

Abstract
Inherently unstable mRNAs contain AU-rich elements (AREs) in the 3′ untranslated regions. Expression of ARE-containing type I interferon transcripts is robustly induced upon viral infection and rapidly shut off thereafter. Their transient accumulation is partly mediated through posttranscriptional regulation. Here we show that mouse embryonic fibroblasts derived from knockout mice deficient in KH-type splicing regulatory protein (KSRP), an RNA-binding protein required for ARE-mediated mRNA decay, produce higher levels of Ifna and Ifnb mRNAs in response to viral infection as a result of decreased mRNA decay. Functional analysis showed that KSRP is required for the decay of Ifna4 and Ifnb mRNAs by interaction with AREs. The increased IFN expression renders Ksrp−/− cells refractory to herpes simplex virus type 1 and vesicular stomatitis virus infection. These findings support a role of a posttranscriptional mechanism in the control of type I IFN expression and highlight the function of KSRP in innate immunity by negatively regulating IFN production.
INTRODUCTION
Type I interferons (IFN-α and IFN-β) play critical roles in the innate immune response against viral infection (17, 45). It is known that expression of type I IFN genes is highly induced upon virus infection and rapidly shut off thereafter (26, 55). Although the expression is primarily controlled at the transcriptional level (25), posttranscriptional regulation exerted on mRNA stability is also suggested to contribute to the transient nature of type I IFN gene expression (26). The posttranscriptional control is mediated through the AU-rich element (ARE)-containing 3′ untranslated regions (3′-UTRs) that are responsible for rapid decay of IFN-α and IFN-β transcripts (26). While the role of transcriptional regulation of type I IFN gene expression in the innate immune response has been studied extensively (25), the importance of posttranscriptional regulation of gene expression against viral infection is unclear.
AREs play an important role in posttranscriptional regulation of gene expression by directing rapid mRNA decay through a process referred to as ARE-mediated mRNA decay (AMD) (3, 4, 14). A number of proteins, collectively named ARE-binding proteins (ARE-BPs), have been described to bind AREs and regulate AMD (6, 8). Decay-promoting ARE-BPs bind ARE-containing mRNAs and target them for decay. Currently, there are at least five RNA-binding proteins required for AMD. The zinc finger proteins tristetraprolin (TTP/ZFP36) and butyrate response factors 1 and 2 (BRF1/ZFP36L1 and BRF2/ZFP36L2) are potent stimulators of AMD. TTP binds the AREs of tumor necrosis factor alpha (TNF-α) as well as other cytokine mRNAs and promotes their deadenylation and decay (9, 27). BRF1 and BRF2 also promote decay of ARE-containing mRNAs (28, 46). In addition, AU-rich binding factor 1 (AUF1) modulates mRNA decay and either stabilizes or destabilizes ARE-containing mRNAs, depending on the experimental system used (30, 41, 43, 58). KSRP (KH-type splicing regulatory protein) is also required for decay of ARE-containing mRNAs (13, 18, 56).
Experimentally, the functions of the above-mentioned ARE-BPs have been largely analyzed by using heterologous reporter mRNAs consisting of various AREs under the condition of either overexpression or downregulation of a given ARE-BP in a transiently transfected cell line. To study the in vivo functions of ARE-BPs, mice deficient in some of these RNA-binding proteins have been generated. Ttp-deficient mice develop a complex syndrome of cachexia, arthritis, and autoimmunity due to overproduction of TNF-α resulting from decreased mRNA turnover (12, 51). TTP appears to play an important role in constitutively targeting TNF-α mRNA for decay. Auf1 knockout mice display symptoms of severe endotoxic shock resulting from overproduction of TNF-α and interleukin-1β (IL-1β), which is due to an inability to rapidly degrade these mRNAs in macrophages upon lipopolysaccharide (LPS) stimulation (31). Brf1 knockout embryos die in utero due to a failure of chorioallantoic fusion (48). It was postulated that lack of Brf1 expression during midgestation results in abnormal stabilization of mRNAs whose encoded proteins lead to abnormal placentation and fetal death. Brf2 knockout mice die within 2 weeks of birth, suffer from intestinal hemorrhage, and exhibit decreased hematopoietic progenitor cells from the fetal liver and yolk sac (47). Finally, mice with deletion of both Brf1 and Brf2 in lymphocytes develop a T cell acute lymphoblastic leukemia, likely due to overexpression of Notch1 (24). Altogether, these findings strongly suggest that tight in vivo control of ARE-mediated mRNA decay by decay-promoting ARE-BPs is critical for normal mouse development and/or for proper cell growth as well as cellular processes.
By using RNA interference (RNAi) depletion in transient-transfection experiments, KSRP was shown to be required for decay of reporter mRNAs containing various AREs from c-fos, TNF-α, and IL-8 (18, 56) and several endogenous cytokine mRNAs and other ARE-containing mRNAs in established cell lines (10, 11, 19, 56). No true physiological KSRP mRNA targets have been identified previously by using cells or tissues derived from Ksrp-deficient mice. To assess the in vivo function of KSRP in AMD and identify its physiological mRNA targets, we generated Ksrp-null mice. Although Ksrp−/− mice are viable and do not exhibit obvious abnormalities, type I IFN gene expression is upregulated in Ksrp−/− cells and in Ksrp−/− mice in response to viral infection. The increased expression of type I IFNs contributes to resistance to herpes simplex virus 1 (HSV-1) and vesicular stomatitis virus (VSV) infection. Biochemical analysis demonstrated that KSRP interacts with Ifna4 and Ifnb transcripts through the ARE-containing 3′-UTRs that are required for their rapid decay. Our data provide genetic and biochemical evidence that KSRP is a critical factor for the posttranscriptional control of type I IFN gene expression via mRNA decay.
MATERIALS AND METHODS
Generation of Ksrp-null mice.
A hygromycin cassette flanked by loxP sites was designed to replace exon 1 to exon 13 of the Ksrp gene. The Ksrp-targeting vector containing genomic fragments upstream of exon 1 (3.7 kb) and downstream of exon 13 (8.0 kb) that were used as short and long homologous arms, respectively, was linearized by restriction enzyme digestion and electroporated into V6.5 mouse embryonic stem (ES) cells. The cells were subsequently selected with hygromycin (250 μg/ml) and ganciclovir (2 μM). Resistant ES colonies were genotyped by Southern blot analysis, and the Ksrp-targeted ES clones were introduced with Cre recombinase to remove the hygromycin cassette; successful removal was confirmed by Southern blotting and PCR analysis. The ES clones were injected into C56BL/6 blastocysts to generate chimeric mice. Male chimeras were further backcrossed with C56BL/6 females for 8 generations and used for this study. All animal studies were conducted in accordance with guidelines for animal use and care established by the University of Alabama at Birmingham Animal Resource Program and the Institutional Animal Care and Use Committee.
Plasmids.
Construction of pTRE-GB was previously described (15). The 3′-UTRs of Ifna4 and Ifnb and subregions of the Ifna4 3′-UTR were PCR amplified and subcloned into the BglII and PvuII sites of pTRE-GB. Primer sequences for PCR amplification are available on request. A fragment encoding FLAG-tagged KSRP was obtained by digesting pcDNA3-FLAG-KSRP (29) with HindIII and XhoI and subcloned into pMSCV-neo (Clontech), a retroviral vector.
Isolation and immortalization of MEFs.
To generate mouse embryonic fibroblasts (MEFs), Ksrp+/− × Ksrp+/− mice were bred. Embryos were recovered on embryonic day 14.5 and subjected to genotyping. MEFs were prepared from the embryos as described previously (57) and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 1% penicillin-streptomycin, 1% l-glutamine, and 110 μM beta-mercaptoethanol. Immortalized Ksrp+/+ and Ksrp−/− MEF lines were generated by serial passage by using a protocol described previously (57) and used for the studies.
Isolation and preparation of BMDCs.
Mice (2 to 3 months old) were euthanized, and femur-derived bone marrow was used for preparation of bone marrow-derived dendritic cells (BMDCs) as previously described (34). A 2% granulocyte-macrophage colony-stimulating factor (GM-CSF) supernatant was included in the culture medium to support the differentiation and growth of BMDCs.
Poly(I·C) treatment.
Two micrograms of poly(I·C) (GE Healthcare) was mixed with 3 μl of Lipofectamine (Invitrogen). The complex was applied to cells (2 μg/ml) for 3 h. Cells were then washed twice with phosphate-buffered saline (PBS) and incubated with culture medium.
mRNA analysis.
Total RNA was extracted by using TRIzol (Invitrogen) according to the manufacturer's protocol. Poly(A)+ mRNA was isolated by using Oligotex (Qiagen) and subjected to Northern blot analysis as described previously (15). A 32P-labeled DNA probe was generated from the mouse Ksrp cDNA, composed of the C-terminal region downstream of the four KH motifs, by a random priming reaction. 32P-labeled RNA probes were generated by in vitro transcription. For real-time quantitative reverse transcription-PCR (qRT-PCR) analysis, total RNA (1 μg) was treated with DNase I and reverse transcribed using a mixture of oligo(dT) and random hexamer, and amplification was performed by using a Roche LC480 LightCycler and the SYBR green system (Roche). The levels of Ifna4 and Ifnb mRNAs were quantitated and normalized to the level of 18S rRNA.
Transfection and mRNA decay analysis.
MEFs and NIH 3T3-B2A2 cells (59) were transfected with globin mRNA reporters, and mRNA decay was analyzed by Northern blotting using a transcriptional pulse-chase assay as previously described (15, 29). For analysis of the stability of endogenous type I IFN mRNAs, wild-type and Ksrp−/− MEFs were treated with poly(I:C) for 6 or 12 h, and RNA was isolated at different time points after the addition of actinomycin D (5 μg/ml). The levels of Ifna and Ifnb mRNAs were analyzed by Northern blotting using RNA probes corresponding to the coding region of Ifna4, which also recognizes other Ifna subtypes, or to the coding region of Ifnb.
RNA-binding assays.
32P-labeled RNA (0.1 ng; 1 × 105 cpm) was incubated with cytoplasmic extract in a 20-μl reaction mixture containing 10 mM HEPES (pH 7.6), 50 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol (DTT), 5% glycerol, 0.1% NP-40, yeast RNA (1 μg), and heparin (5 mg/ml) at room temperature for 20 min. Next, RNase T1 (50 U; Ambion) was added and incubated for 10 min at room temperature. RNA-protein complexes were subjected to a UV cross-linking reaction and SDS-PAGE as previously described (18). UV cross-linking assays followed by immunoprecipitation were conducted as previously described (18).
RNP immunoprecipitation and RT-PCR analysis.
Poly(I:C)-stimulated cells were fixed with 1% formaldehyde in PBS for 10 min at room temperature, neutralized by 0.25 M glycine (pH 7.0) for another 5 min, and disrupted by radioimmunoprecipitation assay (RIPA) buffer followed by brief sonication. Soluble supernatants were incubated with protein A/G beads conjugated with polyclonal antibodies against KSRP or preimmune serum for 1.5 h at room temperature. The protein A/G beads were washed with high-stringency buffer (RIPA buffer containing 1 M urea) 6 times. The cross-linked RNA-protein complexes in the immunoprecipitates were reversed by incubation with a reducing buffer (50 mM Tris-Cl [pH 7.4], 5 mM EDTA, 10 mM DTT, 1% SDS) at 70°C for 45 min. RNA was extracted by using TRIzol and reverse transcribed to cDNA. Ifna4 and Ifnb transcripts were detected by PCR with specific primers corresponding to the 3′-UTRs.
Immunoblot analysis.
Proteins were extracted by using RIPA buffer (1% NP-40, 0.5% deoxycholate, 0.1% SDS in PBS [pH 7.4]) containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1% protease inhibitor cocktail (Sigma) and subjected to SDS-PAGE and immunoblot analysis. Rabbit polyclonal anti-KSRP serum and a mouse monoclonal anti-KSRP antibody were previously described (18, 21).
Virus infection, ELISA, and plaque assay.
MEFs were infected with either HSV-1(F) strain or VSV Indiana strain. Briefly, cells (1.5 × 105 cells/well) were seeded in 24-well plates, incubated with HSV-1 for 2 h in DMEM plus 1% FBS or with VSV for 1 h in DMEM plus 2% FBS using various multiplicities of infection (MOIs), washed with PBS, and refreshed with growth medium. Infected cells were collected for RNA analysis, and culture medium was collected for enzyme-linked immunosorbent assay (ELISA) and virus titer analysis. ELISA plates to measure mouse IFN-α and IFN-β were purchased from PBL Biomedical Laboratories. Virus titers were determined by plaque assays using Vero cells (for HSV-1) or HeLa cells (for VSV) as described previously (22, 33). BMDCs were seeded in 24-well plates (5 × 105 cells/well) and infected with HSV-1 (MOI, 3). After infection, supernatants and cells were collected at the indicated time points for ELISA and RNA analyses, respectively.
KSRP rescue assay.
A retroviral expression vector (pMSCVneo) expressing FLAG-tagged human KSRP or a vector expressing green fluorescent protein (GFP) was packed in 293T packaging cells. Virus-containing medium was collected and filtered 48 h after transfection. The viral supernatants were then used to transduce MEFs in the presence of Polybrene (8 μg/ml; Sigma-Aldrich). The infected cells were selected with G418 (400 μg/ml) for 7 days and infected with VSV or HSV-1.
Downregulation of Ifnb by short hairpin RNA.
Four distinct premade lentiviral vectors expressing short hairpin RNAs (shRNA) targeting mouse Ifnb and a control construct targeting GFP were purchased from Open Biosystems. Lentiviruses were generated in 293T cells from a pool of the four lentiviral vectors and used to infect Ksrp−/− MEFs. Two days after infection, transduced cells were selected with puromycin (2 μg/ml) for 2 weeks and then infected with VSV.
Animal study.
Four-week-old male mice were used for the viral study. Mice were injected intracranially with HSV-1(F) (300 PFU/per mouse), and the survival rate was monitored daily for 28 days as previously described (38, 39). Infected brains were harvested at day 3 for ELISA analysis and at day 6 for plaque assays.
Statistical analysis.
For statistical analysis of survival of HSV-1-infected mice, plots of Kaplan-Meier survival estimates were constructed. The significance of the survival difference between wild-type and Ksrp knockout mice was assessed by log-rank (Mantel-Cox) pairwise comparison. All other data are presented as the means and standard errors of three independent experiments. Statistical significance (P value) was calculated by using Student's t test within Microsoft Excel software.
RESULTS
Generation of Ksrp knockout mice.
To study the in vivo function of KSRP, we generated Ksrp knockout (Ksrp−/−) mice by homologous recombination in mouse ES cells. A selection cassette containing the hygromycin resistance gene (HygR) flanked by 34-bp Cre recombinase recognition sites (loxP) was used to replace exon 1 to exon 13 of the Ksrp gene (Fig. 1 A). The inserted HygR cassette was further removed by transient expression of Cre recombinase in Ksrp gene-targeted ES cells. The targeted Ksrp allele was identified by Southern blotting and PCR analysis, using genomic DNA extracted from ES cells (data not shown) and F2 mouse offspring (Fig. 1B and C). Full-length Ksrp transcripts were not detected in Ksrp−/− liver by Northern blot analysis (Fig. 1D). The protein product was undetectable in Ksrp−/− tissues when we used either a monoclonal anti-KSRP antibody or polyclonal antiserum against the full-length protein (Fig. 1E and data not shown), suggesting a successful disruption of the Ksrp allele. Ksrp−/− offspring showed no distinguishable gross phenotypes compared with their wild-type littermates. Major organs were examined by histological examination but showed no obvious abnormalities in adult Ksrp−/− mice (data not shown). Breeding of Ksrp+/− mice generated Ksrp−/− offspring with the expected Mendelian ratio (+/+, 26.6%; −/−, 24.3%; +/−, 49.1%; n = 519). These results suggest that disruption of the Ksrp gene neither causes embryonic lethality nor interferes with mouse development.
Fig. 1.

Generation of Ksrp knockout mice. (A) The wild-type Ksrp allele, the targeting vector, the targeted allele, and the genotyping methods are described. A hygromycin selection cassette with loxP sites flanked at both ends was designed to replace exon 1 to exon 13 of the mouse Ksrp allele. The hygromycin cassette in the targeted Ksrp allele was further excised by expression of Cre recombinase in ES cells. EcoRV sites, a 3′ external probe, and PCR primers used for genotyping are indicated. (B) Southern blot analysis shows that the Ksrp allele is targeted. Genomic DNA from the second generation of mouse offspring was digested with EcoRV and hybridized with 32P-labeled 3′ external probe. (C) Genotyping by PCR analysis. Genomic DNA was analyzed by PCR using P1, P2, and P3 primers to specifically amplify the wild-type (300-bp) and the targeted (600-bp) Ksrp alleles. (D) Northern blot analysis shows undetectable Ksrp mRNA expression in Ksrp-null mice. Poly(A)+ mRNA was isolated from wild-type and Ksrp−/− livers and analyzed by using a 32P-labeled Ksrp cDNA probe. The blot was reprobed with a Gapdh probe. (E) Immunoblot analysis shows no detection of KSRP expression. Total protein was extracted from wild-type and Ksrp−/− livers and subjected to immunoblotting using a monoclonal anti-KSRP antibody. The same blot was rehybridized with antibodies against α-tubulin.
Type I interferon gene expression is upregulated in Ksrp−/− cells as the result of increased mRNA stability.
In a parallel analysis to identify potential mRNA targets, KSRP expression was reduced by RNA interference in HeLa cells, and expression of mRNAs was analyzed by microarray analysis. Expression of several IFN-stimulating genes (ISGs), including IFIT1, IFIT2, IFI44, and G1P2, was increased upon KSRP downregulation, and their upregulation upon KSRP knockdown was confirmed in another human cell line, HT-1080 fibrosarcoma cells (data available from authors upon request). The increase of these ISGs in KSRP-depleted cells by RNAi was not caused by the alteration of mRNA stability, as mRNA half-life analysis failed to show change in mRNA decay rate (data available on request). These data suggest that these ISGs are not direct targets of KSRP, i.e., they are not targeted for decay by interaction with KSRP, and that it is likely that the expression of genes in the IFN signaling pathway is upregulated in response to KSRP downregulation. Interestingly, previous reports have shown that type I IFN genes contain AREs in their 3′-UTRs and AREs derived from human IFN-β direct rapid mRNA decay in a reporter assay (26, 40, 55). Thus, type I IFN genes are likely upregulated upon KSRP knockdown, leading to increased expression of these ISGs.
To examine whether expression of the type I IFN gene is increased upon KSRP downregulation, HT-1080 cells were transfected with a KSRP small interfering RNA (siRNA) and further treated with poly(I·C), a synthetic double-stranded RNA molecule that induces type I IFN expression. IFNB expression was further increased at early time points (6 h and 9 h) after poly(I·C) stimulation in cells with reduced KSRP expression, compared to the control cells (Fig. 2 A). Using MEFs isolated from Ksrp−/− mice, the levels of both Ifna and Ifnb mRNAs were also further increased at early time points (3 h and 6 h) in Ksrp−/− MEFs treated with poly(I·C) compared with Ksrp+/+ MEFs (Fig. 2B), but at 12 h their levels were similar. Consistent with the increased mRNA level, poly(I·C)-treated Ksrp−/− MEFs produced significantly more IFN-β and IFN-α proteins than did Ksrp+/+ MEFs (Fig. 2C and D). Similarly, the further increase in IFN gene expression was observed when we used another congenic line of Ksrp+/+ and Ksrp−/− MEFs (data not shown). To determine whether the increased expression is caused by altered mRNA stability, the decay of Ifna and Ifnb mRNAs was analyzed. Since the expression of type I IFNs returned to the basal level in both Ksrp+/+ and Ksrp−/− MEFs between 12 h and 24 h (Fig. 2B), we monitored their decay rates 6 h and 12 h after poly(I·C) stimulation. The half-lives of Ifna and Ifnb mRNAs were similarly prolonged (2- to 4-fold increase) in 6-h and 12-h poly(I·C)-treated Ksrp−/− MEFs compared to that in Ksrp+/+ MEFs (Fig. 2E), suggesting that the decrease of Ifna and Ifnb mRNA levels at later times (between 12 h and 24 h) is likely due to shutoff of IFN transcription as well as mRNA decay mediated by additional decay-promoting ARE-BPs (see Discussion). Similar stabilization of Ifna and Ifnb mRNAs was also observed with another congenic line of Ksrp+/+ and Ksrp−/− MEFs (data not shown). To further show that the increased Ifna and Ifnb mRNA stability was primarily due to a Ksrp deficiency, we examined the expression levels of several ARE-BPs and found no altered expression of TTP, AUF1, and HuR in Ksrp−/− MEFs (data not shown).
Fig. 2.

Increased type I IFN gene expression in Ksrp−/− MEFs. (A) Human fibrosarcoma HT-1080 cells were transfected with a control siRNA or a KSRP siRNA. At 48 h after transfection, cells were stimulated with poly(I·C) (10 μg/ml). Ifnb mRNA levels were analyzed by Northern blotting at different time points after poly(I·C) treatment. (B) Wild-type or Ksrp-deficient immortalized MEFs were treated with poly(I·C) (2 μg/ml). Total RNA was isolated at different time points and analyzed with probes against the coding region of Ifnb or Ifna4. The Ifna4 probe also recognizes other Ifna subtypes, due to their high homologies. (C and D) IFNB and IFNA levels in wild-type and Ksrp−/− MEFs were analyzed by ELISA 12 h and 24 h after poly(I·C) treatment. Data are mean ± standard errors of the means of three experiments (IFNB, P < 0.01 at 12 h and P < 0.001 at 24 h; IFNA, P < 0.001 at 24 h). (E) Elevated IFN gene expression in Ksrp−/− MEFs is due to increased mRNA stability. Wild-type or Ksrp-deficient MEFs were treated with poly(I·C) for 6 h or 12 h, and RNA was isolated at different time points after the addition of actinomycin D. mRNA levels of Ifna and Ifnb were analyzed by Northern blotting and quantitated with a phosphorimager. The calculated t1/2 values are shown as means ± standard deviations (n = 3).
We next examined Ifna and Ifnb expression in DCs, the major source of type I IFN in vivo (1, 2, 16). BMDCs derived from Ksrp+/+ and Ksrp−/− mice were infected with HSV-1. The expression of Ifna4, the first α-subtype expressed in response to viral infection (32), and Ifnb was analyzed by qRT-PCR. Figure 3 shows consistently higher Ifna4 and Ifnb mRNA expression in Ksrp−/− BMDCs after HSV-1 infection than in HSV-1-infected Ksrp+/+ BMDCs (Fig. 3A and B) and that higher IFNA and IFNB proteins were produced in Ksrp−/− BMDCs (Fig. 3C and D). Collectively, these data demonstrate that type I IFN genes are upregulated in Ksrp−/− MEFs and Ksrp−/− BMDCs due to decreased mRNA decay in the absence of KSRP.
Fig. 3.

Elevated IFN expression in primary Ksrp−/− BMDCs. (A and B) BMDCs derived from Ksrp+/+ and Ksrp−/− mice were infected with HSV-1 (MOI, 3), and total RNA was isolated at different time points. Quantitative RT-PCR was used to analyze the levels of Ifna4 (A) and Ifnb (B). Data are means ± standard errors of the means (n = 3; Ifna4, P < 0.01 at 3 h, P < 0.02 at 6 h; Ifnb, P < 0.02 at 6 h). (C and D) IFNA and IFNB levels in the supernatants of Ksrp+/+ and Ksrp−/− BMDCs were analyzed by ELISA 12 h after HSV-1 infection. Data are means ± standard errors of the means of three experiments (IFNA, P < 0.001 at 12 h; IFNB, P < 0.01 at 12 h).
KSRP promotes decay of Ifna4 and Ifnb mRNAs through interaction with their 3′-UTRs.
KSRP promotes mRNA decay through direct binding to the 3′-UTRs of its target mRNAs. We first examined whether KSRP interacts with Ifna4 and Ifnb mRNAs by RNP immunoprecipitation analysis. Poly(I:C)-stimulated Ksrp+/+ and Ksrp−/− MEFs were treated with formaldehyde to induce the formation of stable RNA-protein complexes. The RNA-KSRP complexes were immunoprecipitated with antiserum against KSRP. The presence of type I IFN transcripts in the immunoprecipitates was analyzed by RT-PCR. Both Ifna4 and Ifnb mRNAs were detected in the anti-KSRP immunoprecipitates from Ksrp+/+ MEFs, but not from Ksrp−/− MEFs (Fig. 4 A), suggesting that KSRP binds these mRNAs in vivo. RNA-binding and UV cross-linking assays showed that RNA-KSRP complexes, which immunoprecipitated with anti-KSRP (Fig. 4B and C, lane 4), were formed with the AREs of Ifna4 and Ifnb when we used extracts of Ksrp+/+ MEFs (Fig. 4B and C, lanes 1 and 5), but it was not detected when we used cell extracts from Ksrp−/− MEFs (Fig. 4B and C, lanes 2 and 6). Additional RNA-binding assays using recombinant protein showed that KSRP directly interacted with the regions composed of the AREs of Ifna4 and Ifnb (data available on request).
Fig. 4.

Regulation of Ifna4 and Ifnb mRNA stability by KSRP through interaction with the 3′-UTRs. (A) Association of Ifna4 and Ifnb transcripts with KSRP. Extracts of wild-type and Ksrp−/− MEFs treated with poly(I·C) were subjected to immunoprecipitation with preimmune serum or anti-KSRP serum. The coprecipitated RNA was isolated, and the presence of Ifna4 and Ifnb transcripts was analyzed by RT-PCR. The presence of Ifna4 and Ifnb transcripts in the input is also shown. The presence of β-actin and Gapdh mRNAs was also analyzed as controls for specificity. (B and C) KSRP interacts with the AREs of Ifnb and Ifna4. Wild-type or Ksrp−/− extracts were incubated with 32P-labeled RNAs composed of the AREs of Ifnb (B) or Ifna4 (C) (sequence available on request) and subjected to UV cross-linking. The RNA-protein complexes were either directly analyzed by SDS-PAGE (lanes 1, 2, 5, and 6) or subjected to immunoprecipitation with preimmune serum (lanes 3 and 7) or anti-KSRP serum (lanes 4 and 8) and then the immunoprecipitates were analyzed by SDS-PAGE and autoradiography. Note some variations of protein-binding levels to Ifna4 ARE in different experiments. (D and E) The 3′-UTRs of Ifna4 and Ifnb confer mRNA instability and regulation by KSRP. Wild-type and Ksrp−/− MEFs were cotransfected with a construct expressing β-globin mRNA reporter containing either the 3′-UTR of Ifna4, GB-Ifna4(I+II) (D) or the 3′-UTR of Ifnb, GB-Ifnb (E) under the control of a tetracycline-responsive promoter, a plasmid expressing a β-globin reporter consisting of the 3′-UTR of Gapdh under the control of the cytomegalovirus promoter (GB-Gapdh), and a plasmid expressing tetracycline-responsive transactivator (tTA). At 6 h after transcriptional induction, doxycycline (2 μg/ml) was added to block transcription. Total RNA was isolated at different time points. The levels of GB-Ifna4(I+II), GB-Ifnb, and GB-Gapdh mRNAs were analyzed by Northern blotting and quantitated. The calculated half-lives of GB-Ifna4(I+II) and GB-Ifnb mRNAs are shown as means ± standard deviations (n = 3). (F) The decay of a globin mRNA containing the non-ARE region of Ifna4, GB-Ifna4(I), was analyzed in Ksrp+/+ or Ksrp−/− MEFs as described for panel D. The calculated half-lives of GB-Ifna4(I) mRNA are shown as means ± standard deviations (n = 3).
We next examined whether the 3′-UTRs of Ifna4 and Ifnb can direct rapid mRNA decay by using heterologous β-globin reporters fused with the 3′-UTRs in a transcriptional pulse-chase assay. While ARE-containing 3′-UTRs of both Ifna4 and Ifnb promoted rapid mRNA decay in Ksrp+/+ MEFs, their decay was impaired in Ksrp−/− MEFs (Fig. 4D and E). The impaired mRNA decay in Ksrp−/− MEFs was specific for AMD, as the decay of a globin mRNA composed of the non-ARE region of Ifna4 (sequence availabe on request), GB-Ifna4(I), was not compromised in Ksrp-deficient cells (Fig. 4F). Using an NIH 3T3 cell line, we also found that decay of mRNA reporters consisting of the 3′-UTRs was impaired when KSRP was downregulated by RNAi and that the AREs of Ifna4 and Ifnb conferred regulation by KSRP (Fig. 5). These data demonstrate that KSRP binds the AREs of Ifna4 and Ifnb mRNAs and promotes their decay.
Fig. 5.

Decay of mRNA reporters consisting of the 3′-UTRs was impaired in KSRP-downregulated NIH 3T3 cells. (A) Immunoblot analysis showed downregulation of KSRP in NIH 3T3-B2A2 cells transfected with a KSRP siRNA. Different amounts of extracts from cells transfected with a control (chloramphenicol [CAT]) siRNA were loaded in lanes 1 to 4 to show the efficiency of knockdown. The levels of KSRP and HuR were analyzed by immunoblotting. (B) Schematic diagram showing the 3′-UTR of Ifna4, which is further divided into two subregions (I and II). Open circles indicate AUUUA motifs. (C and D) NIH 3T3-B2A2 cells, stably expressing a tetracycline-responsive transactivator, were transfected with constructs expressing globin reporters composed of the 3′-UTR of Ifnb (C) or different regions of the Ifna4 3′-UTR (D) under the control of a Tet regulatory promoter and a construct constitutively expressing GB-Gapdh mRNA under the control of the cytomegalovirus promoter. Total RNA was isolated at different time points after addition of doxycycline (Dox). The levels of reporter mRNAs were analyzed by Northern blotting. The calculated half-lives (t1/2) of reporter mRNAs are shown (n = 3).
Ksrp−/− MEFs are refractory to viral infection due to enhanced type I IFN expression.
We examined whether the enhanced expression of type I IFN in Ksrp−/− cells is able to repress viral replication. We first chose a double-stranded DNA virus, HSV-1, to examine the induced expression of IFN and viral susceptibility of Ksrp−/− MEFs. While the induction profiles were transient compared to that with poly(I·C) treatment (compared Fig. 6 A and B with 2B), the levels of Ifna4 and Ifnb mRNAs were significantly increased upon HSV-1 infection in Ksrp−/− MEFs compared with Ksrp+/+ MEFs (Fig. 6A and B), with higher production of IFN-β in Ksrp−/− MEFs (Fig. 6C). Importantly, HSV-1 replication was significantly decreased (10- to 50-fold reduction, depending on the MOI used) in Ksrp−/− MEFs compared with Ksrp+/+ MEFs (Fig. 6D). We next examined whether Ksrp−/− MEFs were also resistant to VSV infection, a single-stranded RNA virus. VSV replication was markedly reduced in Ksrp−/− MEFs compared to Ksrp+/+ MEFs (Fig. 6E).
Fig. 6.

Replication levels of HSV-1 and VSV are markedly reduced in Ksrp−/− MEFs. (A and B) Ksrp+/+ and Ksrp−/− MEFs were infected with HSV-1 (MOI, 3). The levels of Ifna4 (A) and Ifnb (B) were analyzed by qRT-PCR. Values represent means ± standard errors of the means (n = 3; Ifna4, P < 0.05 at 3 h and P < 0.001 at 6 h; Ifnb, P < 0.005 at 3 h and P < 0.001 at 6 h). (C) IFNB levels in culture supernatants of Ksrp+/+ and Ksrp−/− MEFs were analyzed by ELISA 24 h after HSV-1 infection. Data are means ± standard errors of the means (n = 3; P < 0.03). (D) Reduced HSV-1 replication in Ksrp−/− MEFs. Ksrp+/+ and Ksrp−/− MEFs were infected with HSV-1 using different MOIs (1, 0.1, or 0.01). At 72 h after infection, viral production was analyzed by plaque assay. Data are means ± standard errors of the means of three independent experiments (MOI, 1 [P < 0.0001]; MOI, 0.1 [P < 0.0002]; MOI, 0.01 [P < 0.001]). (E) Reduced VSV replication in Ksrp−/− MEFs. Ksrp+/+ and Ksrp−/− MEFs were infected with VSV at an MOI of 0.2. At 24 h after infection, viral production was analyzed by plaque assay. Data are means ± standard errors of the means of three experiments (P < 0.004).
To show that the reduction of viral replication is indeed due to Ksrp deficiency, we reexpressed KSRP in Ksrp−/− MEFs (Fig. 7 A) and analyzed viral production. While the expression levels of Ifna4 and Ifnb mRNAs were markedly increased in Ksrp−/− MEFs compared with Ksrp+/+ MEFs upon VSV infection, the expression was dramatically decreased (70% reduction) in Ksrp−/− MEFs expressing KSRP (Fig. 7B and C). More importantly, this resulted in a marked increase of VSV titer compared to that in the control Ksrp−/− MEFs (Fig. 7D). The lack of complete recovery of VSV replication to the level observed in Ksrp+/+ MEFs was likely due to the poor KSRP expression in Ksrp−/− MEFs (Fig. 7A). A direct comparison indicated that, while the reexpressed KSRP level in Ksrp−/− MEFs was much lower than that in Ksrp+/+ MEFs (Fig. 7A), its level was still significantly higher than the remaining KSRP level in HT 1080 cells treated with a KSRP siRNA in Fig. 2A (data not shown). Although there was a trend that reexpression of KSRP in Ksrp−/− MEFs restored HSV-1 replication compared to the control Ksrp−/− MEFs, the increase in HSV-1 titer did not reach a significant level (data not shown). The discrepancy of viral replication rescued by KSRP expression between VSV and HSV-1 is likely due to their differential sensitivities to type I IFN-mediated inhibition.
Fig. 7.

The viral resistance of Ksrp−/− MEFs is associated with a lack of KSRP. (A) Reexpression of KSRP in Ksrp−/− MEFs. Protein samples from control retrovirus (GFP)-transduced Ksrp+/+ and Ksrp−/− MEFs, or KSRP-expressing retrovirus-transduced Ksrp−/− MEFs, were analyzed by immunoblotting using anti-KSRP antibody. The blot was rehybridized with anti-α-tubulin antibody. (B and C) Decreased IFN mRNA expression in Ksrp−/− MEFs reexpressing KSRP. MEFs were infected with VSV (MOI, 1). The mRNA levels of Ifna4 (B) and Ifnb (C) were analyzed by qRT-PCR 2 h and 4 h after infection. Values represent means ± standard errors of the means (n = 3). (D) Increased VSV replication in Ksrp−/− MEFs reexpressing KSRP. MEFs were infected with VSV (MOI, 1). VSV production was analyzed 24 h after infection by plaque assay. Data are means ± standard errors of the means of three experiments.
To examine whether the enhanced IFN expression is responsible for the viral resistance of Ksrp−/− MEFs, we attenuated IFN signaling by incubation with neutralizing antibodies against both IFNA and IFNB proteins or against IFN receptor 1 (IFNAR1). Addition of both anti-IFNA and anti-IFNB antibodies or anti-IFNAR1 antibodies, but not control serum, significantly restored VSV replication in Ksrp−/− MEFs (Fig. 8 A and B). We next downregulated Ifnb expression by RNAi, as downregulation of all 14 Ifna subtypes was not practical, and examined viral susceptibility. VSV replication was restored in Ksrp−/− MEFs when Ifnb expression was downregulated by RNAi (Fig. 8C and D). Taken together, these data strongly suggest that the enhanced IFN expression contributes to the increased viral resistance observed in Ksrp−/− MEFs and is directly associated with the absence of KSRP.
Fig. 8.

Enhanced IFN expression is responsible for the viral resistance in Ksrp−/− MEFs. (A and B) Ksrp−/− MEFs were infected with VSV (MOI, 0.01) and incubated with neutralizing antibodies against both IFNA and IFNB (A) or against IFNAR1 (B) or control IgG. VSV production was analyzed 24 to 36 h after infection by plaque assay. Data are means ± standard errors of the means of three experiments (Ksrp−/− [PI] versus Ksrp−/− [anti-IFN], P < 0.001; Ksrp−/− [IgG] versus Ksrp−/− [IFNAR1], P < 0.001). (C and D) Ksrp−/− MEFs were infected with lentiviral shRNAs against Ifnb or shRNA against GFP. Transduced cells were infected with VSV (MOI, 1). Expression of Ifnb was analyzed by qRT-PCR 2 h postinfection, and the virus titer was analyzed 24 h after infection. Data are means ± standard errors of the means of three experiments (P < 0.005).
Increased HSV-l resistance in Ksrp−/− mice.
We further examined whether Ksrp−/− mice are refractory to HSV-1 following intracranial infection. The infected mice were monitored daily for 28 days. Ksrp−/− mice displayed a significantly increased survival rate compared to wild-type mice (70% versus 30%; P < 0.01) (Fig. 9 A). We also noticed a significant difference in median survival time between Ksrp+/+ and Ksrp−/− mice (Ksrp+/+, 9 days, versus Ksrp−/−, >28 days). The increased survival of Ksrp−/− mice correlated with the increased production of IFNB protein as well as the decreased HSV-1 replication in infected Ksrp−/− brains compared to that in Ksrp+/+ brains (Fig. 9B and C). Thus, Ksrp−/− mice are protected against intracranial HSV-1 infection.
Fig. 9.
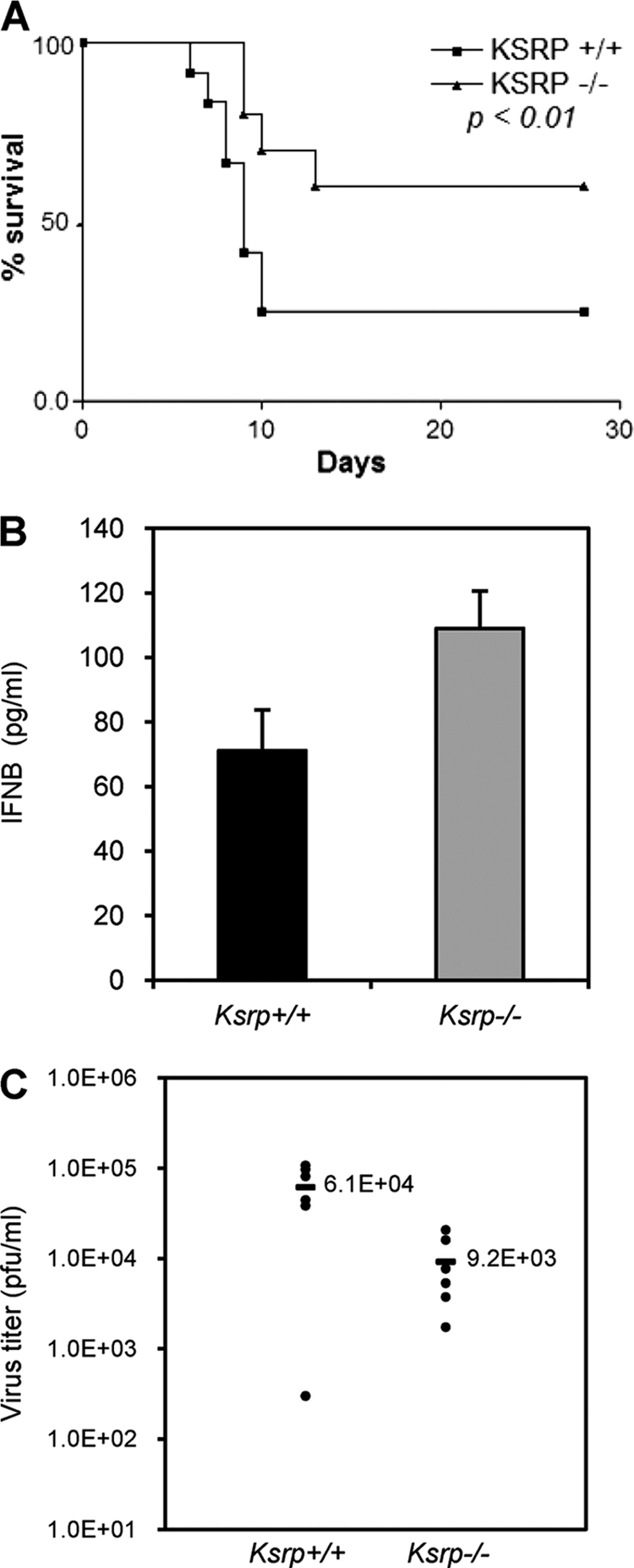
Ksrp−/− mice are resistant to HSV-1 infection. (A) Ksrp+/+ and Ksrp−/− mice were intracranially infected with 300 PFU of HSV-1. The survival of the infected mice was monitored for 28 days. Kaplan-Meier survival curves are shown (animals surviving at the end of experiment: Ksrp+/+, 5/17; Ksrp−/−, 11/16; P < 0.01, log-rank test). The median survival was 9 days for Ksrp+/+ mice and >28 days for Ksrp−/− mice. (B) HSV-1-infected Ksrp+/+ and Ksrp−/− brains were harvested 3 days after infection. IFNB levels were analyzed by ELISA. Values are means ± standard errors of the means (n = 3; P < 0.05). (C) HSV-1-infected Ksrp+/+ and Ksrp−/− brains were harvested 6 days after infection. Virus production was analyzed by plaque assay (Ksrp+/+, n = 6; Ksrp−/−, n = 6; P < 0.02).
DISCUSSION
Although previous studies suggested that several endogenous mRNAs are targeted for decay by KSRP (10, 19, 42, 56), the mRNA targets of KSRP at the physiological and organismal level have not been identified. In the present study, we generated Ksrp knockout mice and demonstrated that type I IFN gene expression is increased in the absence of Ksrp, as the result of decreased mRNA decay. Biochemical analysis showed that KSRP binds the 3′-UTRs of Ifna4 and Ifnb mRNAs, thereby triggering mRNA decay. While we have only examined the decay of one of the Ifna subtypes, Ifna4, there are at least 14 Ifna genes in mice (25, 37) and most of the other Ifna subtypes contain AREs in the 3′-UTRs (26), we suggest that mRNAs encoding some of the other Ifna subtypes are likely targeted for decay by KSRP.
We showed that the stability of Ifnb and Ifna4 mRNAs was increased by 2- to 4-fold in the absence of KSRP, and they were still relatively unstable (Fig. 2, 4, and 5). These data suggest that additional decay-promoting ARE-BPs may be involved in the decay. Indeed, we have found that downregulation of TTP also decreases Ifnb and Ifna mRNA decay and “simultaneous downregulation” of TTP and KSRP additively increases mRNA stability (unpublished data), indicating that KSRP and TTP play redundant and independent roles in Ifn mRNA decay. Thus, we suggest that some of the other decay-promoting ARE-BPs, such as BRF1, BRF2, and AUF1, may be involved in Ifn mRNA decay.
We also showed that KSRP was able to repress IFN expression by targeted mRNA decay in both nonstimulated cells (Fig. 4 and 5) and stimulated cells (Fig. 2). Since IFN expression is constantly low in nonstimulated cells and is robustly induced upon viral infection, and as we showed that deletion of Ksrp had a beneficial role in viral resistance by decreasing Ifn mRNA decay, it was of interest to test whether the decay activity of KSRP was regulated during viral infection. We examined this by monitoring GB-Ifnb mRNA decay in cells treated with poly(I·C) and found no difference in the decay rates between nonstimulated and stimulated cells (data not shown). These data support that KSRP is capable of constitutively targeting Ifn mRNA decay in nonstimulated cells.
Type I IFNs are rapidly and transiently induced in response to viral infection, which requires activation of pattern recognition receptors followed by activation of IRF3 and IRF7 (25, 37, 53). The transient nature of type I IFN expression acts through repression of transcription and rapid mRNA decay by the AU-rich elements following induction. However, the factors that are involved in the control of type I IFN mRNA decay remain unknown (26). Our findings demonstrate that KSRP is the critical negative factor that plays an important role in the posttranscriptional regulation of type I IFN gene expression and that disruption of the decay of IFN mRNAs enhances their expression, rendering increased viral resistance in Ksrp−/− cells. The reversed phenotype observed in Ksrp−/− cells through the blocking of IFN signaling with neutralizing antibodies or RNAi suggests that the enhanced IFN expression is the mechanism for the increased viral resistance. However, we cannot formally exclude other mechanisms that are likely to confer viral resistance in Ksrp−/− cells and Ksrp−/− mice. Nevertheless, our findings strongly suggest that inhibition of IFN mRNA decay provides an additional mechanism to combat viral infection.
Whereas the robust induction is necessary for cell defense, the temporal and transient accumulation of type I IFN is also critical in preventing a sustained expression that sensitizes cells to apoptosis and induces prolonged translational repression in targeted cells (45, 50, 52). Prolonged type I IFN signaling also increases the risk for the formation of autoreactive T cells and autoantibodies. Thus, type I IFNs have been considered a risk factor for the development of autoimmune disease (5, 50). IRF2-deficient mice display disruption of homeostatic erythropoiesis and develop spontaneous skin inflammatory disease, symptoms similar to patients with psoriasis, due to increased type I IFN signaling (23, 44). Systemic lupus erythematosus (SLE) patients show increased serum levels of type I IFN and an upregulated IFN transcriptome signature in their peripheral blood mononuclear cells (5, 7). In addition, arthritis is a well-documented side effect in hepatitis C and multiple sclerosis patients treated with therapeutic type I IFN (49). In the animal model of arthritis, in which the onset of severe autoimmune syndrome is induced by Borrelia burgdorderi infection, the blockage of systematic IFN signaling was shown to effectively reduce arthritis severity, further indicating the involvement of type I IFN in the progression of infection-triggered autoimmunity (35). The negative regulation of type I IFN expression by KSRP suggests an interesting, exciting mechanism for the involvement of posttranscriptional regulation in the pathophysiological progress of these autoimmune diseases. An understanding of the regulation will provide a means to develop a therapeutic approach to restrict type I IFN expression, thereby intervening with the onset or progression of such diseases.
Previous studies have suggested that KSRP selectively targets mRNAs encoding several cytokines and developmental regulators for decay in established cell culture lines (10, 19, 20, 56). The availability of Ksrp−/− mice provides us an invaluable tool to identify the physiological mRNA targets of KSRP at the organismal level and to characterize the phenotype associated with Ksrp deficiency due to dysregulated mRNA decay. Further study of Ksrp−/− mice will shed new light on the functions of KSRP in the regulation of mRNA metabolism, including mRNA decay, splicing, and microRNA maturation (18, 36, 54).
Currently, several mouse models in which individual decay-promoting ARE-BPs are lacking have been generated to investigate the importance of ARE-mediated mRNA decay in vivo (24, 31, 47, 48, 51). Each knockout mouse model exhibits a unique phenotype due to the dysregulation of specific mRNA targets for each ARE-BP. It is currently unknown whether each ARE-BP targets its mRNA targets for decay globally, i.e., in all cell types where they are expressed, or in a cell-type/tissue-specific manner, i.e., only certain mRNAs in certain cell types, whereas previous studies have also suggested that some of these ARE-BPs are functionally redundant (18, 24). To address these questions, it is necessary to globally characterize the target mRNAs for each decay-promoting ARE-BP by analyzing these knockout animal models individually and, additionally, with an intercross strategy. These studies will allow further insights into the in vivo physiological functions played by distinct ARE-BPs.
In summary, we have provided biochemical and genetic evidence that KSRP is a critical negative factor for the posttranscriptional control of type I IFN gene expression in the innate immune response, which may serve as a target for therapeutics to combat virus infection. As type I IFNs also play a critical role in the pathogenesis of certain autoimmune diseases, elucidating the role of KSRP in restricting the expression of type I IFNs may lead to the development of therapeutic strategies for these diseases.
ACKNOWLEDGMENTS
We are indebted to Myriam Gorospe and Ashish Lal for their help in identifying KSRP mRNA targets in HeLa cells by microarray analysis. These results inspired us to analyze type I IFN gene expression in Ksrp−/− cells. We also thank colleagues, especially Joe Sun, Jinxiang Ren, and Yi-Sing Lai, in the laboratory of Tim Townes for their tremendous help to generate Ksrp-null mice, members in our laboratory for their support, Kaimao Liu for lentiviral shRNA against GFP, Craig Maynard for providing a protocol for isolation and preparation of BMDCs and GM-CSF, Ann-Bin Shyu for the gift of NIH 3T3-B2A2 cells, Douglas Black for providing anti-KSRP monoclonal antibody, and Casey Morrow for comments on this report.
This work was supported by NIH grant GM068758 (C.C.) and grants from AIRC (IG 2010-2012) and AICR (10-0527) to R.G.
Footnotes
Published ahead of print on 20 June 2011.
REFERENCES
- 1. Asselin-Paturel C., et al. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150 [DOI] [PubMed] [Google Scholar]
- 2. Asselin-Paturel C., Trinchieri G. 2005. Production of type I interferons: plasmacytoid dendritic cells and beyond. J. Exp. Med. 202:461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bakheet T., Williams B. R., Khabar K. S. 2003. ARED 2.0: an update of AU-rich element mRNA database. Nucleic Acids Res. 31:421–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bakheet T., Williams B. R., Khabar K. S. 2006. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 34:D111–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banchereau J., Pascual V. 2006. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25:383–392 [DOI] [PubMed] [Google Scholar]
- 6. Barreau C., Paillard L., Osborne H. B. 2005. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33:7138–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bennett L., et al. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bevilacqua A., Ceriani M. C., Capaccioli S., Nicolin A. 2003. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J. Cell. Physiol. 195:356–372 [DOI] [PubMed] [Google Scholar]
- 9. Blackshear P. J. 2002. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 30:945–952 [DOI] [PubMed] [Google Scholar]
- 10. Briata P., et al. 2005. p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol. Cell 20:891–903 [DOI] [PubMed] [Google Scholar]
- 11. Briata P., et al. 2003. The Wnt/beta-catenin→Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol. Cell 12:1201–1211 [DOI] [PubMed] [Google Scholar]
- 12. Carballo E., Lai W. S., Blackshear P. J. 1998. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001–1005 [DOI] [PubMed] [Google Scholar]
- 13. Chen C. Y., et al. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107:451–464 [DOI] [PubMed] [Google Scholar]
- 14. Chen C. Y., Shyu A. B. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465–470 [DOI] [PubMed] [Google Scholar]
- 15. Chou C. F., et al. 2006. Tethering KSRP, a decay-promoting AU-rich element-binding protein, to mRNAs elicits mRNA decay. Mol. Cell. Biol. 26:3695–3706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colonna M., Trinchieri G., Liu Y. J. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226 [DOI] [PubMed] [Google Scholar]
- 17. Garcia-Sastre A., Biron C. A. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882 [DOI] [PubMed] [Google Scholar]
- 18. Gherzi R., et al. 2004. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell 14:571–583 [DOI] [PubMed] [Google Scholar]
- 19. Gherzi R., et al. 2006. The RNA-binding protein KSRP promotes decay of beta-catenin mRNA and is inactivated by PI3K-AKT signaling. PLoS Biol. 5:e5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Graham J. R., Hendershott M. C., Terragni J., Cooper G. M. 2010. mRNA degradation plays a significant role in the program of gene expression regulated by phosphatidylinositol 3-kinase signaling. Mol. Cell. Biol. 30:5295–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hall M. P., Huang S., Black D. L. 2004. Differentiation-induced colocalization of the KH-type splicing regulatory protein with polypyrimidine tract binding protein and the c-src pre-mRNA. Mol. Biol. Cell 15:774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hellums E. K., et al. 2005. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neurol. Oncol. 7:213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hida S., et al. 2000. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity 13:643–655 [DOI] [PubMed] [Google Scholar]
- 24. Hodson D. J., et al. 2010. Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat. Immunol. 11:717–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Honda K., Takaoka A., Taniguchi T. 2006. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360 [DOI] [PubMed] [Google Scholar]
- 26. Khabar K. S., Young H. A. 2007. Post-transcriptional control of the interferon system. Biochimie 89:761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lai W. S., et al. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lai W. S., Carballo E., Thorn J. M., Kennington E. A., Blackshear P. J. 2000. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J. Biol. Chem. 275:17827–17837 [DOI] [PubMed] [Google Scholar]
- 29. Lin W. J., Duffy A., Chen C. Y. 2007. Localization of AU-rich element-containing mRNA in cytoplasmic granules containing exosome subunits. J. Biol. Chem. 282:19958–19968 [DOI] [PubMed] [Google Scholar]
- 30. Loflin P., Chen C. Y., Shyu A. B. 1999. Unraveling a cytoplasmic role for hnRNP D in the in vivo mRNA destabilization directed by the AU-rich element. Genes Dev. 13:1884–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu J. Y., Sadri N., Schneider R. J. 2006. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 20:3174–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marie I., Durbin J. E., Levy D. E. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez I., Rodriguez L. L., Jimenez C., Pauszek S. J., Wertz G. W. 2003. Vesicular stomatitis virus glycoprotein is a determinant of pathogenesis in swine, a natural host. J. Virol. 77:8039–8047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maynard C. L., et al. 2009. Contrasting roles for all-trans retinoic acid in TGF-beta-mediated induction of Foxp3 and IL10 genes in developing regulatory T cells. J. Exp. Med. 206:343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller J. C., et al. 2008. A critical role for type I IFN in arthritis development following Borrelia burgdorferi infection of mice. J. Immunol. 181:8492–8503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Min H., Turck C. W., Nikolic J. M., Black D. L. 1997. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 11:1023–1036 [DOI] [PubMed] [Google Scholar]
- 37. Noppert S. J., Fitzgerald K. A., Hertzog P. J. 2007. The role of type I interferons in TLR responses. Immunol. Cell Biol. 85:446–457 [DOI] [PubMed] [Google Scholar]
- 38. Parker J. N., et al. 2000. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. U. S. A. 97:2208–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parker J. N., et al. 2006. Genetically engineered herpes simplex viruses that express IL-12 or GM-CSF as vaccine candidates. Vaccine 24:1644–1652 [DOI] [PubMed] [Google Scholar]
- 40. Paste M., Huez G., Kruys V. 2003. Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3′-untranslated region and an instability sequence in the coding region. Eur. J. Biochem. 270:1590–1597 [DOI] [PubMed] [Google Scholar]
- 41. Raineri I., Wegmueller D., Gross B., Certa U., Moroni C. 2004. Roles of AUF1 isoforms, HuR and BRF1 in ARE-dependent mRNA turnover studied by RNA interference. Nucleic Acids Res. 32:1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruggiero T., et al. 2007. Identification of a set of KSRP target transcripts upregulated by PI3K-AKT signaling. BMC Mol. Biol. 8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sarkar B., Lu J. Y., Schneider R. J. 2003. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J. Biol. Chem. 278:20700–20707 [DOI] [PubMed] [Google Scholar]
- 44. Sato T., et al. 2009. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med. 15:696–700 [DOI] [PubMed] [Google Scholar]
- 45. Stetson D. B., Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381 [DOI] [PubMed] [Google Scholar]
- 46. Stoecklin G., et al. 2002. Functional cloning of BRF1, a regulator of ARE-dependent mRNA turnover. EMBO J. 21:4709–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stumpo D. J., et al. 2009. Targeted disruption of Zfp36l2, encoding a CCCH tandem zinc finger RNA-binding protein, results in defective hematopoiesis. Blood 114:2401–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stumpo D. J., et al. 2004. Chorioallantoic fusion defects and embryonic lethality resulting from disruption of Zfp36L1, a gene encoding a CCCH tandem zinc finger protein of the Tristetraprolin family. Mol. Cell. Biol. 24:6445–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Swiecki M., Colonna M. 2010. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 234:142–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takaoka A., Taniguchi T. 2003. New aspects of IFN-alpha/beta signalling in immunity, oncogenesis and bone metabolism. Cancer Sci. 94:405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor G. A., et al. 1996. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445–454 [DOI] [PubMed] [Google Scholar]
- 52. Terenzi F., Hui D. J., Merrick W. C., Sen G. C. 2006. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem. 281:34064–34071 [DOI] [PubMed] [Google Scholar]
- 53. Thompson A. J., Locarnini S. A. 2007. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell Biol. 85:435–445 [DOI] [PubMed] [Google Scholar]
- 54. Trabucchi M., et al. 2009. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459:1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Whittemore L. A., Maniatis T. 1990. Postinduction turnoff of beta-interferon gene expression. Mol. Cell. Biol. 10:1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Winzen R., et al. 2007. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol. Cell. Biol. 27:8388–8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu J. 2005. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr. Protoc. Mol. Biol. Chapter 28:Unit 28 21. [DOI] [PubMed] [Google Scholar]
- 58. Xu N., Chen C. Y., Shyu A. B. 2001. Versatile role for hnRNP D isoforms in the differential regulation of cytoplasmic mRNA turnover. Mol. Cell. Biol. 21:6960–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu N., Loflin P., Chen C. Y., Shyu A. B. 1998. A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res. 26:558–565 [DOI] [PMC free article] [PubMed] [Google Scholar]