

Abstract
Accurate and rapid diagnosis of malaria infections is crucial for implementing species-appropriate treatment and saving lives. Molecular diagnostic tools are the most accurate and sensitive method of detecting Plasmodium, differentiating between Plasmodium species, and detecting subclinical infections. Despite available whole-genome sequence data for Plasmodium falciparum and P. vivax, the majority of PCR-based methods still rely on the 18S rRNA gene targets. Historically, this gene has served as the best target for diagnostic assays. However, it is limited in its ability to detect mixed infections in multiplex assay platforms without the use of nested PCR. New diagnostic targets are needed. Ideal targets will be species specific, highly sensitive, and amenable to both single-step and multiplex PCRs. We have mined the genomes of P. falciparum and P. vivax to identify species-specific, repetitive sequences that serve as new PCR targets for the detection of malaria. We show that these targets (Pvr47 and Pfr364) exist in 14 to 41 copies and are more sensitive than 18S rRNA when utilized in a single-step PCR. Parasites are routinely detected at levels of 1 to 10 parasites/μl. The reaction can be multiplexed to detect both species in a single reaction. We have examined 7 P. falciparum strains and 91 P. falciparum clinical isolates from Tanzania and 10 P. vivax strains and 96 P. vivax clinical isolates from Venezuela, and we have verified a sensitivity and specificity of ∼100% for both targets compared with a nested 18S rRNA approach. We show that bioinformatics approaches can be successfully applied to identify novel diagnostic targets and improve molecular methods for pathogen detection. These novel targets provide a powerful alternative molecular diagnostic method for the detection of P. falciparum and P. vivax in conventional or multiplex PCR platforms.
INTRODUCTION
Malaria continues to be a leading cause of morbidity and mortality worldwide. It is responsible for 200,000 to 300,000 diagnosed cases and 600,000 to 900,000 deaths in 2009 alone (40). Early detection and accurate diagnosis are the best tools for saving lives in regions of endemicity. Correct species identification and accurate diagnosis of mixed infections are of particular importance for proper treatment in regions where multiple parasite species are endemic. Of the five species within the genus Plasmodium known to infect humans, Plasmodium falciparum is the most deadly, followed by Plasmodium vivax, which also causes significant morbidity and some mortality (2, 10, 14, 23, 29, 38). P. falciparum and P. vivax also have wider global distributions than other species. The remaining three species (which are not the subject of this paper), P. malariae, P. ovale, and P. knowlesi, have different global distributions (with P. malariae being found primarily in South America and Asia and P. ovale and P. knowlesi being found primarily in Asia) and different levels of morbidity and mortality.
Light microscopy remains the gold standard of malaria diagnosis in regions of endemicity. While microscopy is cost-effective and requires little equipment, a well-trained microscopist is essential. A highly trained and experienced microscopist can typically detect parasitemias of as low as 90 to 200 parasites/μl. Misdiagnosis may still occur due to low parasitemia or mixed infection. Immunochromatographic rapid diagnostic tests (RDTs) are increasingly being implemented in case management and control programs. RDTs identify the parasite antigens HRP2, pLDH, and pAldolase and may be pan-specific (for all Plasmodium species), P. falciparum specific, or both, depending on the test. RDTs are not effective for the full diagnosis of mixed infections, as they can only distinguish P. falciparum and indicate the presence or absence of another Plasmodium species. While they can detect parasitemia at levels as low as 100 parasites/μl, they are not quantitative (21). Additionally, the HRP2 antigen can persist in blood after parasite clearance, leading to false-positive diagnoses. It has also been reported that up to 40% of P. falciparum parasites in some parts of South America have HRP-2 gene deletions, increasing concerns about false-negative diagnoses (8).
The use of molecular diagnostic tools is the most accurate and sensitive method for detecting malaria parasite species. Their current use, however, is restricted to reference laboratories or research studies, since there are limitations associated with the use of molecular tools in regions of endemicity for routine diagnostic use (including infrastructure problems, prohibitive costs, a refrigerated or frozen supply cold chain, and the requirement for trained personnel). Despite these limitations, molecular methods are the best methods for detecting multiple species and subclinical infections (4, 7), making them invaluable for malaria parasite detection. Molecular methods will become increasingly important given the proposed eradication/elimination goals and the need to detect subclinical infections (12).
PCR-based amplification methods, including multiplex PCR, real-time PCR, and, more recently, the loop-mediated DNA amplification method (LAMP), have been developed to detect malaria parasite species (11, 24, 25, 30, 31, 35, 37). Molecular methods offer the advantage of highly specific differentiation of Plasmodium species. Recently, molecular techniques confirmed the natural infection of humans with the zoonotic P. knowlesi in Southeast Asia (33). This simian malaria parasite species had not previously been found in humans in great numbers, and a similar morphology resulted in an incorrect P. malariae diagnosis by microscopy.
The most widely used molecular target for the detection of Plasmodium and diagnosis of malaria was developed prior to the completion of any Plasmodium genome sequence. The target is the 18S rRNA gene(s) (11, 16, 30, 32, 34). This target was a logical choice given its high sequence conservation, the availability of universal primer sequences for its amplification, and the fact that it was known to exist in multiple copies in all organisms that had been examined at the time. The availability of complete Plasmodium genome sequences presents a great opportunity for improving the existing molecular diagnostic tools by identifying new targets for more sensitive and specific detection. The P. falciparum genome was completed in 2002 (9), and P. vivax and P. knowlesi have since been sequenced (5, 26). Despite the existence of genomic information for three of the five human-infecting malaria parasites for many years, the majority of molecular diagnostic tools still rely on 18S rRNA. Subsequent examination of Plasmodium genome sequences has revealed that the 18S rRNA target is present in only 4 to 8 divergent, nontandem copies, depending upon the species, in contrast to the case for other eukaryotic genomes that have hundreds of tandem copies of rRNA gene clusters (18, 19). In addition, the few 18S rRNA sequences that are present are not identical in sequence and are variably expressed during the parasite life cycle (15). As PCR sensitivity is greatly influenced by the starting target molecule copy number, a low target copy number limits the detection capabilities of these assays, especially if the parasitemia is low.
The 18S rRNA gene target also presents challenges for effective multiplex platforms. The design of multiple primers to the same target can result in primer competition and decrease the efficiency of the assay. While multiplex assays for simultaneous detection of malaria parasite species do exist (25, 31, 37), they show decreased sensitivity, particularly in detecting the minor species (20). Rubio et al. (31) designed a seminested two-tube multiplex PCR, with an initial genus-specific amplification followed by a secondary amplification using a universal Plasmodium primer and species-specific reverse primers. Padley et al. (25) designed a one-tube multiplex assay, using species-specific primers. However, both of these methods have been shown to perform less effectively than the standard nested PCR method (20). Taylor et al. (37) designed a multiplex real-time platform, relying on the increased sensitivity of both novel targets and fluorescent probes. However, this assay was most effective in duplex format and not as a true four-species multiplex assay.
To address the limitations of existing molecular diagnostic tools, we have mined Plasmodium genome sequence data and identified new target DNA sequences for improved molecular diagnostic applications. Here we detail the method used to identify these targets in P. falciparum and P. vivax, and we show that they provide increased sensitivity in a single-step PCR and increased efficacy in multiplex assays.
MATERIALS AND METHODS
Data harvesting.
Assembled genome sequence data for P. falciparum (3D7 strain) and P. vivax (Sal-1 strain) were obtained from PlasmoDB (release 5.5). The P. falciparum genome data consist of 14 sequences (23,264,338 bp), and the P. vivax genome data consist of 2,747 sequences (27,007,990 bp). Differences in the numbers of sequences between species reflect the more advanced state of P. falciparum genome assembly relative to P. vivax. There are 14 highly assembled chromosomes for each species and 2,733 unassigned contigs for P. vivax.
CRS screens and copy number determination.
The pipeline shown in Fig. 1 and described below was constructed using custom PERL scripts. RepeatScout (version 1.0.5, default parameters) (28) was used to identify genomic consensus repeat sequences (CRS). Totals of 418 P. falciparum and 428 P. vivax CRS were generated. The Tandem Repeat Finder program (TRF) version 4.0 (3) was used to eliminate CRS with internal tandem repeats that could potentially interfere with PCR amplification. Repeats containing vector sequences introduced during genome sequencing were identified by a comparison with the NCBI UniVec database (build 5.2; http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html) (with WU-BLAST [blastn ver. 2.0; http://blast.wustl.edu]) with an E-value cutoff of 1E−10. To ensure that targets were not also present in the human genome, CRS were compared to human genome sequences (RefSeq, Primary Reference Assembly, build 37, version 1) with BLAST (1) (version 2.2.22, blastn), with an E-value cutoff of 1E−10. Screens were applied in parallel to all CRS. Any sequence failing a screen was removed from further consideration. A total of 165 P. falciparum sequences and 331 P. vivax sequences passed all screens. All P. falciparum and P. vivax CRS were compared (WU-BLAST) to all available Plasmodium sequence data, and the results were manually inspected to ensure species specificity. To allow sufficient space for primer design and the evaluation of repeat family conservation, CRS smaller than 300 bp were not considered further. CRS were used to calculate the copy number of each repeat. Each screened repeat was used to search (WU-BLAST) against the species' genome from which it was derived. Repeat copies were required to hit to the CRS with an E value of less than 1E−50 for P. vivax. The stringency for P. falciparum was relaxed to 1E−10 because lower E-value requirements did not produce sufficient candidates for screening. A minimum distance of 100 bp between copies was required to remove potential amplification complications. Repeat families with at least 6 copies were considered for further testing, yielding totals of 21 P. falciparum and 68 P. vivax candidates.
Fig. 1.
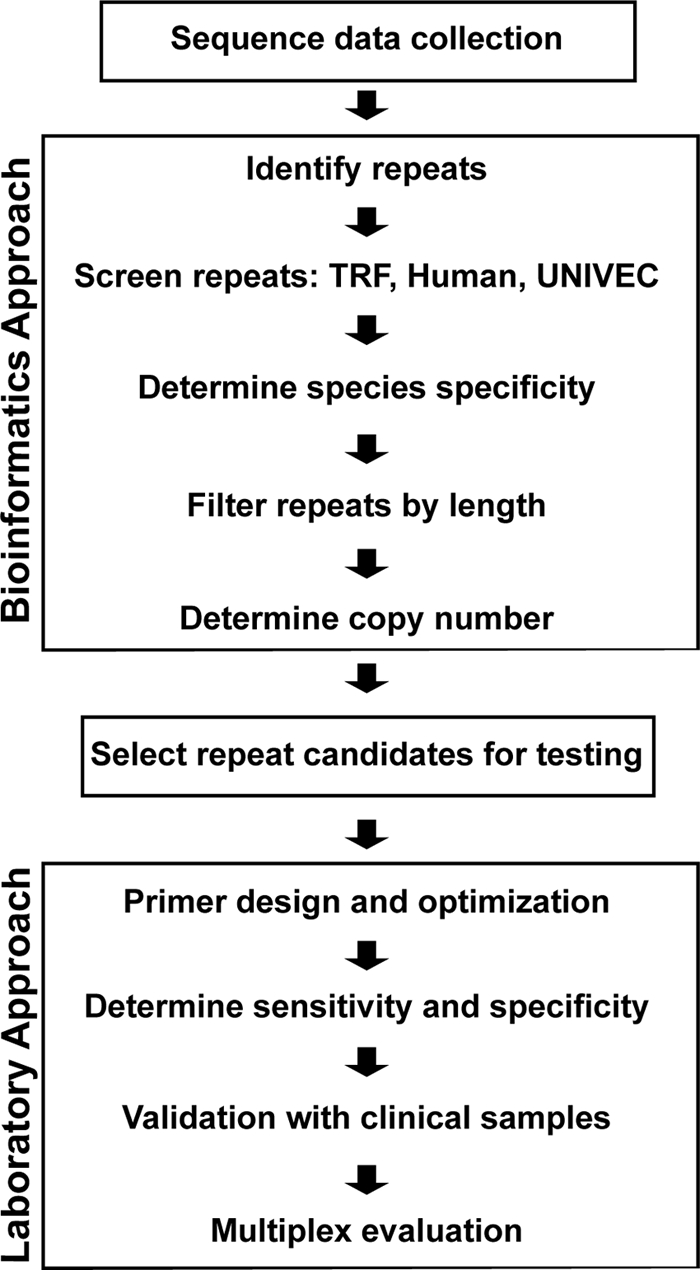
Schematic of diagnostic target screening and development pipeline. All genomic sequences for P. vivax and P. falciparum were downloaded from PlasmoDB. Data were mined for repeats using the RepeatScout algorithm to construct consensus repeat sequences (CRS) for each identified repeat family. CRS were then screened in parallel for tandem repeats, similarity to human sequences, and vector sequences. Any CRS failing these screens were removed from further consideration. CRS that were not species specific or less than 300 bp long were eliminated. Family copy numbers for the remaining candidates were determined via comparison of the CRS against the appropriate genome data. Candidate repeat families containing 6 or more copies separated by at least 100 bp were considered for further testing. For additional information and clinical sample validation, see Materials and Methods.
Target validation.
Primers were designed to test six P. falciparum and seven P. vivax CRS families. Primers were designed manually to candidate targets and screened for GC content, melting temperature, secondary structure, and primer dimer-forming potential using Primer Explorer version 2.0 (http://primerexplorer.jp/e/). Primer pairs were optimized using gradient PCR cycling on Bio-Rad iCycler machines to determine the optimum annealing temperature, with additional adjustments to primer concentration (concentrations from 0.25 μM to 1.0 μM were tested) and master mix components (MgCl2 concentrations from 2.0 mM to 4.0 mM were tested) (see below for final conditions). Primers were further tested for species specificity using laboratory cultures of P. falciparum (3D7) or DNA stocks of P. vivax (SV4), P. malariae, P. ovale, and P. knowlesi.
Plasmodium parasites.
P. falciparum strains 3D7, W2, V1-S, Dd2, HB3, D6, and FCR3 were cultured in our laboratory. DNA stocks of P. vivax (Sal-1, SV4, and NAM/CDC), P. ovale, P. malariae, and P. knowlesi and filter paper blood spots of additional P. vivax strains (from Thailand, North Korea, Vietnam, India, Miami [FL], New Guinea, South Vietnam, and Brazil) were all provided by John Barnwell (CDC). DNA was isolated using commercially available QIAamp DNA minikits (Qiagen, Valencia CA), following the manufacturer's instructions.
Nested PCR.
Nested PCR for malaria parasite detection (as described by Singh et al. [32]) was used as the standard method for comparison.
Amplification of CRS targets by PCR.
Amplification of CRS targets was performed in a 25-μl reaction mixture containing 1× Taq buffer (contains 10 mM Tris-HCl, 50 mM KCl, and 1.5 mM MgCl2; New England BioLabs, Ipswich, MA), 4 mM MgCl2, 200 μM each deoxynucleoside triphosphate (dNTP), 500 nM each oligonucleotide primer, 1.25 units of Taq DNA polymerase (New England BioLabs), and 1 μl of DNA template. Oligonucleotide primers for P. falciparum candidate Pfr364 and P. vivax candidate Pvr47 are shown in Table 1. Separate reactions were performed for P. falciparum and P. vivax with the following cycling parameters: initial denaturation at 95°C for 2 min and then 35 cycles of 95°C for 30 s, 57°C (for P. falciparum) or 54°C (for P. vivax) for 30 s, and 72°C for 45 s, followed by final extension at 72°C for 5 min. PCR products were visualized by gel electrophoresis on a 2% agarose gel.
Table 1.
New target primer sequencesa
| Primer | Sequence |
|
|---|---|---|
| P. falciparum Pfr364 | P. vivax Pvr47 | |
| Forward | 5′-CCATTTTACTCGCAATAACGCTGCAT | 5′-CTGATTTTCCGCGTAACAATG |
| Reverse | 5′-CTGAGTCGAATGAACTAGTCGCTAC | 5′-CAAATGTAGCATAAAAATCYAAG |
| Alt-Forward | 5′-CCGGAAATTCGGGTTTTAGAC | |
| Alt-Reverse | 5′-GCTTTGAAGTGCATGTGAATTGTGCAC | |
Primer sequences designed to targets Pfr364 and Pvr47. The alternate (Alt) primer pair for P. falciparum was used in multiplex reactions only. The P. vivax primer set was the same for both single-species and multiplex PCRs.
Serial dilutions of quantified parasite DNA, isolated from laboratory cultures, were used to determine the detection limits (DNA concentrations ranging from 10,000 parasites/μl to 0.01 parasites/μl were tested). Final validation of targets was performed with P. falciparum and P. vivax clinical samples from Tanzania (n = 91; median parasitemia, 3,200 parasites/μl) and Venezuela (n = 96; no parasitemia data are available), respectively, as well as with additional geographically diverse strains for both targets (for Pfr364, P. falciparum strains W2, V1-S, Dd2, HB3, D6, and FCR3; for Pvr47, P. vivax isolates from Thailand, North Korea, Vietnam, India, Miami, New Guinea, South Vietnam, and Brazil).
Multiplex PCR.
The multiplex PCR platform was optimized by gradient PCR cycling to determine the annealing temperature, with additional adjustments to primer concentrations (0.25 to 1.0 μM were tested) and master mix component concentrations (MgCl2 from 2.0 mM to 4.0 mM, dNTPs from 200 μM to 400 μM each, and Taq DNA polymerase from 1.25 units to 2.5 units were all tested). Multiplex PCR for detecting P. falciparum and P. vivax was performed in a 25-μl reaction mixture containing 1× Taq buffer (New England BioLabs, Ipswich MA; contains 10 mM Tris-HCl, 50 mM KCl, and 1.5 mM MgCl2), 4 mM MgCl2, 400 μM each dNTP, 1,000 nM each P. falciparum primer, 600 to 800 nM each P. vivax primer, 2.5 units of Taq DNA polymerase (New England BioLabs, Ipswich, MA), and 1 μl of DNA template. The alternate P. falciparum oligonucleotide primer sequences (Table 1) were used in the multiplex assay. The P. vivax primers were the same as used in the conventional PCR described above. The reaction was carried out under the following cycling parameters: initial denaturation at 95°C for 2 min and then 35 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 45 s, followed by final extension at 72°C for 5 min. All possible combinations of 10-fold dilutions ranging from 10,000 parasites/μl to 0.01 parasites/μl for each species were tested. PCR products were visualized by gel electrophoresis on a 2% agarose gel.
Sensitivity and specificity calculations.
Sensitivity and specificity (95% confidence interval) were calculated using the nested 18S rRNA PCR as the gold standard for distinguishing a true positive from a false positive (Table 2).
Table 2.
Sensitivities and specificities of new PCR assays compared to standard nested 18S rRNA PCRa
| Species | Result with: |
Sensitivity (%) | Specificity (%) | ||
|---|---|---|---|---|---|
| 18S rRNA nested PCR (n) | New primers |
||||
| No. positive | No. negative | ||||
| P. falciparum | Positive (91) | 91 | 0 | 100 | 100 |
| Negative (9) | 0 | 9 | |||
| P. vivax | Positive (96) | 95 | 1 | 98.9 | 100 |
| Negative (13) | 0 | 13 | |||
The sensitivities and specificities of the new PCR assays were compared to those of standard nested 18S rRNA PCR (32). For conventional PCR, sensitivity and specificity were calculated using 96 P. vivax samples from Venezuela and 91 P. falciparum samples from Tanzania. DNA from nonmalarious patients was included as a negative control.
RESULTS
Repeat mining and screening of diagnostic candidates.
A semiautomated bioinformatics pipeline was constructed for genome repeat mining and in silico candidate screening (Fig. 1) (see Materials and Methods). Six P. falciparum and seven P. vivax putative targets were identified for validation. Over 50 primer pairs were designed to these targets and empirically tested in conventional PCR amplification assays and multiplex assays. Of these targets, the most effective were P. falciparum candidate Pfr364 and P. vivax candidate Pvr47, as these targets consistently performed with the greatest sensitivity and specificity. The functions of Pfr364 and Pvr47 are not known. Neither sequence is annotated or encodes protein. However, regions of Pfr364 are expressed according to PlasmoDB. Full-length sequence alignments and repeat coordinates can be found in Fig. S1 and S2 and Table S1 in the supplemental material.
Diagnostic targets: copy number and distribution.
At least one putative target from each species was found to significantly improve existing diagnostic capabilities. Pfr364 exists in 41 copies, each of which is localized to the SB2 subtelomeric repeat region found on most chromosome ends (Fig. 2). The size of the SB2 region of P. falciparum chromosomes is variable (1 to 3 kb, though it may contain up to 6 kb of additional sequence) and is composed of different repeat types (9). Many regions were found to contain two proximal copies of Pfr364, and chromosome 6 contains three copies at its 3′ end (data not shown). Multiple alignment reveals significant subfamily structure resulting in two related alignment groups, which we have designated subfamilies 1 and 2 (Fig. 3A; see Fig. S2 and Table S1 in the supplemental material). Interestingly, when multiple copies of Pfr364 are found at chromosome ends, there is one member of each subfamily present (Fig. 2).
Fig. 2.

Spatial distribution of Pfr364 family members across the 14 P. falciparum chromosomes. Tick marks indicate 200 kb of sequence. Pfr364 family members occur in two proximal copies at most chromosome ends. Black lines represent the outermost copies (subfamily 1), and gray lines represent the innermost copies (subfamily 2). Chromosome 6 has three copies at its 3′ end (only two are shown). Circos 0.51 (http://mkweb.bcgsc.ca/circos/) was used to generate this map.
Fig. 3.

Alignments of Pfr364 and Pvr47 family members with PCR primers. (A) Pfr364 with primers. Arrows represent locations of PCR primers in context of the full alignment. The full alignment is 1,538 positions in length; here a partial alignment is shown. Vertical black lines indicate where the sequence alignment has been truncated to enable viewing of all 4 primer locations. The alignment shows two subfamilies within Pfr364. We have designated the upper 22 sequences subfamily 1 and the lower 19 sequences subfamily 2. Forward and reverse primer pairs used for multiplex and conventional PCR are, respectively, the last two sequence pairs in the alignment. (B) Pvr47 with primers. Arrows represent locations of PCR primers in context of the full alignment. The full alignment is 1,070 positions; here only positions 433 to 776 are shown. Vertical lines indicate where the sequence alignment has been truncated for easier viewing. Forward and reverse primers are, respectively, the last two sequences in the alignment.
Pvr47 is found in 14 copies (Fig. 3B; see Fig. S1 and Table S1 in the supplemental material). All members are located on contigs that have not yet been assigned to chromosome scaffolds. The majority of these members map to small (<16-kb) subtelomeric contigs that could not be assembled onto chromosomes due to their repetitive nature (5). Two of these family members are located proximal to annotated vir genes, while a third is located proximal to the subtelomeric transmembrane protein Pvstp1 (6).
Detection of P. vivax and P. falciparum.
Primers designed to Pfr364 and Pvr47 (Table 1) specifically identified P. falciparum and P. vivax, respectively. Other Plasmodium species, including P. malariae, P. ovale, and P. knowlesi, were not amplified. No amplification was observed using human nonmalaria DNA (data not shown). Using known quantities of laboratory-cultured parasites, we were able to consistently detect parasites in concentrations of as low as 10 to 0.1 parasites/μl, compared to 10 to 1 parasites/μl detected with the standard method (Fig. 4 and Table 3). P. falciparum candidate Pfr364 detected between 10 and 0.1 parasites/μl of DNA (detected 0.1 parasites/μl twice and 10 parasites/μl once). For each repeat target, single amplified products were clearly defined on a 2% agarose gel stained with ethidium bromide.
Fig. 4.
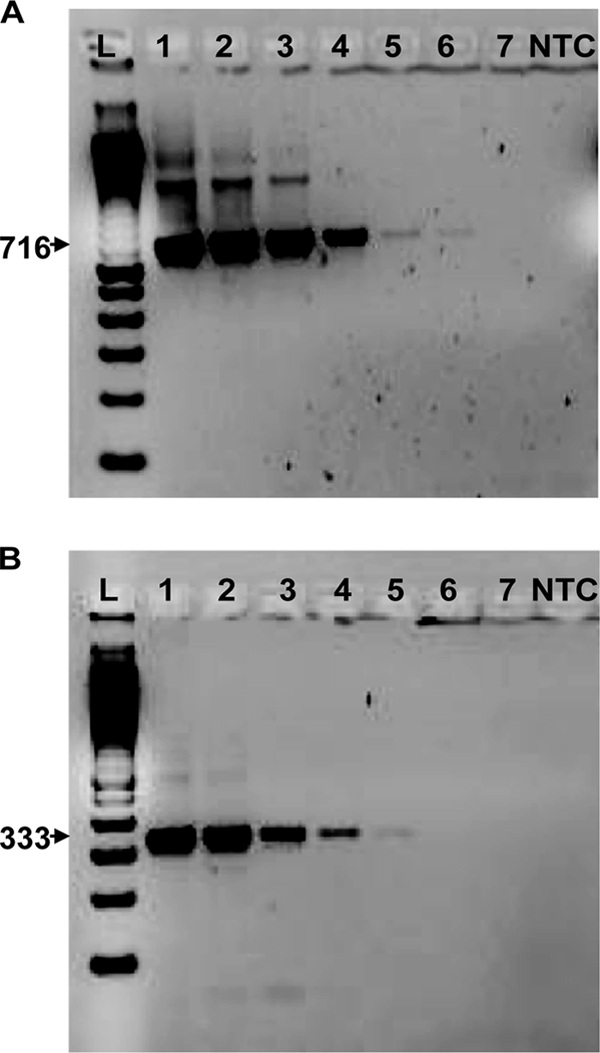
Limits of detection for conventional PCR assays. Primers to novel targets P. falciparum Pfr364 (A) and P. vivax Pvr47 (B) were used to amplify parasite DNA of the appropriate species. DNA was quantified, and 10-fold serial dilutions from 10,000 parasites/μl (lane 1) to 0.01 parasites/μl (lane 7) were used to determine the limit of detection. A 100-bp standard ladder (L) and a no-template control (NTC) were included.
Table 3.
Detection limits of new diagnostic targets
| Replicate | Detection limit (parasites/μl)a |
|
|---|---|---|
| P. falciparum | P. vivax | |
| 1 | 0.1 | 10 |
| 2 | 0.1 | 1 |
| 3 | 10 | 10 |
Calculated using 10-fold serial dilutions of P. falciparum and P. vivax DNAs (see Fig. 4).
Specificity and sensitivity.
The targets were further validated in three ways. First, microscopically determined P. vivax samples from Venezuela (n = 96) and P. falciparum samples from Tanzania (n = 91) were used. In comparison to standard nested 18S rRNA PCR, Pvr47 had 98.9% sensitivity and 100% specificity, and Pfr364 had 100% sensitivity and 100% specificity. Second, target amplification in 7 P. falciparum strains and 10 P. vivax strains from around the world was assessed. The target was successfully amplified in each case (Fig. 5). Finally, PlasmoDB was queried to assess the number and distribution of single-nucleotide polymorphisms (SNPs) in the 41 P. falciparum repeats using data reported previously (13, 22, 39). These data represent information from 21 P. falciparum strains. There are an average of 50 polymorphic sites along the ∼1,500-nucleotide (nt) length of each of the Pfr364 repeats, for an average of 3% each. An average of 2 different nucleotides are observed at each polymorphic position (see Table S2 in the supplemental material).
Fig. 5.
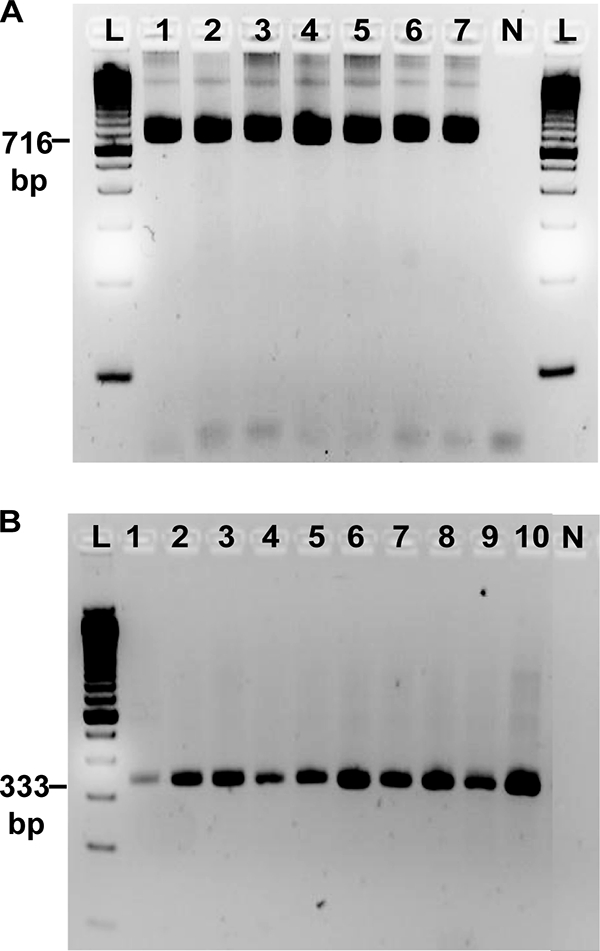
Evaluation of Pfr364 and Pvr47 primers on geographically diverse field isolates. (A) Pfr364 primers tested on various P. falciparum isolates. Lanes: 1, 3D7; 2, W2; 3, V1-S; 4, Dd2; 5, Hb3; 6, D6; 7, FCR3. A 100-bp standard ladder (L) and a no-template control (N) were included. (B) Pvr47 primers tested on various P. vivax field isolates. Lanes: 1, Thailand; 2, North Korea; 3,Vietnam; 4, India; 5, NAM/CDC; 6, Miami; 7, New Guinea; 8, Sal-1; 9, South Vietnam; 10, Brazil. The Pfr364 and Pvr47 primers clearly detect all of the tested isolates. Pfr364 primers detected an additional 91/91 (100%) P. falciparum isolates from Tanzania, and Pvr47 primers detected an additional 95/96 (98.9%) P. vivax isolates from Venezuela (not shown).
Multiplex assay.
The multiplex PCR assay with combined Pvr47 and Pfr364 specifically detected P. vivax and P. falciparum and correctly identified both single- and mixed-species infections. An alternative P. falciparum primer was used to make the PCR products similar in size to increase efficiency (see Materials and Methods) (Table 1). The limit of detection for the multiplex platform was determined using “mock mixed” infections of P. falciparum and P. vivax laboratory cultures. This method had a limit of detection of 10 parasites/μl for each species (Fig. 6). P. falciparum DNA was also detected at 1.0 parasites/μl when P. vivax was present at the same concentration (P. vivax was not detected). Clinical mixed P. falciparum-P. vivax samples from Venezuela (n = 11) were detected with 90.9% sensitivity and 100% specificity in comparison to the standard nested PCR method, which was performed as separate reactions for the different species.
Fig. 6.
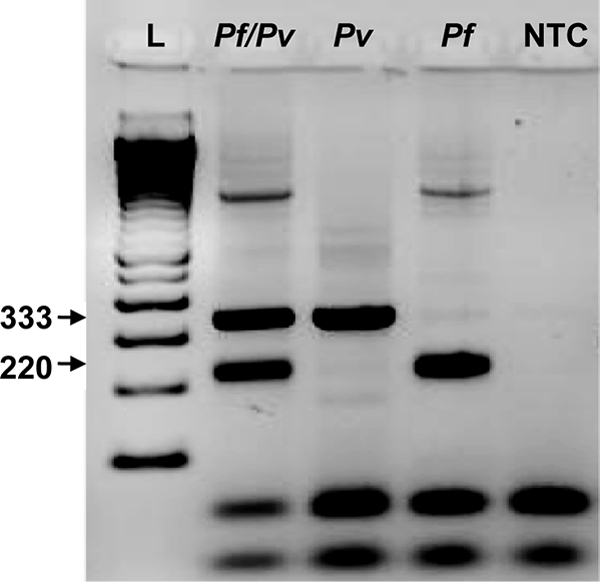
Multiplex PCR. The multiplex method clearly identified mock mixed P. falciparum and P. vivax infections (lane Pf/Pv). Single-species infections (lanes Pf and Pv) were also detected. The P. falciparum band appears at 220 bp and the P. vivax band at 333 bp. A 100-bp standard ladder (L) and a no-template control (NTC) were used.
DISCUSSION
Here we show the value of applying bioinformatics methods and mining genomic data to answer biological questions that address practical needs. This approach can be applied to additional pathogens or used to improve existing molecular diagnostic tools (LAMP, real-time PCR, etc.). Increasing the sensitivity and specificity of molecular assays will facilitate greater high-throughput detection of pathogens.
Discovery of the exact locations of Pvr47 repeats will depend on the continued refinement of the P. vivax genome assembly and improved annotation. The presence of some members near genes known to be located in subtelomeric regions (see above), combined with the known subtelomeric location of Pfr364, points to an interesting role in Plasmodium chromosome end biology for use in diagnostic target development. There has been no comprehensive, systematic study of the genomic repeats of the genus Plasmodium. Our understanding of the organization and content of subtelomeric regions is largely restricted to what is known in P. falciparum, where it has been shown that these regions contain genes responsible for host immune evasion and antigenic variation (9). Given the biological importance of these regions and the useful diagnostic targets that they contain, it is critical that we increase our understanding of their repeat content and organization.
While there is evidence for their location and distribution, the biological functions of Pfr364 and Pvr47 are not yet established. Combined with their repetitive, potentially nongenic nature, this necessitates a thorough evaluation of their robustness as diagnostic targets. Sequences with no coding potential often evolve more quickly than coding regions (17). However, we show that these families are highly conserved (<3% variation at the nucleotide level in P. falciparum), which is indicative of selection. Further, assays designed to the targets were able to detect infections across as large a range of field isolates (7 P. falciparum strains and 10 P. vivax strains) as the standard nested 18S rRNA PCR. These observations suggest that these targets are as robust to evolutionary change as the 18S rRNA target, despite the uncertainty about their biological roles.
Pfr364 and Pvr47 are not necessarily the most abundant repeats in these genomes. We tested only a few repetitive sequences resulting from our data mining for their potential as diagnostic targets. It is possible that more sensitive targets exist. As we have noted above, there has been no comprehensive investigation of the genomic repeat content of these organisms, and our analysis is ongoing.
Amplification of the novel targets presented here was highly sensitive and specific. Both assays have a detection limit 10-fold lower than the historic standard and utilize a single, as opposed to nested, PCR. This is an important improvement, as single-round, unnested PCRs have fewer steps, decrease the chances of contamination or error, decrease the overall cost in materials, and require less time to complete. The standard nested protocol requires two separate reactions, and the amplified product of the first reaction must be transferred to a second tube prior to the second reaction. Opening the tubes increases the risks of contamination and human error and also increases the time and costs for necessary reagents and consumables.
The targets produced clean products, clearly visible on an agarose gel stained with ethidium bromide, at 716 bp and 333 bp for P. falciparum and P. vivax, respectively. There were no nonspecific bands in clinical samples, including negative samples, as was sometimes found with the standard PCR method (data not shown). While DNA amplified from laboratory cultures using the standard nested PCR method showed clean bands on the agarose gel, clinical samples often produced nonspecific bands with sizes similar to those of the expected bands when the 18S rRNA gene-based method was used. This can be especially confusing when interpreting the results, and additional time was required to fully separate the bands by electrophoresis. The nonspecific bands appeared when P. falciparum samples were tested to amplify field samples with the P. vivax-specific primers (unpublished observation). Additionally, sometimes several rounds of repetition of the standard method described by Singh et al. (32) were necessary to confirm the results for the clinical samples tested. We found that PCR amplification with the newly identified targets yielded consistent clear results with no spurious bands among the clinical samples tested in this study.
One-step multiplex reactions will offer a great improvement to existing Plasmodium diagnostics. Efficient, high-throughput pathogen detection will decrease the time to results and appropriate treatment. Mixed infections naturally occur in regions where multiple parasite species are found, and these present a challenge for diagnosis. To validate our multiplex method, we tested all possible combinations of 10-fold DNA concentrations (from 10,000 parasites/μl to 1 parasite/μl) to cover all the range of naturally occurring mixed infections (data not shown). The limit of detection (10 parasites/μl for P. falciparum and P. vivax) compares favorably to those for other multiplex methods. In mixed-species infections, one major species will frequently dominate over another that is present in relatively low concentrations during PCR amplification (27, 36). In contrast, the current method detects both major and minor species of mixed infection, providing another advantage of using this method for diagnosis.
In conclusion, the findings from this study demonstrate that using bioinformatics to identify novel genetic targets for diagnostic application is a valid approach. This methodology will be extended to identify additional targets from other Plasmodium species for diagnostic assays when the genome sequences become available. Our results demonstrate that the newly identified Pfr364 and Pvr47 targets are valuable tools to improve and simplify molecular diagnostic methods for field use.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jatan Patel and Zubin Mehta for their bioinformatics assistance in screening putative target sequences.
This work was supported in part by a CDC-UGA seed grant (OPHR no. 8212) awarded to J.C.K. and V.U. This study was supported in part by resources and technical expertise from the University of Georgia Research Computing Center, a partnership between the Office of the Vice President for Research and the Office of the Chief Information Officer. A.D. was supported by an EID Fellowship from the Association of Public Health Laboratories and the CDC. N.W.L. and A.D. (after the EID Fellowship) were supported by the Atlanta Research and Education Foundation, Atlanta, GA. J.D. was supported by NIH grant R01 AI068908 awarded to J.C.K.
Footnotes
Supplemental material for this article may be found at http://jcm.asm.org/.
Published ahead of print on 27 April 2011.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2. Barcus M. J., et al. 2007. Demographic risk factors for severe and fatal vivax and falciparum malaria among hospital admissions in northeastern Indonesian Papua. Am. J. Trop. Med. Hyg. 77:984–991 [PubMed] [Google Scholar]
- 3. Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bronzan R. N., McMorrow M. L., Kachur S. P. 2008. Diagnosis of malaria: challenges for clinicians in endemic and non-endemic regions. Mol. Diagn. Ther. 12:299–306 [DOI] [PubMed] [Google Scholar]
- 5. Carlton J. M., et al. 2008. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455:757–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. del Portillo H. A., et al. 2001. A superfamily of variant genes encoded in the subtelomeric region of Plasmodium vivax. Nature 410:839–842 [DOI] [PubMed] [Google Scholar]
- 7. Erdman L. K., Kain K. C. 2008. Molecular diagnostic and surveillance tools for global malaria control. Travel Med. Infect. Dis. 6:82–99 [DOI] [PubMed] [Google Scholar]
- 8. Gamboa D., et al. 2010. A large proportion of P. falciparum isolates in the Amazon region of Peru lack pfhrp2 and pfhrp3: implications for malaria rapid diagnostic tests. PLoS One 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gardner M. J., et al. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419:498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Genton B., et al. 2008. Plasmodium vivax and mixed infections are associated with severe malaria in children: a prospective cohort study from Papua New Guinea. PLoS Med. 5:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han E. T., et al. 2007. Detection of four Plasmodium species by genus- and species-specific loop-mediated isothermal amplification for clinical diagnosis. J. Clin. Microbiol. 45:2521–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harris I., et al. 2010. A large proportion of asymptomatic Plasmodium infections with low and sub-microscopic parasite densities in the low transmission setting of Temotu Province, Solomon Islands: challenges for malaria diagnostics in an elimination setting. Malar. J. 9:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jeffares D. C., et al. 2007. Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat. Genet. 39:120–125 (Erratum, 39:567.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kochar D. K., et al. 2009. Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am. J. Trop. Med. Hyg. 80:194–198 [PubMed] [Google Scholar]
- 15. Li J., et al. 1997. Regulation and trafficking of three distinct 18 S ribosomal RNAs during development of the malaria parasite. J. Mol. Biol. 269:203–213 [DOI] [PubMed] [Google Scholar]
- 16. Li J., et al. 1995. Plasmodium: genus-conserved primers for species identification and quantitation. Exp. Parasitol. 81:182–190 [DOI] [PubMed] [Google Scholar]
- 17. Li W.-H., Graur D. 1991. Fundamentals of molecular evolution. Sinauer Associates, Inc., Sunderland, MA [Google Scholar]
- 18. Long E. O., Dawid I. B. 1980. Repeated genes in eukaryotes. Annu. Rev. Biochem. 49:727–764 [DOI] [PubMed] [Google Scholar]
- 19. Mercereau-Puijalon O., Barale J. C., Bischoff E. 2002. Three multigene families in Plasmodium parasites: facts and questions. Int. J. Parasitol. 32:1323–1344 [DOI] [PubMed] [Google Scholar]
- 20. Mixson-Hayden T., Lucchi N., Udhayakumar V. 2010. Evaluation of three PCR-based diagnostic assays for detecting mixed Plasmodium infection. BMC Res. Notes 3:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moody A. 2002. Rapid diagnostic tests for malaria parasites. Clin. Microbiol. Rev. 15:66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mu J. B., et al. 2010. Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat. Genet. 42:268–U113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mueller I., et al. 2009. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 9:555–566 [DOI] [PubMed] [Google Scholar]
- 24. Notomi T., et al. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Padley D., Moody A. H., Chiodini P. L., Saldanha J. 2003. Use of a rapid, single-round, multiplex PCR to detect malarial parasites and identify the species present. Ann. Trop. Med. Parasitol. 97:131–137 [DOI] [PubMed] [Google Scholar]
- 26. Pain A., et al. 2008. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455:799–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Polz M. F., Cavanaugh C. M. 1998. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64:3724–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Price A. L., Jones N. C., Pevzner P. A. 2005. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl. 1):i351–i358 [DOI] [PubMed] [Google Scholar]
- 29. Price R. N., Douglas N. M., Anstey N. M. 2009. New developments in Plasmodium vivax malaria: severe disease and the rise of chloroquine resistance. Curr. Opin. Infect. Dis. 22:430–435 [DOI] [PubMed] [Google Scholar]
- 30. Rougemont M., et al. 2004. Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR asays. J. Clin. Microbiol. 42:5636–5643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rubio J. M., et al. 2002. Alternative polymerase chain reaction method to identify Plasmodium species in human blood samples: the semi-nested multiplex malaria PCR (SnM-PCR). Trans. R. Soc. Trop. Med. Hyg. 96(Suppl. 1):S199–S204 [DOI] [PubMed] [Google Scholar]
- 32. Singh B., et al. 1999. A genus- and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am. J. Trop. Med. Hyg. 60:687–692 [DOI] [PubMed] [Google Scholar]
- 33. Singh B., et al. 2004. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 363:1017–1024 [DOI] [PubMed] [Google Scholar]
- 34. Snounou G., Viriyakosol S., Jarra W., Thaithong S., Brown K. N. 1993. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol. Biochem. Parasitol. 58:283–292 [DOI] [PubMed] [Google Scholar]
- 35. Snounou G., et al. 1993. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol. Biochem. Parasitol. 61:315–320 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki M. T., Giovannoni S. J. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62:625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taylor S. M., et al. 2010. High-throughput pooling and real-time PCR-based strategy for malaria detection. J. Clin. Microbiol. 48:512–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tjitra E., et al. 2008. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med. 5:e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkman S. K., et al. 2007. A genome-wide map of diversity in Plasmodium falciparum. Nat. Genet. 39:113–119 [DOI] [PubMed] [Google Scholar]
- 40. WHO 2010. World malaria report 2008. World Health Organization, Geneva, Switzerland [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.