

Abstract
Amino acid substitutions conferring resistance to quinolones in Mycobacterium tuberculosis have generally been found within the quinolone resistance-determining regions (QRDRs) in the A subunit of DNA gyrase (GyrA) rather than the B subunit of DNA gyrase (GyrB). To clarify the contribution of an amino acid substitution, E540V, in GyrB to quinolone resistance in M. tuberculosis, we expressed recombinant DNA gyrases in Escherichia coli and characterized them in vitro. Wild-type and GyrB-E540V DNA gyrases were reconstituted in vitro by mixing recombinant GyrA and GyrB. Correlation between the amino acid substitution and quinolone resistance was assessed by the ATP-dependent DNA supercoiling assay, quinolone-inhibited supercoiling assay, and DNA cleavage assay. The 50% inhibitory concentrations of eight quinolones against DNA gyrases bearing the E540V amino acid substitution in GyrB were 2.5- to 36-fold higher than those against the wild-type enzyme. Similarly, the 25% maximum DNA cleavage concentrations were 1.5- to 14-fold higher for the E540V gyrase than for the wild-type enzyme. We further demonstrated that the E540V amino acid substitution influenced the interaction between DNA gyrase and the substituent(s) at R-7, R-8, or both in quinolone structures. This is the first detailed study of the contribution of the E540V amino acid substitution in GyrB to quinolone resistance in M. tuberculosis.
INTRODUCTION
A major human infectious disease, tuberculosis (TB) is estimated to affect approximately one-third of the world's population, and 95% of cases occur in developing countries (15, 30, 31). Current estimates show that approximately 9.4 million new cases and nearly 1.7 million deaths from TB occur each year, and TB remains a major cause of premature death (36).
The increased incidence of multidrug-resistant (MDR) TB (TB resistant to more than two anti-TB drugs, including rifampin and isoniazid [35]) has hampered the treatment and control of TB and is associated with an increase in mortality rates in people with TB (3, 37, 40). Consequently, the required drug dosage for the treatment of TB has dramatically increased (38), and fluoroquinolones (FQs) are now considered to be important second-line anti-TB agents (13, 20).
FQs are a large and widely used class of synthetic antibacterial agents (10, 11, 21, 39) which are frequently used in treating patients infected with MDR TB (6, 17, 22). The target of the FQs in Mycobacterium tuberculosis is DNA gyrase, which consists of two subunits, GyrA and GyrB, that form the catalytically active GyrA2GyrB2 heterotetrameric structure (7, 9, 18). DNA gyrase is an ATP-dependent enzyme that transiently cleaves and unwinds double-stranded DNA (9) to catalyze the negative supercoiling of DNA and is thus essential for efficient DNA replication, transcription, and recombination (7, 24, 29). Most eubacteria, such as Escherichia coli, have two DNA topoisomerases, DNA gyrase and topoisomerase IV. A few bacteria, however, such as M. tuberculosis, have only DNA gyrase (8), which is therefore the sole target of quinolones.
The quinolone-binding sites in DNA gyrase have been found to be in the quinolone resistance-determining regions (QRDRs) in the GyrA subunit (amino acids Gly-88 to Asp-94 in M. tuberculosis) and the GyrB subunit (amino acids Asp-500 to Asn-538 in M. tuberculosis), which contain the majority of the amino acid substitutions that confer quinolone resistance (Fig. 1) (2, 19, 27, 32, 33). A recent study using three-dimensional structure analysis, however, has suggested that QRDRs of M. tuberculosis gyrase are located at amino acids Ser-73 to Gln-113 of the GyrA subunit and Asn-493 to Asn-540 of the GyrB subunit (28). The QRDRs in GyrB are thought to interact with those in GyrA and DNA strands to form a quinolone-binding pocket (QBP).
Fig. 1.
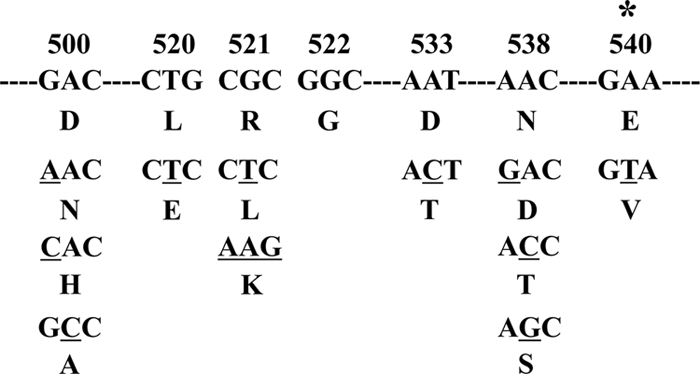
Amino acid substitutions found within the QRDR of GyrB in FQ-resistant M. tuberculosis. The amino acid substitutions are shown below the nucleotide sequence. The target nucleotide substitution of this study is denoted by the asterisk, and mutated bases are underlined.
In this study, we elucidated the contribution of an amino acid substitution located at position 540 that is found in a quinolone-resistant clinical isolate by in vitro DNA supercoiling and cleavage assays in the presence or absence of FQs. We also propose the mechanism of interaction between substituents of FQs and amino acid residues in the QBP of GyrB.
MATERIALS AND METHODS
Reagents and kits.
Gatifloxacin (GAT), levofloxacin (LVX), ciprofloxacin (CIP), sparfloxacin (SPX), and enoxacin (ENX) were purchased from LKT Laboratories, Inc. (St. Paul, MN); sitafloxacin (SIX) was from Daiichi Pharmaceutical, Co., Ltd. (Tokyo, Japan); norfloxacin (NOR) was from Wako Pure Chemical Industries, Ltd. (Tokyo, Japan); moxifloxacin (MXF) was from Toronto Research Chemicals Inc. (Ontario, Canada); and ampicillin was from Meiji Seika Kaisha, Ltd. (Tokyo, Japan). Oligonucleotide primers were synthesized by Life Technologies (Carlsbad, CA). TOPO TA cloning (pCR 4-TOPO) and Ni-nitrilotriacetic acid protein purification kits were purchased from Life Technologies. Restriction enzymes were obtained from New England BioLabs, Inc. (Ipswich, MA). The supercoiling assay kit and supercoiled and relaxed pBR322 DNA were purchased from John Innes Enterprises Ltd. (Norwich, United Kingdom). Protease inhibitor cocktail (Complete Mini, EDTA free) was purchased from Roche Applied Science (Mannheim, Germany).
Bacterial strains and plasmids.
E. coli strain TOP-10 (Life Technologies) was used as the host for cloning purposes. E. coli strains Rosetta-gami 2 and BL21(DE3)/pLysS were purchased from Merck KGaA (Darmstadt, Germany) and used for protein expression. Vector plasmids pET-20b (+) and pET-19b (Merck KGaA) were used to construct expression plasmids for M. tuberculosis proteins GyrA and GyrB, respectively.
Construction of wild-type (WT) and GyrB-E540V DNA gyrase expression vectors.
The construction of WT DNA gyrase expression vectors is shown in Fig. S1 in the supplemental material. A nucleotide substitution was introduced into the M. tuberculosis WT gyrB gene by PCR with pairs of complementary primers containing the nucleotide substitution of interest (Table 1). The gyrB-M cassettes with mutated bases were amplified from the WT gyrB gene cassette (see Fig. S1C in the supplemental material) and ligated into the TA cloning plasmid. Recombinant plasmids were recovered from the colonies, and a nucleotide substitution in QRDRs in the 294-base PCR products was confirmed. The gyrB-M cassettes were digested with SacI and HindIII, ligated into pTB-B digested with same restriction endonucleases, and transformed into E. coli TOP-10 to obtain GyrB-E540V expression plasmids. Recombinant clones were selected from the resistant colonies on LB agar plates containing ampicillin (100 μg/ml).
Table 1.
Oligonucleotide sequences of primers used in PCR
| Primer | Sequence (nucleotide position), underlined element(s) | Comment |
|---|---|---|
| ON-873 | 5′-CCCATATGACAGACACGACGTTGCCGCC-3′ (1–23), NdeI site | WT gyrA |
| ON-874 | 5′-GTTAACCGGGCTTCGGTGTACCTCATCG-3′ (377–404), HpaI site | WT gyrA |
| ON-875 | 5′-GGTTAACCCCGTTGGCGATGGAGATGC-3′ (398–424), HpaI site | WT gyrA |
| ON-876 | 5′-GGCTCGAGTTAATGATGATGATGATGATGATTGCCCGTCTGGTCTGCGCCG-3′ (2493–2517), XhoI site and 6-histidine tag, respectively | WT gyrA |
| ON-35 | 5′-CCCCCCCATATGGGTAAAAACGAGGCCAGAAG-3′ (1–23), NdeI site | WT gyrB |
| ON-882 | 5′-CACGAGCTCTCGTGCCTTACGTGCCGCGATACG-3′ (1366–1398), SacI site | WT gyrB |
| ON-883 | 5′-GAGAGCTCGTGCGGCGTAAGAGCGCCACCG-3′ (1388–1417) | WT gyrB |
| ON-884 | 5′-CGATCTTGTGGTAGCGAAGCTTGCCGATATCGA-3′ (1667–1699), HindIII site | WT gyrB |
| ON-885 | 5′-CGATATCGGCAAGCTTCGCTACCACAAGATCG-3′ (1668–1699) | WT gyrB |
| ON-886 | 5′-GGCTCGAGTTAGACATCCAGGAACCGAACATCC-3′ (2130–2145), XhoI site | WT gyrB |
| ON-40 | 5′-AAAGAACACCGTAGTTCAGGCGA-3′ (1607–1630)a | Mutant gyrB |
| ON-41 | 5′-TCGCCTGAACTACGGTGTTCTTT-3′ (1607–1630)a | Mutant gyrB |
Mutated codon shown in bold type.
Sequencing of products.
PCR products were purified by agarose gel electrophoresis. The agarose blocks containing the bands of interest were sliced out of the agarose gel, frozen at −80°C for 30 min, and centrifuged at 20,400 × g at 4°C for 10 min to collect the supernatants. Supernatants having DNA concentrations between approximately 10 and 20 ng/μl were used directly as templates for cycle sequencing in both directions with corresponding primers (0.10 μmol) using the ABI Prism BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA). The sequencing reactions were performed according to the manufacturer's instructions. Cycle sequencing products were subsequently analyzed on an ABI PRISM 3130x automated genetic analyzer (Applied Biosystems). The sequences generated with the program were compared to their respective WT and GyrB-E540V sequences using BioEdit software.
Recombinant expression and purification of DNA gyrase.
DNA gyrase subunits were purified as previously described, with the following modifications (1, 2). Expression vectors carrying the gyrA and gyrB genes of M. tuberculosis were transformed into E. coli Rosetta-gami 2 and BL21(DE3)/pLysS, respectively. Expression of GyrA and GyrB was induced with the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Wako Pure Chemicals Ltd.), followed by further incubation at 14°C for 20 h or at 23°C for 5 h, respectively. Combined elution fractions resulting from the addition of elution buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 250 mM imidazole) to nickel-nitrilotriacetic acid agarose resin (Invitrogen) were dialyzed twice overnight at 4°C against 1 liter of gyrase dilution buffer (50 mM Tris-HCl [pH 7.5], 100 mM KCl, 2 mM dithiothreitol, 1 mM EDTA). After dialysis, the eluates were added to glycerol to yield 50% (wt/vol) and stored at −80°C until use. The protein fractions were examined by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis.
DNA supercoiling assay and inhibition by FQs.
ATP-dependent DNA supercoiling and quinolone-inhibited supercoiling assays were carried out as previously described (1, 2, 25). DNA supercoiling activity was tested with a combination of purified M. tuberculosis GyrA and GyrB proteins. The reaction mixture (total volume, 30 μl) consisted of DNA gyrase assay buffer, relaxed pBR322 DNA (0.3 μg), and WT and GyrB-E540V gyrase proteins (3 μM). Reactions were run at 37°C for 1 h and stopped by the addition of 30 μl of chloroform-isoamyl alcohol (24:1 mixture) and 6 μl of 5× stop and loading solution. The total reaction mixtures were subjected to electrophoresis in 1% agarose gels in 0.5× Tris-borate-EDTA (TBE) buffer. The gels were run for 1 h at 50 mA and stained with ethidium bromide (0.7 μg/ml). E. coli DNA gyrase (John Innes Enterprises Ltd.) was used as a positive control for the assay procedures and buffer. The inhibitory effects of quinolones on DNA gyrase were assessed by determining the drug concentrations required to inhibit the supercoiling activity of the enzyme by 50% (IC50s) in the presence or absence of serial 2-fold increases in the concentrations of the eight FQs. Supercoiling activity was assessed by tracing the brightness of the bands corresponding to the supercoiled pBR322 DNA with the Molecular Analyst software ImageJ (http://rsbweb.nih.gov/ij). To allow direct comparison, all incubations with WT and GyrB-E540V enzymes were carried out and processed in parallel on the same day under identical conditions. All enzyme assays were performed at least three times to confirm reproducibility.
Quinolone-mediated DNA cleavage assay.
DNA cleavage assays were carried out as previously described (1, 2, 25). Supercoiled, rather than relaxed, pBR322 DNA was used as the substrate for cleavage assays. The reaction mixture (total volume, 30 μl) contained DNA gyrase assay buffer, purified DNA gyrase subunits, supercoiled pBR322 DNA (0.3 μg), and increasing concentrations of GAT, LVX, CIP, MXF, SPX, or SIX. After incubation for 1 h at 37°C, 3 μl of 2% SDS and 3 μl of proteinase K (1 mg/ml) were added to the reaction mixture. After additional incubation for 30 min at 37°C, reactions were stopped to allow relaxation activity by the addition of 3 μl of 0.5 mM EDTA, 30 μl of chloroform-isoamyl alcohol (24:1 mixture), and 3 μl of 10× DNA loading solution. Plasmid pBR322 linearized by BamHI digestion was used as a marker for cleaved DNA. The total reaction mixtures were subjected to electrophoresis in 0.8% agarose gels in 0.5× TBE buffer. The gels were run for 1.5 h at 50 mA, stained with ethidium bromide (0.7 μg/ml), and photographed under UV transillumination. The extent of DNA cleavage was quantified with the Molecular Analyst software ImageJ (http://rsbweb.nih.gov/ij). The quinolone concentrations required to induce 25% of the maximum DNA cleavage (CC25s) were determined for the eight FQs.
RESULTS
Expression and purification of recombinant gyrase A and B proteins.
The WT gyrA and gyrB genes were amplified from M. tuberculosis H37Rv (1, 2, 34). The full-length gyrA and gyrB genes were inserted downstream of the T7 promoter in expression vectors pET-20b (+) and pET19b, respectively, for expression as His-tagged recombinant proteins since the His tag has been previously shown not to interfere with the catalytic functions of GyrA and GyrB (14). Resulting plasmids pTB-A (gyrA in pET-20b) and pTB-B (gyrB in pET-19b) were used to transform E. coli Rosetta-gami 2 (DE3)/pLysS and BL21(DE3)/pLysS, respectively. Expression of the WT gyrA and gyrB genes in E. coli strains by induction with IPTG and subsequent purification of the corresponding proteins by Ni-nitrilotriacetic acid affinity purification resulted in 2 and 5 mg of soluble His-tagged 93-kDa and 79-kDa proteins from 200-ml cultures, respectively. We used mutagenesis of WT gyrB to introduce the desired amino acid substitution and purified the corresponding GyrB-E540V protein by the same procedure as that used for WT GyrB. All recombinant subunits were obtained at high purity (>95%) in milligram amounts (see Fig. S2 in the supplemental material) and free of contaminating E. coli topoisomerase activity, as assessed by the lack of supercoiling activity of either GyrA or GyrB alone (Fig. 2, lanes 4 and 5).
Fig. 2.

Recombinant WT GyrA and GyrB subunits of M. tuberculosis generate an ATP-dependent DNA supercoiling activity. Relaxed pBR322 DNA (0.3 μg) was incubated with WT DNA gyrase reconstituted from GyrA (3 μM) and GyrB (3 μM) in the presence and absence of 1 mM ATP. The reactions were stopped, and the DNA products were separated by electrophoresis in 1% agarose gels. DNA was stained with ethidium bromide and photographed under UV illumination. Lane 1, relaxed pBR322 DNA; lane 2, relaxed pBR322 DNA and E. coli DNA gyrase; lane 3, relaxed pBR322 DNA and both recombinant GyrA and GyrB proteins; lane 4, relaxed pBR322 DNA and only GyrA protein; lane 5, relaxed pBR322 DNA and only GyrB protein; lane 6, absence of ATP. R and SC, relaxed and supercoiled pBR322 DNA, respectively.
DNA supercoiling activity of WT and GyrB-E540V DNA gyrases.
Combinations of the WT GyrA and GyrB subunits were examined for DNA supercoiling activity with relaxed pBR322 DNA as the substrate in the presence and absence of ATP (Fig. 2). A combination of GyrA and GyrB at 3 μM each was sufficient for the conversion of 100% of 0.3 μg of relaxed plasmid pBR322 DNA to its supercoiled form and was used for all DNA supercoiling experiments. Since the combination of the GyrA and GyrB subunits at 3 μM led to plasmid supercoiling in the presence of ATP, reconstituted DNA gyrase was considered functional (Fig. 2, lane 3). Neither subunit alone exhibited DNA supercoiling activity in the presence of 1 mM ATP (Fig. 2, lanes 4 and 5), and no supercoiling activity was observed when ATP was absent from the reaction mixture (Fig. 2, lane 6), indicating that both subunits and ATP were essential for DNA supercoiling activity. The activity of a GyrB-E540V enzyme (designated GyrB-E540V) was also determined by DNA supercoiling assay in the presence of complementary WT GyrA (see Fig. S3 in the supplemental material).
Determination of IC50s of quinolones.
The inhibitory effects of quinolones on the WT and GyrB-E540V enzymes were elucidated by quinolone-inhibited DNA supercoiling assay. A set of representative data showing the inhibitory effect of CIP is shown in Fig. 3, and data for the other FQs are presented in Fig. S4 in the supplemental material. Each of the quinolones showed dose-dependent inhibition of the WT and GyrB-E540V enzymes. Inhibitory effects of quinolones against recombinant gyrases are presented as IC50s ordered from low to high in Table 2. The gyrase bearing the E540V amino acid substitution in GyrB was highly resistant to inhibition by quinolones (Fig. 3; Table 2; see Fig. S4 in the supplemental material). The IC50 of SIX was 10 μg/ml, those of GAT, LVX, MXF, and SPX were 37 to 82 μg/ml, and those of CIP, NOR, and ENX were 251, 274, and >320 μg/ml, respectively.
Fig. 3.

Inhibitory activities of CIP on the supercoiling activities of WT and GyrB-E540V M. tuberculosis DNA gyrases. Relaxed pBR322 DNA (0.3 μg) was incubated with WT (A) or GyrB-E540V (B) DNA gyrase in the presence of the concentrations of CIP indicated. The reactions were stopped, and the DNA products were analyzed by electrophoresis in 1% agarose gels. R and SC denote relaxed and supercoiled pBR322 DNA, respectively.
Table 2.
IC50s and CC25 of FQs against WT and mutant DNA gyrasesa

The structures of the compounds used (A, basic quinolone; B, CIP; C, MXF; D, SIX) and the locations of the substituents are shown at the top.
NO, not observed.
Quinolone-mediated DNA cleavage complex formation by WT and GyrB-E540V DNA gyrase.
To examine the effects of FQs on cleavage complex formation by WT and GyrB-E540V DNA gyrase, cleavage assays were performed in which supercoiled pBR322 was incubated with WT or GyrB-E540V DNA gyrase in the presence or absence of increasing concentrations of quinolones. Figure 4 shows the results of a representative cleavage assay using CIP. Table 2 presents the CC25s of the other FQs. The CC25s of FQs for WT gyrase ranged from 2 to 25 μg/ml, while those for the GyrB-E540V enzyme ranged from 3 to 317 μg/ml (Table 2).
Fig. 4.

CIP-mediated DNA cleavage complex by WT and GyrB-E540V gyrases of M. tuberculosis. Supercoiled pBR322 DNA (0.3 μg) was incubated with WT (A) or GyrB-E540V (B) DNA gyrase in the presence of the concentrations of CIP indicated. After addition of SDS and protease K, the reactions were stopped and the mixture samples were analyzed by electrophoresis in 0.8% agarose gels. R, L, and SC denote relaxed, BamHI-linearized, and supercoiled pBR322 DNA, respectively.
DISCUSSION
In light of the increased demand for a new treatment regimen for MDR TB, FQs have started to be used as anti-TB agents (13, 20). Although the main target of FQs is known to be bacterial DNA gyrase, the molecular details of quinolone-gyrase interactions are not yet fully understood. In this study, we examined the E540V amino acid substitution we recently found in a clinical isolate from Bangladesh (unpublished data). The same amino acid substitution has also been reported in a clinical isolate from Vietnam (12). Even though the E540 residue of GyrB, equivalent to the E466 residue in E. coli, has been suggested to be located in the QRDR in M. tuberculosis by X-ray crystallography (28), there was no experimental confirmation of this. We examined the effect of the amino acid substitution on FQ resistance at the molecular level by using purified recombinant gyrase subunits. Supercoiling and cleavage assays in the presence of several FQs demonstrated the significant contribution of the E540V amino acid substitution to quinolone resistance (Fig. 2 and 3; Table 2; see Fig. S3 and S4 in the supplemental material). These results support the model proposed by Piton et al. (28).
The structure-activity relationship between FQs and WT and GyrB-E540V gyrases were analyzed. All eight FQs studied have a substituent, pyrrolidine, piperazine, or azabicyclo, at R-7 which has been suggested to be associated with E540 on GyrB (28). NOR and ENX, with an ethyl residue at R-1, have high IC50s (102 and 84 μg/ml, respectively), whereas the other FQs, with a cyclopropyl at R-1 or an N1-C8 bridge, have significantly lower IC50s (4 to 22 μg/ml) for WT gyrase, suggesting that a cyclopropyl at R-1 or an N1-C8 bridge contributes to FQ activity. Although higher IC50s of all FQs were observed for the GyrB-E540V enzyme than for the WT enzyme, the difference in the two IC50s was lowest for SIX. In particular, this FQ has a fluorinated cyclopropyl ring at R-1 while four other FQs, GAT, SPX, MXF, and CIP, have a cyclopropyl. In contrast, the difference between the CIP IC50s for the E540V and WT enzymes was significantly high. The only apparent difference between CIP and the other effective FQs was the absence of a substituent at R-8 (Table 2). We have also attempted to elucidate the effects of the E540V amino acid substitution in GyrB using DNA cleavage assays (Fig. 4 and Table 2) to confirm the results obtained by quinolone-inhibited supercoiling assays.
Amino acid residues in GyrB located close to E540 (E501 in reference 28), including N538 (N499) and T539 (T500) on the α2 helix and R482 (R521) on β2, have been proposed to interact with the GyrA subunit and DNA strands to form the QBP (28). It has been suggested that the β1-α1 loop (residues 498 to 501 in GyrB of M. tuberculosis) interacts with the R-1 group, the β2-DBL (DNA-binding loop, residues 519 to 525) interacts with the R-7 and R-8 groups, and the beginning of α2 (residues 537 to 541) interacts with the R-7 group (28). A substitution of the glutamic acid at position 540 with a valine may therefore lead to a conformational change in QBP geometry in GyrB (28).
Based on the crystal structure and current data obtained with several FQs (5, 16, 28), we hypothesize the mechanism of FQ resistance conferred by the E540V amino acid substitution on GyrB as shown in Fig. 5. The hydrogen bonding network involving E540 could play a pivotal role in the recognition of quinolones by the GyrB subunit on QBP. One of the FQs, CIP, exerts potent inhibitory activity against WT DNA gyrase (IC50 = 7 μg/ml) and may bind tightly to GyrB through two hydrogen bonds (O-H—N and O−—H-N) involving a hydroxyl group (OH) of the T539 residue and a carboxylate (CO2−) of the E540 residue (Fig. 5A, left; Table 2). In contrast, the efficacy of CIP against the GyrB-E540V gyrase is drastically lower (IC50 = 251 μg/ml). The E540V amino acid substitution replaces glutamic acid, which bears a carboxyl group, with valine, which is nonpolar and sterically demanding. The subsequent loss of the hydrogen bonding interactions with E540 could induce a substantial conformational change, which would disrupt binding with CIP (Fig. 5A, right). The efficacy of other FQs carrying a substituent at R-8 against the E540V enzyme was shown to be higher than that of CIP (Table 2). For example, MXF showed inhibitory activities against WT GyrB and E540V GyrB, with IC50s of 16 and 61 μg/ml, respectively. MXF and CIP vary structurally with regard to the substituents at R-7 and R-8: MXF has a bulky azabicyclo group at R-7 and a methoxy group at R-8, whereas CIP has a simple piperazine group at R-7 and no substituent at R-8 (Table 2). In the binding of MXF with WT GyrB (Fig. 5B, left), the substituents at R-7 and R-8 may efficiently interact with residues of the GyrB subunit through three hydrogen bonding networks involving T539 (O-H—N), E540 (O−—H-N), and R521 (H-N-H—O-Me). With regard to the GyrB E540V gyrase (Fig. 5B, right), however, MXF could still retain a substantial affinity for the GyrB subunit through hydrogen bonding with the substituents at R-7 and R-8 (O-H—N and H-N-H—O-Me), whereas CIP could not retain such an affinity due to the absence of hydrogen bonding with the substituent at R-8 and would thus be more sensitive to the amino acid substitution. Apart from MXF and CIP, SIX carries a pyrrolidine at R-7 and a halide (chlorine) at R-8 and exhibits potent activities toward the WT (IC50 = 4 μg/ml) and GyrB-E540V (IC50 = 10 μg/ml) gyrase enzymes. As shown in Fig. 5C, the substituents at R-7 and R-8 of SIX may interact with the residues of the WT GyrB subunit through three hydrogen bonds and van der Waals forces and/or halogen bonding (4) involving T539 (O-H—N), E540 (O−—H-N), N538 (O-O—H-N), and R521 (van der Waals radius zone between residue R521 and the R-8 halide group). It seems likely that interactions between R521 and the substituents at R-1 and R-8 are sufficient to make up for the loss of hydrogen bonding with the GyrB-E540V gyrase and thus play important roles in the binding of SIX.
Fig. 5.

Hypothetical models of interactions of WT and E540V GyrB with quinolones with substituents at R-7 and R-8. The models show the hydrogen bonding network relationship between residues of the WT and E540V gyrases and the R-7 and R-8 groups of CIP (A). Panels B and C show the relationships of MXF and SIX with the QBP of the DNA GyrB subunit of M. tuberculosis. The left and right panels show the WT and GyrB-E540V gyrase activities for hydrogen interaction with quinolones. Position 540 is indicated by bold type.
In summary, our study indicates that the E540V amino acid substitution in GyrB is involved in the resistance of M. tuberculosis against quinolones, although E540 is reported to be outside GyrB QRDRs. We thus propose that the E540 residue be included in the QRDR in M. tuberculosis as shown in Streptococcus pneumoniae (23, 26). Moreover, we also demonstrated an association between structural features of quinolones and activities against the WT and E540V forms of the GyrB DNA gyrase subunit of M. tuberculosis. The interaction between FQs and GyrB, which is composed of hydrogen bonding networks involving substituents of FQs and amino acid residues of QBP in GyrB, plays an important role in the inhibitory activity of FQs. Further studies investigating the contributions of other amino acid substitutions may also help gain a comprehensive understanding of the mechanism by which FQ-resistant TB emerges.
Supplementary Material
ACKNOWLEDGMENTS
We thank Haruka Suzuki, Yukari Fukushima, and Aiko Ohnuma for their technical support with several of the experiments and Yusuke Suzuki and Kyeong Hwa Bae for their helpful comments.
This work was supported by a grant from U.S.-Japan Cooperative Medical Science Programs, the Global Center of Excellence (COE) Program, Establishment of International Collaboration Centers for Zoonosis Control, Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan, in part by J-GRID, the Japan Initiative for Global Research Network on Infectious Diseases from MEXT to Y.S., and by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) to Y.S. and C.N.
Footnotes
Supplemental material for this article may be found at http://aac.asm.org/.
Published ahead of print on 6 June 2011.
REFERENCES
- 1. Aubry A., Pan X. S., Fisher L. M., Jarlier V., Cambau E. 2004. Mycobacterium tuberculosis DNA gyrase: interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 48:1281–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aubry A., et al. 2006. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: functional analysis of mutant enzymes. Antimicrob. Agents Chemother. 50:104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aziz M. A., et al. 2006. Epidemiology of antituberculosis drug resistance (the Global Project on Anti-tuberculosis Drug Resistance Surveillance): an updated analysis. Lancet 368:2142–2154 [DOI] [PubMed] [Google Scholar]
- 4. Barnard F. M., Maxwell A. 2001. Interaction between DNA gyrase and quinolones: effects of alanine mutations at GyrA subunit residues Ser83 and Asp87. Antimicrob. Agents Chemother. 45:1994–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bax B. D., et al. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–940 [DOI] [PubMed] [Google Scholar]
- 6. Blumberg H. M., et al. 2003. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167:603–662 [DOI] [PubMed] [Google Scholar]
- 7. Champoux J. J. 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70:369–413 [DOI] [PubMed] [Google Scholar]
- 8. Cole S. T., et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 9. Corbett K. D., Berger J. M. 2004. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 33:95–118 [DOI] [PubMed] [Google Scholar]
- 10. Drlica K. 1999. Mechanisms of fluoroquinolone action. Curr. Opin. Microbiol. 2:504–508 [DOI] [PubMed] [Google Scholar]
- 11. Drlica K., Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61:377–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duong D. A., et al. 2009. Beijing genotype of Mycobacterium tuberculosis is significantly associated with high-level fluoroquinolone resistance in Vietnam. Antimicrob. Agents Chemother. 53:4835–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fattorini L., et al. 1999. Activity of 16 antimicrobial agents against drug-resistant strains of Mycobacterium tuberculosis. Microb. Drug Resist. 5:265–270 [DOI] [PubMed] [Google Scholar]
- 14. Freydank A. C., Brandt W., Dräger B. 2008. Protein structure modeling indicates hexahistidine-tag interference with enzyme activity. Proteins 72:173–183 [DOI] [PubMed] [Google Scholar]
- 15. Frieden T. R., et al. 1993. The emergence of drug-resistant tuberculosis in New York City. N. Engl. J. Med. 328:521–526 [DOI] [PubMed] [Google Scholar]
- 16. Fu G. S., et al. 2009. Crystal structure of DNA gyrase B′ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 37:5908–5916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gillespie S. H., Kennedy N. 1998. Fluoroquinolones: a new treatment for tuberculosis? Int. J. Tuberc. Lung Dis. 2:265–271 [PubMed] [Google Scholar]
- 18. Gore J., et al. 2006. Mechanochemical analysis of DNA gyrase using rotor bead tracking. Nature 439:100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guillemin I., Jarlier V., Cambau E. 1998. Correlation between quinolone susceptibility patterns and sequences in the A and B subunits of DNA gyrase in mycobacteria. Antimicrob. Agents Chemother. 42:2084–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hatfull G. F., Jacobs W. R., Jr 2000. Molecular genetics of mycobacteria, p. 235–256 American Society for Microbiology, Washington, DC [Google Scholar]
- 21. Heeb S., et al. 2011. Quinolones: from antibiotics to autoinducers. FEMS Microbiol. Rev. 35:247–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Infectious Diseases Society of the Republic of China; Society of Tuberculosis, Taiwan; Medical Foundation in Memory of Deh-Lin Cheng; Foundation of Wei-Chuan Hsieh for Infectious Diseases Research and Education; C Y Lee's Research Foundation for Pediatric Infectious Diseases and Vaccines 2004. Guidelines for chemotherapy of tuberculosis in Taiwan. J. Microbiol. Immunol. Infect. 37:382–384 [PubMed] [Google Scholar]
- 23. Laponogov I., et al. 2010. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5:e11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levine C., Hiasa H., Marians K. J. 1998. DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1400:29–43 [DOI] [PubMed] [Google Scholar]
- 25. Pan X. S., Yague G., Fisher L. M. 2001. Quinolone resistance mutations in Streptococcus pneumoniae GyrA and ParC proteins: mechanistic insights into quinolone action from enzymatic analysis, intracellular levels, and phenotypes of wild-type and mutant proteins. Antimicrob. Agents Chemother. 45:3140–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan X. S., Gould K. A., Fisher L. M. 2009. Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob. Agents Chemother. 53:3822–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pitaksajjakul P., et al. 2005. Mutations in the gyrA and gyrB genes of fluoroquinolone-resistant Mycobacterium tuberculosis from TB patients in Thailand. Southeast Asian J. Trop. Med. Public Health 36(Suppl. 4):228–237 [PubMed] [Google Scholar]
- 28. Piton J., et al. 2010. Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS One 5:e12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reece R. J., Maxwell A. 1991. DNA gyrase: structure and function. Crit. Rev. Biochem. Mol. Biol. 26:335–375 [DOI] [PubMed] [Google Scholar]
- 30. Sepkowitz K. A., Telzak E. E., Recalde S., Armstrong D. 1994. Trend in the susceptibility of tuberculosis in New York City, 1987-1991. Clin. Infect. Dis. 18:755–759 [DOI] [PubMed] [Google Scholar]
- 31. Sharma S. K., Mohan A. 2004. Multidrug-resistant tuberculosis. Indian J. Med. Res. 120:354–376 [PubMed] [Google Scholar]
- 32. Sun Z., et al. 2008. Comparison of gyrA gene mutations between laboratory-selected ofloxacin-resistant Mycobacterium tuberculosis strains and clinical isolates. Int. J. Antimicrob. Agents 31:115–121 [DOI] [PubMed] [Google Scholar]
- 33. Takiff H. E., et al. 1994. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob. Agents Chemother. 38:773–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Veziris N., et al. 2007. Treatment failure in a case of extensively drug-resistant tuberculosis associated with selection of a GyrB mutant causing fluoroquinolone resistance. Eur. J. Clin. Microbiol. Infect. Dis. 26:423–425 [DOI] [PubMed] [Google Scholar]
- 35. World Health Organization 2010. Multidrug and extensively drug-resistant TB (M/XDR-TB). Global report on surveillance and response. Document WHO/HTM/TB/2010.3. World Health Organization, Geneva, Switzerland [Google Scholar]
- 36. World Health Organization 2010. Global tuberculosis control: WHO report. Document WHO/HTM /TB/2010.7. World Health Organization, Geneva, Switzerland [Google Scholar]
- 37. World Health Organization 2008. Guidelines for the programmatic management of drug-resistant tuberculosis. WHO/HTM/TB/2008.402. World Health Organization, Geneva, Switzerland [Google Scholar]
- 38. World Health Organization 2008. Anti-tuberculosis drug resistance in the world fourth global report. The WHO/IUATLD Global Project on Anti-tuberculosis Drug Resistance Surveillance 2002-2007 WHO/HTM/TB/2008.394. World Health Organization, Geneva, Switzerland [Google Scholar]
- 39. Zhou J., et al. 2000. Selection of antibiotic-resistant bacterial mutants: allelic diversity among fluoroquinolone-resistant mutations. J. Infect. Dis. 182:517–525 [DOI] [PubMed] [Google Scholar]
- 40. Zignol M., et al. 2006. Global incidence of multidrug-resistant tuberculosis. J. Infect. Dis. 194:479–485 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.