin was inactivated by addition of Iscove's modified Dulbecco's medium (IMDM; Invitrogen, Carlsbad, CA) containing 20% fetal calf serum (FCS; HyClone, Thermo Scientific, Logan, UT), and the cells in the supernatant were collected by centrifugation. This process was repeated 3 times, and the PCK cells were pooled and resuspended in IMDM containing 10% FCS. Chicken peripheral blood mononuclear cells (PBMCs) were prepared as previously described (18). Whole blood was collected aseptically by venipuncture of the wing vein and was diluted 1:1 with CMF-HBSS at 4°C. PBMCs were isolated by density gradient centrifugation using Polymorphprep (Fresenius Kabi, Oslo, Norway). After being washed with CMF-HBSS, the cells were resuspended in IMDM containing 10% fetal bovine serum (FBS), 1× nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell viability, determined by trypan blue exclusion, was consistently >90%.
Preparation of chicken spleen lymphocytes.
Three-week-old chickens were randomly divided into two groups. Chickens in group I were orally inoculated with 200 μl of phosphate-buffered saline (PBS) as uninfected controls. Chickens in group II were kept in a separate room and were orally inoculated with 200 μl of PBS containing 1.0 × 104 E. tenella sporulated oocysts. One week after primary infection, chickens in group I were still given PBS and chickens in group II were given a secondary oral infection with 1.0 × 105 E. tenella sporulated oocysts in PBS. Splenic lymphocytes from uninfected and infected animals were isolated as previously described (7), with modifications. Spleens were obtained aseptically at 7 days post-secondary infection, and single-cell suspensions were prepared by passage through a wire mesh in IMDM containing 5% FBS, 10 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were passed through a nylon wool column to remove clumps and debris, and splenic lymphocytes were enriched by density gradient centrifugation through Polymorphprep as described above. Purified lymphocytes were resuspended in IMDM containing 10% FBS, 1× nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell viability, determined by trypan blue exclusion, was consistently >90%.
Egress assay.
PCK cells and PBMCs were seeded in 24-well plates containing glass slides (2.0 × 107 cells/well) and cultured overnight at 41°C, and nonadherent cells were removed by washing with CMF-HBSS. Two to 3 days later, the cells were incubated overnight at 41°C with E. tenella strain BJ (for PCK cells) or strain WLR-1 (for PBMCs) sporozoites at a multiplicity of infection of 1.0 (parasite/cell ratio of 1:1). Free parasites were removed by washing with CMF-HBSS at 41°C. Following sporozoite infection, the cells were left untreated or were cocultured with splenic lymphocytes from uninfected or E. tenella-infected animals for 1, 3, or 5 h at 41°C, using splenocyte:PCK cell or splenocyte:PBMC ratios of 25:1, 50:1, and 100:1. In some experiments, sporozoite-infected PBMCs were cocultured with spleen cells in Transwell membrane inserts (8-μm pore size; Corning, Lowell, MA) to assess the role of cell-cell contact in parasite egression. Following coculture incubation, the splenocytes were removed by washing with CMF-HBSS, and the infected cells were fixed with methanol for 10 min at 4°C and permeabilized with 0.05% Triton X-100 in PBS for 10 min at 4°C. The cells were incubated with mouse anti-Eimeria antibodies for 30 min at 4°C, washed, and incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibody (Invitrogen) in 0.1 μg/ml of Evan's blue for 30 min at 4°C. Intracellular parasites were counted in 30 randomly selected fields by use of a fluorescence microscope (Carl Zeiss, Thornwood, NY, or Olympus). The percentage of parasite egress was calculated using the following formula: % egress = [(N1 − N2)/N1] × 100, where N1 is the number of intracellular parasites in cells cocultured with splenocytes from primed or naive chickens and N2 is the number of intracellular parasites in cells cocultured with IMDM. Each experiment was repeated 3 times.
Determination of sporozoite viability and infectivity.
E. tenella sporozoite-infected PCK cells were cocultured with splenocytes in Transwell membrane inserts. After 5 h of incubation, cell culture medium which contained egressed E. tenella sporozoites was collected, and the viability of egressed sporozoites was determined using trypan blue dye exclusion. To evaluate the infectivity of egressed sporozoites, they were added to the monolayer of PCK cells overnight. The freshly prepared E. tenella sporozoites were used as a control. Free parasites were removed by washing with CMF-HBSS at 41°C, and the intracellular parasites were quantified by counting a minimum of 10 fields per well (×40 objective; Olympus IX71 microscope). Infection rates were calculated by dividing the number of intracellular parasites by the total number of inoculated parasites.
Host cell viability evaluation.
After overnight incubation of the PCK cells with sporozoites, PCK cells were washed and cocultured with E. tenella-primed or naive splenocytes at a lymphocyte-to-PCK-cell ratio of 100:1 for 3 h at 41°C in a 5% CO2 incubator. At each time point, PCK cells were washed and viability was assessed using an annexin V-fluorescein isothiocyanate (annexin V-FITC) apoptosis detection kit (Nanjing Keygen Biotechnology Co. Ltd., China). Briefly, the adherent PCK cells were washed in cold PBS, gently trypsinized, and washed again twice in PBS, and single cells were resuspended in 500 μl binding buffer. Next, 2 μl of annexin V-FITC and 5 μl of propidium iodide (PI) were added and incubated at room temperature for 15 min in the dark. The dead cells were visualized by fluorescence microscopy (Olympus IX71) with appropriate filters. Viability of host cells after parasite egression was calculated according to the following formula: % viability = (% egress − % death)/% egress, where % death means the total number of dead host cells divided by the total number of host cells.
Magnetic bead cell subpopulation depletion.
Spleen lymphocyte subpopulations were depleted using a commercial cell separation system (BD IMag cell separation system; BD Biosciences, San Jose, CA). Splenocytes were incubated with mouse monoclonal antibodies detecting chicken B cells or CD4+ or CD8+ T cells (21) in CMF-HBSS containing 3% heat-inactivated FCS and 0.01% sodium azide (staining buffer) for 30 min at 4°C. The cells were washed with staining buffer and incubated with biotinylated goat anti-mouse IgG antibody for 30 min at 4°C. The cells were washed, resuspended in BD IMag buffer containing a pretitrated volume of streptavidin-labeled magnetic particles, and exposed to a magnetic field to remove the labeled cells, and the cells remaining in the supernatant were cocultured with sporozoite-infected cells for the egress assay. The efficiency of each depleted cell population was verified by flow cytometry. The cell depletion efficiency was routinely >95%, with >90% cell viability.
Effects of Eimeria antibody or chicken cytokines on parasite egress.
Sporozoite-infected PBMCs were preincubated with recombinant chicken cytokines (interleukin-2 [IL-2], IL-15, and gamma interferon [IFN-γ]) or with recombinant swine IL-13 or chicken anti-Eimeria IgY antibody or nonimmune IgY as a negative control for 5 h at 41°C, the cells were washed, and egress was calculated as described above. Cytokines were produced in COS-7 cells as previously described (22, 34). Anti-Eimeria IgY and nonimmune IgY antibodies were prepared from egg yolks of specific-pathogen-free hens hyperimmunized and not immunized, respectively, with live oocysts of E. tenella, E. acervulina, and E. maxima as described previously (16, 17). Antibody concentrations were determined using the Bradford reagent (Sigma), with bovine serum albumin as the standard.
Effect of PLC inhibition or parasite motility on egress.
U-73122 (phospholipase C [PLC] inhibitor; Sigma) and cytochalasin D (motility inhibitor; Sigma) were used to investigate the roles of PLC and actin-dependent parasite motility on sporozoite egress. Stock solutions of 1.0 mM U-73122 and cytochalasin D were prepared in dimethyl sulfoxide (DMSO; Sigma). E. tenella sporozoite-infected host cells were pretreated for 1 h at 41°C with DMSO vehicle alone or with 2.0 or 10 μM drug in IMDM. The cells were washed with IMDM and cocultured with splenocyte suspensions (100:1) or incubated with IgY antibody (200 μg/ml) for 3 h at 41°C, and egress was calculated as described above.
Statistical analysis.
Each sample was analyzed in triplicate. Statistical analyses were performed using GraphPad Prism 4 or SPSS software (SPSS 15.0 for Windows; SPSS, Chicago, IL), and all data are expressed as means with standard errors of the means (SEM). Comparisons of the mean values were performed by Student's t test, and differences between means were considered statistically significant for P values of <0.05.
RESULTS
Sporozoite egress from PCK cells cocultured with E. tenella-primed splenocytes.
Initially, we determined the effect of E. tenella-immune splenocytes on sporozoite egress from E. tenella-infected PCK cells. To quantify egress, splenocytes from naive or E. tenella-primed chickens were cocultured with infected host cells and the number of intracelluar sporozoites was determined by immunofluorescence microscopy of permeabilized cells (8, 15, 26, 38). Infected PCK cells that had been incubated for 1, 3, or 5 h with primed splenocytes from E. tenella-infected chickens at a ratio of 100:1 (splenocytes:PCK cells) displayed increased parasite egress compared with PCK cells cocultured with naive splenocytes from uninfected chickens (Fig. 1).
Fig. 1.

Sporozoite egress from PCK cells cocultured with E. tenella-primed splenocytes. A PCK cell culture was infected overnight with E. tenella BJ strain sporozoites at a parasite-to-PCK-cell ratio of 1:1. Free parasites were removed by washing, and the infected cells were incubated for 1, 3, or 5 h with spleen lymphocytes from uninfected (naive) or E. tenella-infected (primed) chickens at a splenocyte-to-PCK-cell ratio of 100:1. The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing primed splenocytes with naive splenocytes at the respective time points.
Sporozoite egress from PBMCs cocultured with E. tenella-primed splenocytes.
In addition to gut epithelial cells, Eimeria sporozoites have been shown to invade chicken blood monocytes (37). Therefore, we next examined parasite egress by using E. tenella-infected PBMCs as host cells. Infected PBMCs that had been incubated for 3 h with splenocytes from E. tenella-infected chickens at a ratio of 100:1 (splenocytes: PBMCs) displayed increased parasite egress compared with cells cocultured with naive splenocytes from uninfected chickens (Fig. 2). Coculture at 25:1 and 50:1 cell ratios did not increase parasite egress.
Fig. 2.

Sporozoite egress from PBMCs cocultured with E. tenella-primed splenocytes. Cells were infected overnight with E. tenella WLR-1 strain sporozoites at a 1:1 ratio (cells:parasites). Free parasites were removed by washing, and the infected cells were incubated for 3 h with spleen lymphocytes from uninfected (naive) or E. tenella-infected (primed) chickens at 25:1, 50:1, and 100:1 ratios (splenocytes:PCK cells). The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing primed splenocytes with naive splenocytes at the respective cell ratios.
Viability and infectivity of egressed sporozoites and viability of host cells.
Egressed E. tenella sporozoites remained viable (>95% viability), as determined by trypan blue exclusion, and could invade freshly prepared host cells, but their invasion efficacy was significantly lower than that of freshly prepared sporozoites (P < 0.01) (Fig. 3A). The effect of sporozoite egress on viability of PCK cells after parasite egression was determined after incubation of splenocytes from control or E. tenella-immune birds with parasite-infected PCK cells. Although a higher level of death (5.3%) was shown for PCK cells incubated with splenocytes from E. tenella-infected chickens than for those incubated with splenocytes from naive chickens (0.2%), the majority of host cells were viable after parasite egression (Fig. 3B).
Fig. 3.

Infectivity of egressed parasites and viability of host cells. (A) Infectivity of egressed parasites. Infectivity of egressed sporozoites was detected by adding egressed sporozoites to PCK cell monolayers overnight. Freshly prepared E. tenella sporozoites were used as a control. Infectivity was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 6). **, P < 0.01, comparing egressed sporozoites with freshly prepared sporozoites. (B) Viability of host cells after parasite egression. Dead cells were assessed using an annexin V-FITC apoptosis detection kit to stain cells with annexin V-FITC and propidium iodide. The viability of host cells was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3).
Spleen T and B lymphocytes mediate sporozoite egress.
CD4+ T cells, CD8+ T cells, and B cells comprise >90% of chicken splenocytes (37). To identify the splenocyte subpopulation(s) responsible for enhanced parasite egress, magnetic cell depletion experiments using lymphocyte subpopulation-specific monoclonal antibodies were performed. Depletion of CD8+ splenic T cells reduced but did not completely eliminate the ability of primed splenocytes to increase parasite egress from infected PCK cells (Fig. 4). In contrast, depletion of CD4+ T cells, CD8+ T cells, and B cells nearly completely eliminated the enhanced egression. In the case of infected PBMCs, depletion of CD8+ T cells plus B cells completely reduced the ability of splenocytes to increase parasite egress, while depletion of CD4+ T cells plus B cells or depletion of CD4+ plus CD8+ T cells had lesser effects. No differences in parasite egress were observed using naive spleen cells from uninfected chickens that had been depleted of the different lymphocyte subpopulations.
Fig. 4.

Spleen T and B lymphocytes mediate sporozoite egress. PCK cells or PBMCs were infected overnight with E. tenella sporozoites at a 1:1 ratio. Free parasites were removed by washing, and the infected cells were incubated for 3 h with spleen lymphocytes, left undepleted (normal) or depleted of CD4+ T cells, CD8+ T cells, and/or B cells, from uninfected (naive) or E. tenella-infected (primed) chickens at a 100:1 ratio. The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing respective cell depletions with no cell depletion.
Splenocyte-enhanced parasite egress is independent of cell-cell contact.
A Transwell coculture system using upper and lower culture chambers separated by a cell-impermeable porous membrane was used to determine whether cell-to-cell contact was required for enhanced parasite egress, a process mediated here by E. tenella-primed splenocytes. Splenocytes in the upper chamber were cocultured with Eimeria-infected PCK cells or PBMCs in the lower chamber, and parasite egress was compared with that in a standard coculture system. No significant differences in parasite egress were observed between cells in the Transwell and standard coculture systems (Fig. 5).
Fig. 5.

Splenocyte-enhanced parasite egress is independent of cell-cell contact. PCK cells (A) or PBMCs (B) were infected overnight with E. tenella sporozoites at a 1:1 ratio. Free parasites were removed by washing, and the infected cells were incubated for 5 h with spleen lymphocytes from uninfected (naive) or E. tenella-infected (primed) chickens at a 100:1 ratio in a standard coculture assay (without Transwells) or in a Transwell coculture system. The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing primed splenocytes with naive splenocytes.
Chicken cytokines and Eimeria-specific antibody increase E. tenella egress from PBMCs.
Based on the cell depletion and Transwell coculture results, we speculated that soluble immune factors derived from T and/or B cells may be involved in sporozoite release from infected cells. Therefore, we evaluated the effects of the T cell factors IFN-γ, IL-2, and IL-15, as well as Eimeria-specific IgY antibody as a B cell factor, on parasite egress. Culture supernatants from COS-7 cells transfected with expression plasmids encoding recombinant chicken IFN-γ, IL-2, or IL-15 dose-dependently increased sporozoite egress from E. tenella-infected PBMCs compared with culture supernatants from nontransfected COS-7 cells or with swine IL-13 as a negative control (Fig. 6). As shown in Fig. 7, hyperimmune Eimeria-specific IgY antibody increased parasite egress from PBMCs compared with nonimmune IgY from unimmunized chickens.
Fig. 6.

Chicken cytokines increase E. tenella egress from PBMCs. Cells were infected overnight with E. tenella sporozoites at a 1:1 ratio, free parasites were removed by washing, and the cells were incubated for 3 h with cell culture supernatants containing recombinant IFN-γ, IL-2, or IL-15, with supernatant from nonexpressing COS-7 cells (control), or with the indicated concentrations of swine IL-13 as an additional negative control. The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing cytokines with the respective dilution of the negative control.
Fig. 7.
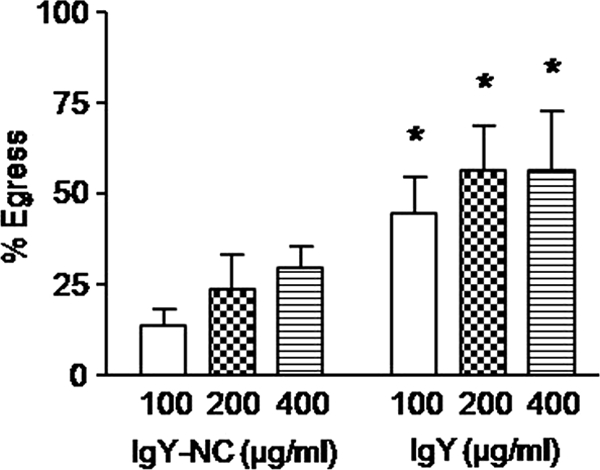
Eimeria-specific antibody increases E. tenella egress from PBMCs. Cells were infected overnight with E. tenella sporozoites at a 1:1 ratio, free parasites were removed by washing, and the cells were incubated for 3 h with the indicated concentrations of purified IgY antibody from chickens immunized with E. tenella, E. acervulina, and E. maxima or with IgY antibody from unimmunized chickens (IgY-NC) as a negative control. The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing IgY with IgY-NC at the respective concentrations.
Sporozoite egress from PBMCs cocultured with E. tenella-primed splenocytes or treated with anti-Eimeria antibody requires PLC activation and parasite motility.
PLC-dependent parasite motility is required for T. gondii egress from host cells (10, 26). Therefore, in order to rule out the possibility that loss of intracellular sporozoites from host cells was due to a mechanism other than parasite egress (e.g., decreased parasite viability), E. tenella-infected PBMCs were left untreated or pretreated with cytochalasin D (motility inhibitor) or U-73122 (PLC inhibitor) prior to coculture with E. tenella-primed splenocytes, and sporozoite egress was quantified. As shown in Fig. 8A, 1-h preexposure of infected PBMCs to cytochalasin D or U-73122 before coculture with splenocytes decreased sporozoite egress in a dose-dependent manner compared with the respective untreated control groups. Moreover, both drug inhibitors also blocked the ability of Eimeria-specific IgY antibody to increase sporozoite egress (Fig. 8B).
Fig. 8.

Sporozoite egress from PBMCs cocultured with E. tenella-primed splenocytes or treated with anti-Eimeria antibody requires PLC activation and parasite motility. Cells were infected overnight with E. tenella sporozoites at a 1:1 ratio, free parasites were removed by washing, and the cells were left untreated or pretreated for 1 h with the indicated concentrations of cytochalasin D or U-73122. The cells were washed and incubated for 3 h with spleen lymphocytes from uninfected (naive) or E. tenella-infected (primed) chickens at a 100:1 ratio or with 200 μg/ml of hyperimmune IgY or nonimmune IgY antibodies. The percentage of egress was calculated as described in Materials and Methods. Each bar represents the mean with SEM (n = 3). *, P < 0.05, comparing cytochalasin D or U-73122 with the respective DMSO control.
DISCUSSION
The results from the current study indicate that in vitro coculture of E. tenella-infected chicken primary kidney cells or peripheral blood mononuclear cells with spleen lymphocytes from E. tenella-infected chickens, but not with those from uninfected chickens, led to premature egress of the intracellular sporozoites. CD4+ T cells, CD8+ T cells, and B cells in the spleen cell population were responsible for this effect, whereas cell-cell contact between the effector and target cells was not required. Antigen-specific antibody and cytokines (IFN-γ, IL-2, and IL-15), derived from B and T lymphocytes, respectively, were also capable of promoting early sporozoite exit. These results suggest a novel, immune-mediated mechanism that may represent an alternative pathway to the use of cytotoxic T lymphocytes (CTLs) and autophagy that the host uses to reduce the number of intracellularly developing parasites.
Coccidian egress from infected cells is a crucial step in the life cycle of Eimeria parasites that plays an important role in propagation and pathogenesis of the microorganism (11). Premature egress at the sporozoite stage prior to differentiation into merozoites is not a natural event of the parasite's life cycle and, as a result, would be expected to interrupt this cycle as the egressed parasites are exposed to the host's hostile microenvironment, where they are targeted by the effector components of innate and adaptive immunity.
The egress assay based upon immunofluorescence quantification of intracellular E. tenella sporozoites that was used in this study has previously been validated in multiple laboratories using a wide range of apicomplexan parasites, including Toxoplasma (15, 26), Plasmodium (38), and Cryptosporidium (8). For this reason, it is highly probable that the reduced presence of intracellular parasites accurately reflects coccidian exit from the infected host cells. Furthermore, our drug inhibition studies are consistent with the notion that PLC-dependent Eimeria motility is required for splenocyte-mediated egression. Therefore, although not formally excluded in the current study, these results do not likely reflect the function of cytotoxic T lymphocytes or NK cells in the spleen cell preparations, as we used outbred chickens in our system. Rather, given that Eimeria-specific antibody and the T cell-derived cytokines IFN-γ, IL-2, and IL-15 can substitute for intact spleen lymphocytes in promoting early sporozoite egress, it is theoretically possible that a plausible mechanism may involve modification of an intracellular process in the infected cells mediated by soluble immune factors. In this regard, IFN-γ, a key cytokine with an inhibitory effect on intracellular pathogens, induced parasite emergence in T. gondii-infected macrophages (28, 33, 36). In chickens, recombinant IFN-γ interrupted the in vitro intracellular development of E. tenella and augmented protective immunity against Eimeria challenge infection when it was given in vivo (20, 25). Similar effects have been reported for chicken IL-2 and IL-15 (22, 25). Our current findings propose an additional role of these cytokines in the host defense against intracellular pathogens.
The role of antigen-specific antibodies in avian coccidiosis is more controversial. In general, no direct correlation between the levels of serum antibodies and coccidiosis protection has been reported. However, antibody-mediated protective immunity has been demonstrated using passively administered hyperimmune, antigen-specific IgY antibodies (18). Additionally, injection of E. tenella sporozoites pretreated with immune serum into the coelomic cavity of chickens significantly reduced the appearance of parasite-containing macrophages compared with administration of untreated coccidia (13). Taken together with the present findings showing that Eimeria-immune IgY enhances egress and that egress decreases upon B cell depletion, these data provide evidence for an important role of antigen-specific antibodies in an immune-mediated pathway involved in parasite egress.
One of the greatest challenges that the immune system faces in dealing with intracellular pathogens is how to eliminate the microorganism without damaging the infected host cells. Recently, autophagy has become recognized as one means that selectively disposes of only intracellular pathogens (1, 19, 23). Our proposed immune-mediated mechanism of premature parasite egression represents a novel process that is relevant to a highly immunogenic pathogen such as Eimeria. This hypothesis is further supported by the observations that (i) postegression host cells remained viable (Fig. 3B), (ii) fewer E. tenella sporozoites were found inside epithelial cells in coccidian-immunized chickens than in naive animals (30), and (iii) externally triggered egress was the major fate of T. gondii during acute infection (36). In the case of Toxoplasma infection, PLC and other Ca2+-related molecules are critical mediators of parasite egress (2, 12, 26). This process is thought to involve an as yet unidentified parasite sensor that detects changes in intracellular Ca2+ levels. Similar observations have been made with E. bovis (3), and the study of possible interactions between PLC and immune cytokines in promoting the egress of Eimeria, as well as other apicomplexans, from host cells is a subject ripe for further investigation.
In conclusion, premature egression of E. tenella sporozoites from chicken cells was induced during in vitro coculture with splenic lymphocytes from E. tenella-infected chickens but not with those from uninfected animals. The egressed E. tenella sporozoites were viable but showed a decreased invasion ability compared with freshly prepared sporozoites. The fact that primed splenic lymphocytes and Eimeria-specific antibodies and cytokines derived from B and T lymphocytes promoted early sporozoite egress suggests that an immune-mediated mechanism is involved in pathogen clearance in coccidiosis. Future studies to further characterize host and parasite factors that signal egress will lead to the development of novel therapeutic approaches to interrupt the normal Eimeria life cycle in vivo.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (project numbers 30871862 and 30671579), the Chinese Universities Scientific Fund (2009-2-02), and the Avian Immunology CRIS project from the Agricultural Research Service-USDA.
We thank Erik P. Lillehoj, University of Maryland School of Medicine, for editorial assistance; Boulard Chantal, Institut National de la Recherche Agronomique, France, for helpful comments; and Margie Nichols for her technical assistance. We thank Xinlei Yan, Yuchen Chen, and Yongshen Ji for assistance with the viability test.
Footnotes
Published ahead of print on 31 May 2011.
REFERENCES
- 1. Andrade R. M., Wessendarp M., Gubbels M. J., Striepen B., Subauste C. S. 2006. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 116:2366–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arrizabalaga G., Boothroyd J. C. 2004. Role of calcium during Toxoplasma gondii invasion and egress. Int. J. Parasitol. 34:361–368 [DOI] [PubMed] [Google Scholar]
- 3. Behrendt J. H., Taubert A., Zahner H., Hermosilla C. 2008. Studies on synchronous egress of coccidian parasites (Neospora caninum, Toxoplasma gondii, Eimeria bovis) from bovine endothelial host cells mediated by calcium ionophore A23187. Vet. Res. Commun. 32:325–332 [DOI] [PubMed] [Google Scholar]
- 4. Black M. W., Arrizabalaga G., Boothroyd J. C. 2000. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol. Cell. Biol. 20:9399–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blackman M. J. 2008. Malarial proteases and host cell egress: an ‘emerging’ cascade. Cell. Microbiol. 10:1925–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caldas L. A., de Souza W., Attias M. 2007. Calcium ionophore-induced egress of Toxoplasma gondii shortly after host cell invasion. Vet. Parasitol. 147:210–220 [DOI] [PubMed] [Google Scholar]
- 7. Chai Y. J., Lillehoj H. S. 1988. Isolation and functional characterization of chicken intestinal intraepithelial lymphocytes showing natural activity against tumour target cell. Immunology 63:111–117 [PMC free article] [PubMed] [Google Scholar]
- 8. Elliott D. A., Clark D. P. 2003. Host cell fate on Cryptosporidium parvum egress from MDCK cells. Infect. Immun. 71:5422–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Endo T., Sethi K. K., Piekarski G. 1982. Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp. Parasitol. 53:179–188 [DOI] [PubMed] [Google Scholar]
- 10. Fruth I. A., Arrizabalaga G. 2007. Toxoplasma gondii: induction of egress by the potassium ionophore nigericin. Int. J. Parasitol. 37:1559–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hammond D. M., Long P. L. 1973. The coccidia: Eimeria, Isospora, Toxoplasma, and related genera. University Park Press, Baltimore, MD [Google Scholar]
- 12. Hoff E. F., Carruthers V. B. 2002. Is Toxoplasma egress the first step in invasion? Trends Parasitol. 18:251–255 [DOI] [PubMed] [Google Scholar]
- 13. Huff D., Clark D. T. 1970. Cellular aspects of the resistance of chickens to Eimeria tenella infections. J. Protozool. 17:35–39 [DOI] [PubMed] [Google Scholar]
- 14. Keeley A., Soldati D. 2004. The glideosome: a molecular machine powering motility and host-cell invasion by Apicomplexa. Trends Cell Biol. 14:528–532 [DOI] [PubMed] [Google Scholar]
- 15. Lavine M. D., Arrizabalaga G. 2008. Exit from host cells by the pathogenic parasite does not require motility. Eukaryot. Cell 7:131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee S. H., et al. 2009. Protective effect of hyperimmune egg yolk IgY antibodies against Eimeria tenella and Eimeria maxima infections. Vet. Parasitol. 163:123–126 [DOI] [PubMed] [Google Scholar]
- 17. Lee S. H., et al. 2009. Induction of passive immunity in broiler chickens against Eimeria acervulina by hyperimmune egg yolk immunoglobulin Y. Poult. Sci. 88:562–566 [DOI] [PubMed] [Google Scholar]
- 18. Lee S. H., et al. 2008. Immune enhancing properties of safflower leaf (Carthamus tinctorius) on chicken lymphocytes and macrophages. J. Poult. Sci. 45:147–151 [Google Scholar]
- 19. Levine B., Deretic V. 2007. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7:767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lillehoj H. S., Choi K. D. 1998. Recombinant chicken interferon-γ mediates inhibition of Eimeria tenella development in vitro and reduction of oocyst production and body weight loss following Eimeria acervulina challenge infection. Avian Dis. 42:307–314 [PubMed] [Google Scholar]
- 21. Lillehoj H. S., Lillehoj E. P., Weinstock D., Schat K. A. 1988. Functional and biochemical characterizations of avian T lymphocyte antigens identified by monoclonal antibodies. Eur. J. Immunol. 18:2059–2065 [DOI] [PubMed] [Google Scholar]
- 22. Lillehoj H. S., et al. 2001. Molecular, cellular, and functional characterization of chicken cytokines homologous to mammalian IL-15 and IL-2. Vet. Immunol. Immunopathol. 82:229–244 [DOI] [PubMed] [Google Scholar]
- 23. Ling Y. M., et al. 2006. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203:2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Long P. L., Joyner L. N., Millard B. J., Norton C. C. 1976. A guide to laboratory techniques used in the study and diagnosis of avian coccidiosis. Folia Vet. Lat. 6:201–217 [PubMed] [Google Scholar]
- 25. Min W., et al. 2002. Adjuvant effects of IL-1β, IL-8, IL-15, IFN-α, IFN-γ, TGF-β and lymphotactin on DNA vaccination against Eimeria acervulina. Vaccine 20:267–274 [DOI] [PubMed] [Google Scholar]
- 26. Moudy R., Manning T. J., Beckers C. J. 2001. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J. Biol. Chem. 276:41492–41501 [DOI] [PubMed] [Google Scholar]
- 27. Ogura H., Fujiwara T. 1987. Establishment and characterization of a virus-free chicken cell line. Acta Med. Okayama 41:141–143 [DOI] [PubMed] [Google Scholar]
- 28. Pfefferkorn E. R., Guyre P. M. 1984. Inhibition of growth of Toxoplasma gondii in cultured fibroblasts by human recombinant gamma interferon. Infect. Immun. 44:211–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roiko M. S., Carruthers V. B. 2009. New roles for perforins and proteases in apicomplexan egress. Cell. Microbiol. 11:1444–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rose M. E., Hesketh P. 1987. Eimeria tenella: effects of immunity on sporozoites within the lumen of the small intestine. Exp. Parasitol. 63:337–344 [DOI] [PubMed] [Google Scholar]
- 31. Santos J. M., Lebru M., Daher W., Soldati D., Dubremetz J. F. 2009. Apicomplexan cytoskeleton and motors: key regulators in morphogenesis, cell division, transport and motility. Int. J. Parasitol. 39:153–162 [DOI] [PubMed] [Google Scholar]
- 32. Schmatz D. M., Crane M. S., Murray P. K. 1984. Purification of Eimeria sporozoites by DE-52 anion exchange chromatography. J. Protozool. 3:181–183 [DOI] [PubMed] [Google Scholar]
- 33. Shirahata T., Shimizu K. 1982. Growth inhibition of Toxoplasma gondii in cell cultures treated with murine type II interferon. Jpn. J. Vet. Sci. 44:865–871 [DOI] [PubMed] [Google Scholar]
- 34. Song K. D., Lillehoj H. S., Choi K. D., Zarlenga D., Han J. Y. 1997. Expression and functional characterization of recombinant chicken interferon-gamma. Vet. Immunol. Immunopathol. 58:321–333 [DOI] [PubMed] [Google Scholar]
- 35. Stommel E. W., Kenneth H. E., Schwartzmann J. D., Kasper L. H. 1997. Toxoplasma gondii: dithiol-induced Ca2+ flux caused egress of parasites from the parasitophorous vacuole. Exp. Parasitol. 87:88–97 [DOI] [PubMed] [Google Scholar]
- 36. Tomita T., Yamada T., Weiss L. M., Orlofsky A. 2009. Externally triggered egress is the major fate of Toxoplasma gondii during acute infection. J. Immunol. 183:6667–6680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trout J. M., Lillehoj H. S. 1995. Eimeria acervulina infection: evidence for the involvement of CD8+ T lymphocytes and macrophages in sporozoite transport and host protection. Poult. Sci. 74:1117–1125 [DOI] [PubMed] [Google Scholar]
- 38. Wickham M. E., Culvenor J. G., Cowman A. F. 2003. Selective inhibition of a two-step egress of malaria parasites from the host erythrocyte. J. Biol. Chem. 278:37658–37663 [DOI] [PubMed] [Google Scholar]