

Summary
Hematopoietic stem cells (HSCs) are used in transplantation therapy to reconstitute the hematopoietic system. Human cord blood (hCB) transplantation has emerged as an attractive alternative treatment option when traditional HSC sources are unavailable, however, the absolute number of hCB HSCs transplanted is significantly lower than bone marrow or mobilized peripheral blood stem cells (MPBSCs). We previously demonstrated that dimethyl-prostaglandin E2 (dmPGE2) increased HSCs in vertebrate models. Here, we describe preclinical analyses of the therapeutic potential of dmPGE2-treatment using human and non-human primate HSCs. dmPGE2 significantly increased total human hematopoietic colony formation in vitro and enhanced engraftment of unfractionated and CD34+ hCB following xenotransplantation. In non-human primate autologous transplantation, dmPGE2-treated CD34+ MPBSCs showed stable multilineage engraftment over one year post-infusion. Together, our analyses indicated that dmPGE2 mediates conserved responses in HSCs from human and non-human primates, and provided sufficient preclinical information to support proceeding to an FDA-approved phase 1 clinical trial.
Introduction
HSCs alone possess the ability to both self-renew and differentiate into all mature blood lineages, thereby maintaining immune function, tissue perfusion and hematopoietic homeostasis throughout the lifetime of the organism. HSCs are therapeutically valuable for transplantation in the treatment of hematologic malignances. They are a rare population in the bone marrow (BM), and methods for direct isolation and expansion of a pure population of functional human HSCs remain elusive. The development of therapeutic options to manipulate and maintain human HSCs is of major clinical interest, however, to date, no such therapy has proven effective in large-scale clinical trails.
HSC transplantation is the only curative option for many patients with leukemia, lymphoma, or BM failure. Stem cells obtained from the BM or peripheral blood (PB) must be human leukocyte antigen (HLA) matched to the patient in order to avoid rejection. Only 25–30% of patients can utilize BM from a related sibling donor, and matched unrelated donors cannot be found in BM registries for all patients, particularly for those from ethnic minorities (Laver et al., 2001). In the past two decades, human cord blood (hCB) stem cells have emerged as an option for unmatched patients, as they are readily obtained in registries and have less stringent requirements for HLA matching (Broxmeyer et al., 1989). The use of hCB transplantation has steadily grown since the first transplant occurred in 1988 to more than 20,000 recipients worldwide (Rocha and Broxmeyer, 2010). In the United States, hCB transplants account for almost 20% of all HSC transplants annually (Broxmeyer et al., 2009); amongst minority populations, the number of hCB transplants reaches 40% (Ballen et al., 2002). Due to limited volume, the absolute number of HSCs available in hCB specimens is only ~10% of that utilized in traditional BM transplants, leading to delayed engraftment and increased peri-transplant complications (Rocha and Broxmeyer, 2010). One approach to alleviate this problem is to transplant two unrelated hCB specimens (Ballen et al., 2007b). While this change correlated with improved adult engraftment rates, the time to engraftment was not shortened; engraftment following a hCB transplant can take > 50% longer than traditional HSC transplants (Broxmeyer et al., 2009). The identification of agents to increase hCB HSC homing, engraftment or total stem cell number is of significant therapeutic value.
Given this important clinical challenge, many investigators have sought to accelerate hCB HSC engraftment and blood count recovery after transplantation. The most clinically advanced approach thus far appears to be short-term culture with the notch ligand Delta (Delaney et al., 2010). This in vitro expansion procedure has been evaluated with significant evidence of success in an ongoing clinical trial; however, Delta treatment may lead to the depletion of long-term engrafting HSCs in the hCB unit, indicating that even this promising approach could need modifications before it can be broadly employed. In vitro expansion potential has also been described for insulin-like growth factor binding protein 2 in xenotransplantation studies (Zhang et al., 2008) and more recently for inhibition of the aryl hydrocarbon receptor (Boitano et al., 2010). Short ex vivo treatment of hCB with a chemical inhibitor of dipeptidylpeptidase IV (CD26) boosts homing to the hematopoietic niche to enhance engraftment in xenotransplantation models (Campbell et al., 2007). Rather than targeting HSCs, parathyroid hormone (PTH) has been used in vivo to enhance engraftment by modifying the murine osteoblastic HSC niche (Adams et al., 2007); PTH has also been used to safely facilitate stem cell mobilization in a clinical trial (Ballen et al., 2007a).
Murine hematopoietic transplantation assays are limited by the relatively short life span of mice compared to humans, enabling the detailed study of HSC function only over a limited time window. This often requires secondary transplant experiments to assess long-term HSC function, including self-renewal. Non-human primates have emerged as a valuable vertebrate model to perform longitudinal HSC transplantation studies. Engraftment and expansion can be examined after autologous transplantation of mobilized peripheral blood stem cells (MPBSCs). Further, efficient viral transduction techniques, utilizing fluorescent markers, allow the direct comparison of differently treated, uniquely labeled cell populations in an in vivo competitive transplantation assay (Donahue et al., 2005; Uchida et al., 2009). Significantly, the life span of non-human primates approaches that of humans, making it possible to study the long-term effects of different treatment modalities on the graft and host under conditions that approximate the demands on the human hematopoietic system (Trobridge and Kiem, 2010).
We previously demonstrated that dmPGE2 increased HSC number in vitro and in vivo (North et al., 2007). Murine limiting dilution competitive transplantation analysis demonstrated a 2 to 4-fold increase in HSC number following short ex vivo dmPGE2 exposure, without impacting multilineage hematopoietic differentiation, or decreasing serial transplantation and self-renewal potential (Hoggatt et al., 2009; North et al., 2007). dmPGE2 functions through cAMP-mediated regulation of the Wnt signaling pathway to control cell proliferation and apoptosis of HSCs in vivo (Goessling et al., 2009). Recent work has expanded these findings (Frisch et al., 2009; Hoggatt et al., 2009) to demonstrate the ability of dmPGE2 to modulate the BM niche and enhance homing.
Here, we present pre-clinical investigations of the safety and therapeutic potential of ex vivo dmPGE2 treatment to enhance human hematopoietic transplantation protocols. We found hCB HSCs differentially express PGE2 receptors, and dmPGE2-treatment elevates cAMP activity in human cells. In vitro, dmPGE2 treatment of CD34+ hCB decreased apoptosis, while significantly increasing HSC proliferation and hematopoietic colony formation. Further, dmPGE2 treatment enhanced the rates of human CD45+ chimerism in NOD/SCID mice xenotransplanted with whole or CD34+ hCB cells. Using CD34+ MPBSCs in a non-human primate (rhesus macaque) competitive autologous transplantation scheme, we showed that dmPGE2 treatment has no negative impact on HSC function, including multilineage repopulation, compared to matched controls at >1 year post infusion. Human and rhesus MPBSCs had reduced expression of EP2 and EP4 compared to hCB HSCs, however, each exhibited dose-responsive increases in cAMP activity after PGE2-treatment. Microarray gene expression analysis of human and rhesus CD34+ MPBSCs revealed conserved regulation of cell cycle, PGE2 pathway and HSC-related genes, representing a potential mechanism of action for dmPGE2; qPCR analysis of hCB samples after dmPGE2-stimulation confirmed similar gene regulation. These results predict dmPGE2 will be safe for use in clinical HSC transplantation protocols.
Results
hCB HSCs express PGE2 receptors and respond to exogenous PGE2 stimulation
We, and others, have shown PGE2 enhances HSC engraftment in isogenic murine transplantation models (Hoggatt et al., 2009; North et al., 2007). To examine the therapeutic potential of dmPGE2 for human HSC transplantation, we investigated the safety and efficacy of ex vivo exposure of hCB HSCs. To assess whether hCB was capable of responding to exogenous PGE2 stimulation, sub-fractionated samples were examined for the presence of each of the four PGE2 receptors. Quantitative PCR (qPCR) was performed using purified cell fractions contained within the umbilical cord: CD34+, CD133+, or mono-nuclear cells (MNC); human umbilical vein endothelial cells (HUVEC) were used as a non-hemogenic control population. The CD34+ population predominantly expressed EP2 and EP4 (Figure 1A); the level of EP4 expression is highly correlated with CD34+ status. EP2 and EP4 are similarly expressed in CD133+ hCB HSCs. Analysis of the total hCB MNC population showed maintenance of EP2 expression, while EP4 was significantly downregulated. In contrast, EP1 and EP3 are not significantly expressed in any hCB populations. SCL and RUNX1 were utilized to confirm that the CD34+ and CD133+ hCB cell fractions contained HSCs (Figure 1B).
Figure 1. hCB expresses prostaglandin receptors and responds to PGE2 signaling (see also Figure S1).
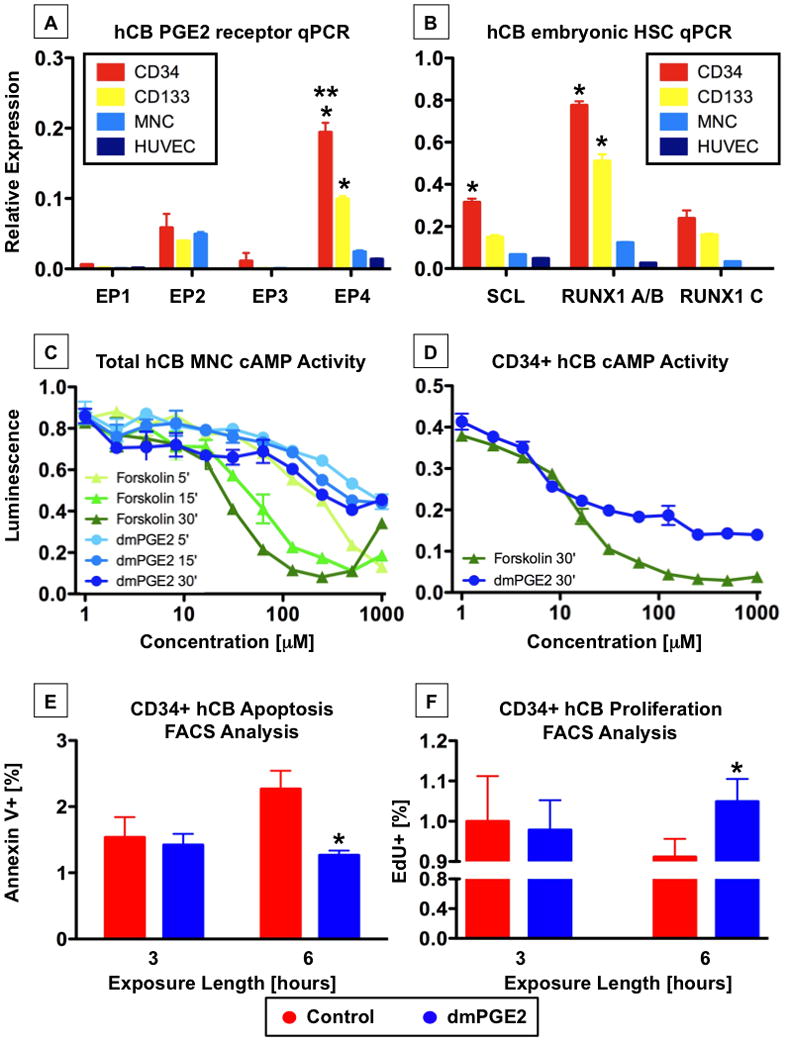
A-CD34+ cells have the highest expression of PGE receptors, particularly EP4. n=3; ANOVA, *p<0.001, compared to other EPs, **p<0.001, compared to other hCB cells
B-CD34+ cells express the immature HSC markers SCL and RUNX1. N=3; ANOVA, *p<0.001, compared to other hCB cells
C,D-Total hCB (C) and CD34+ hCB (D) cells were incubated with increasing concentrations of dmPGE2 or forskolin and subjected to a luminescence-based cAMP assay. cAMP levels increased in response to treatment (mean ±SD of 3–5 samples).
E-Annexin V FACS analysis revealed dmPGE2 exposure significantly decreased apoptotic CD34+ cells at 6hrs post treatment compared to controls. n=8, t-test; *p=0.024.
F-EdU incorporation measured by FACS was significantly enhanced in CD34+ cells in response to dmPGE2 exposure. n=8; t-test, *p=0.006.
Having established the distribution of PGE2 receptors in hCB, the intracellular response to dmPGE2 stimulation was examined. Upon ligand binding, EP2 and EP4, Gαs-coupled protein receptors, enhance intracellular cAMP levels (Regan et al., 1994) to initiate signaling cascades. We have recently shown that PGE2-mediated cAMP elevation regulates HSC number by modification of Wnt activity in vitro and in vivo (Goessling et al., 2009). To determine if cAMP elevation would occur in response to dmPGE2, whole hCB cells were exposed to increasing doses of dmPGE2 for 5, 15, and 30 minutes. dmPGE2 caused a dose-dependent increase in cAMP in hCB cells (Figure 1C) which is similar to the cAMP activator forskolin. Given the enrichment of EP2 and EP4 receptors in the CD34+ population, we directly examined cAMP responsiveness in this cell fraction; interestingly, the effective dose response occurred at a lower concentration in the CD34+ population (Figure 1D), possibly reflecting the high level of expression of EP4 on purified hCB HSCs. Importantly, both unfractionated and CD34+ hCB cells elicited rapid responses to dmPGE2 stimulation over the dose range tested.
PGE2 treatment was previously reported to have an anti-apoptotic effect on murine HSCs (Goessling et al., 2009; Hoggatt et al., 2009); to further analyze the conserved impact of dmPGE2 exposure, apoptosis and cell proliferation studies were conducted. Pooled CD34+ hCB samples were thawed, split for parallel processing, and treated with 1μM dmPGE2 or DMSO vehicle control. At 3 and 6 hours (hrs) after exposure, apoptosis was measured by AnnexinV FACS analysis. Both treatment cohorts demonstrated similar levels of apoptotic cell death at 3 hrs, however, by 6hrs cells treated with dmPGE2 showed a significant reduction in apoptosis compared to controls (Figure 1E, Supplemental Figure 1A). Complementary results were observed for cellular proliferation, where PGE2 caused significant changes in EdU incorporation at 6hrs post dmPGE2 treatment (Figure 1F, Supplemental Figure 1B).
dmPGE2 enhances hCB HSC function in vitro and in vivo
dmPGE2 was previously found to increase hematopoietic colony formation of murine ES cells (Goessling et al., 2009; North et al., 2007). To determine if dmPGE2 exposure impacted the functional potential of hCB samples, pooled CD34+ hCB cells were exposed to DMSO or dmPGE2 (1μM) for fixed incubation periods (15, 30, 60, 180, 360mins) then plated in triplicate on H4434 methylcellulose at limiting dilution (2000, 800, 320) (Figure 2A, Supplemental Table 1). The majority of hematopoietic colony types increased in response to dmPGE2; in particular, the GEMM population was significantly elevated after only 1 hr of dmPGE2 treatment. When colony numbers were combined and normalized for initial plating density, differences in total colonies across all replicates of the limiting dilution and dose-response assay demonstrated a 1.4-fold enhancement following dmPGE2 stimulation (Figure 2B). Together, the in vitro CFU-C data strongly suggest that dmPGE2 enhances colony forming potential of CD34+ cells.
Figure 2. dmPGE2 enhances hCB proliferation in vitro and engraftment in vivo (see also Figures S2-5 and Table S1).
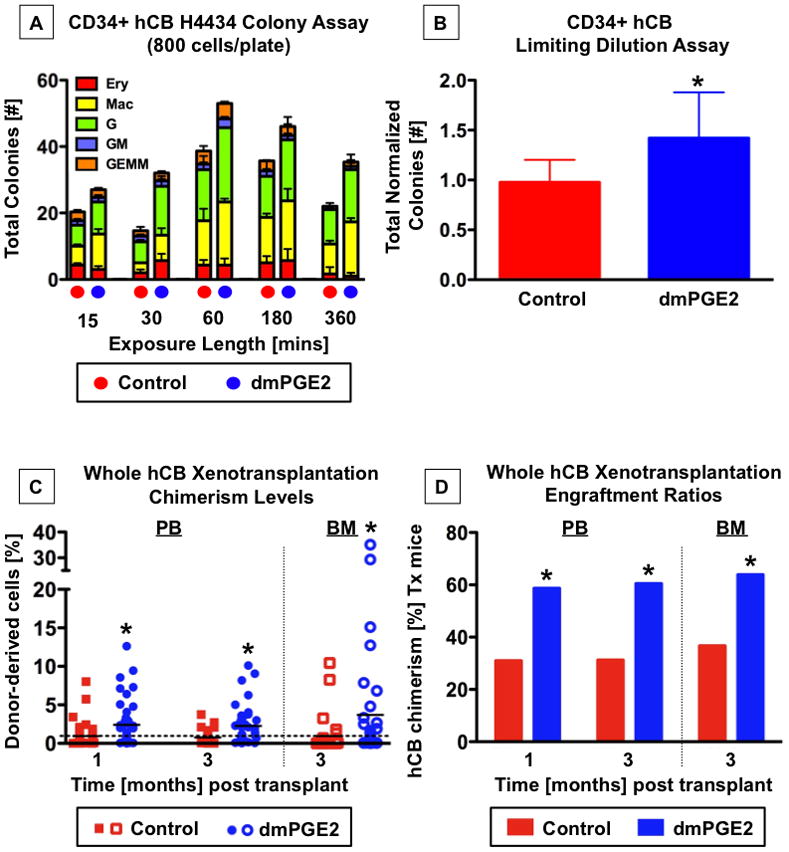
A,B-CD34+ hCB cells were exposed to 1μM dmPGE2 over a matrix of cell densities and exposure times.
A-Colony formation at day 13 was enhanced following dmPGE2 exposure for 15min to 6hrs; shown are the results of 800 cells/plate (n=3).
B-Normalization and combination of cell counts over all exposure times and cell densities revealed a significant increase in total colony number after dmPGE2 exposure (n=45; t-test, *p=0.018).
C,D-Whole hCB was transplanted into sublethally irradiated NOD/SCID mice. PB was analyzed at 1 and 3 months, and recipient BM at ≥3 months post transplantation. *=statistically different from control.
C-Individual hCD45+ chimerism values for mice receiving control (red squares) or dmPGE2-treated hCB (blue circles). The mean is indicated by a solid horizontal line; a dashed line shows the 1% cut-off for positive engraftment. PB-1 month: control 0.94±0.2414, dmPGE2 2.41±0.4118; t-test, p=0.0017; 3 months: control 0.76±0.1418, dmPGE2 2.27±0.3778; t-test, p=0.0004. BM-control 0.95±0.4373, dmPGE2 3.70±1.310; t-test, p=0.0348.
D-Cumulative summary of the % of NOC/SCID recipients engrafted with hCB. PB-1 month: control 13/42, dmPGE2 27/46; Fisher’s exact, p=0.011; 3 months: control 10/32, dmPGE2 23/28; Fisher’s exact, p=0.018; BM-control 11/30, dmPGE2 23/36; Fisher’s exact, p=0.047.
In limiting dilution competitive transplantation assays, dmPGE2 significantly increased the multi-lineage serial transplantable long-term repopulating ability of murine HSCs (Hoggatt et al., 2009; North et al., 2007). We examined the ability of dmPGE2 to enhance hematopoietic engraftment of hCB stem cells in vivo using a xenotransplantation model. To mimic clinical transplantation protocols, which primarily utilize unmanipulated (non-lineage depleted, non-NK/T-cell depleted, non-CD34 enriched) samples, and to assess the potential impact on all cell populations contained within a hCB unit, whole hCB samples were initially employed for xenotransplantation. Individual hCB units were split to account for cord-to-cord alterations in HSC content and viability, and treated in parallel ex vivo with dmPGE2 (10μM) or DMSO for 1hr in dextran/albumin suspension media; alterations in incubation time and media from our original studies (North et al., 2007) did not impact the effect of dmPGE2 stimulation in murine short-term CFU-S12 evaluations (Supplemental Figure 2A,B). To best mimic clinical conditions, recipient NOD/SCID mice were not conditioned to enhance engraftment beyond a standard regimen to clear the hematopoietic niche. Following sublethal irradiation (6.5Gy), NOD/SCID mice were transplanted with either 20 million matched dmPGE2-treated or control-treated whole hCB cells; this dose was chosen to detect positive effects of dmPGE2 on human chimerism in xenotransplant recipients based on prior whole hCB studies (Trowbridge et al., 2006). Human PB chimerism was evaluated by FACS analysis for hCD45; antibody reactivity >1% was used to identify positively engrafted recipients (Supplemental Figure 3A). At 1 and 3 months post transplant, more recipients of hCB cells treated ex vivo with dmPGE2 exhibited human CD45+ hematopoietic repopulation than recipients of matched controls, with overall higher average levels of PB chimerism (Figure 2C,D). hCD45+ BM engraftment >0.2% at 3 months post transplantation was also higher in recipients of dmPGE2-treated hCB (Figure 2C,D, Supplemental Figure 3B). Multi-lineage analysis of mice with hCB BM engraftment values >1% revealed contribution to each of the major blood lineages and the absence of lineage skewing in recipients of dmPGE2-treated cells; significant increases in the overall percentage of hCB derived phenotypic stem/progenitor cells, myeloid and T-cell lineages were observed compared to matched controls (Supplemental Figure 4A–C). Importantly, we were unable to detect any toxicity related to ex vivo dmPGE2 treatment over the duration of analyses: secondary transplants showed PB repopulation by PGE2-treated hCB cells; no excessive cell death, leukemic transformation, or disproportional loss of murine hosts were observed between recipients of dmPGE2-treated hCB and matched controls; and histological analysis of tissue (skin, liver, spleen, intestine, and bone) taken at the time of sacrifice showed no differences in cellular morphology, architecture or vascularity from controls (data not shown).
To confirm whether the enhanced hCB engraftment was due to direct effects on CD34+ HSCs and progenitors, fresh hCB units were enriched by MACS for hCD34 and split for parallel treatment with either dmPGE2 or DMSO control as indicated above. 2500 CD34+ cells were transplanted per recipient; this dose can provide repopulation without cytokine supplementation or the injection of “helper” cells, while testing the lower end of engraftment efficiency. As seen with whole hCB, dmPGE2 treatment led to a higher percentage of mice exhibiting hCD45+ PB and BM chimerism (Supplemental Figure 5A,B). Our results suggest that ex vivo treatment of whole hCB units with dmPGE2 will be safe and effective in achieving expansion of HSCs for transplantation in the clinical setting, due to preferential targeting of the CD34+ population.
Human and non-human primate mobilized peripheral blood stem cells can functionally respond to dmPGE2 treatment
To further assess the long-term safety of ex vivo dmPGE2 exposure for clinical transplantation protocols, a non-human primate model was employed. As CB samples are not routinely harvested from primates, safety evaluations were conducted using CD34+ MPBSCs isolated following a G-CSF/SCF conditioning protocol optimized for rhesus macaques (Donahue et al., 2005). To assess of the inherent ability of rhesus MPBSCs (rhMPBSC) to respond to PGE2 stimulation, sub-fractionated CD34+ samples were examined for the presence of each of the four PGE2 receptors by qPCR; results were compared to that observed on similarly mobilized human MPBSC (hMPBSC) samples. CD34+ hMPBSCs predominantly expressed EP2 and EP4 (Figure 3A), however, expression of EP4 in hMPBSC was relatively equal to that of EP2, and unlike hCB HSCs (see Figure 1), EP1 also was prominently expressed (Figure 3A). qPCR analysis of rhMPBSCs revealed 5-fold lower EP receptor expression in non-human primate HSCs than hMPBSCs, with EP2 particularly underrepresented (Figure 3B). However, using the luminescent assay described above, dose-dependent responses to dmPGE2 were observed for both MPBSCs populations; hMPBSC showed a comparable response to CD34+ hCB, while rhMPBSC required a slightly higher dose for cAMP activation (Figure 3C,D). These data indicate that human and rhesus CD34+ HSCs can react to dmPGE2.
Figure 3. PGE2 receptors and cAMP response are preserved between human and non-human primate peripheral blood stem cells.
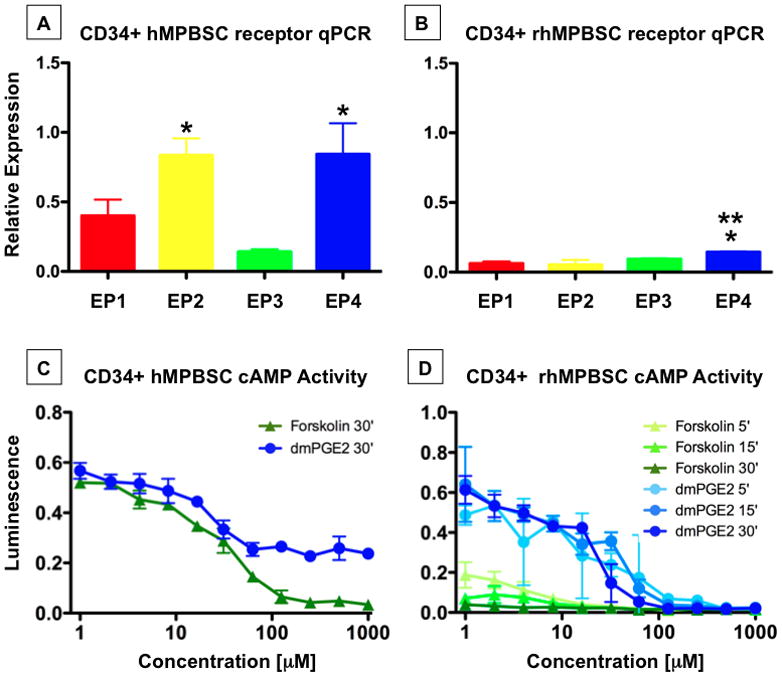
PB stem cells were mobilized from humans and non-human primates.
A-qPCR analysis showed EP2 and EP4 are equally and predominantly expressed in hMPBSC. n=3; ANOVA, *p<0.001 EP 2 or 4 vs EP 1 and 3.
B-qPCR revealed low EP receptor expression on rhesus MPBSC, with EP4 predominant. n=3–6; ANOVA, * p<0.001 vs. EP1, 2; **p=0.01 vs. EP3.
C,D-The cAMP response to dmPGE2 is preserved in human and rhesus MPBSC.
dmPGE2-treated rhMPBSCs show stable long-term multilineage engraftment in non-human primate competitive transplantation study
Receptor distribution and response assays suggested that the non-human primate model would be relevant to assess the potential long-term safety of the therapeutic use of dmPGE2. To examine the functional impact of dmPGE2 exposure on rhMBPSCs, a competitive autologous transplant study was performed. A two-pronged design scheme was developed to internally control for compound treatment and vector transduction parameters; reporter vectors utilized and transduction protocols were previously described in rhesus macaques (Uchida et al., 2009). CD34+ rhMPBSCs were split for parallel processing, and treated ex vivo with 10μM dmPGE2 or X-Vivo10 for 1hr either prior to or after transduction; dmPGE2 dose was selected based on prior murine (North et al., 2007) and xenotransplant data. Retroviral vectors expressing either EGFP or EYFP were transduced in order to differentiate the treatment groups in vivo; treatment/vector pairings were alternated between individuals to control for potential protocol-related bias. Transduction efficiencies varied between primates but were similar for EGFP and EYFP in each experiment (Supplemental Table 2). Treated, differentially-labeled cells were combined and reinfused into each donor for competitive repopulation analysis; PB samples were taken at fixed intervals beginning at >2-weeks post-transplantation, and the contribution of each labeled population followed via multi-lineage FACS analysis. Neither pre- nor post-transduction treatment with dmPGE2 negatively affected the engraftment potential of rhMPBSCs; both the percentage of total PB contribution of dmPGE2-treated cells, as well as that of each lineage remained present, stable and comparable to controls at all time points examined up to 1 year post-reinfusion (Figure 4A,B, Supplemental Figure 6). In all transplant recipients, reconstitution was uneventful and no toxicity was observed; cells transduced with EGFP showed greater PB engraftment then cells expressing EYFP, regardless of treatment. dmPGE2 was demonstrated to be safe to the long-term functional potential of rhMPBSCs.
Figure 4. dmPGE2 exposure of non-human primate MPBSC shows long-term safety (see also Figure S6 and Table S2).
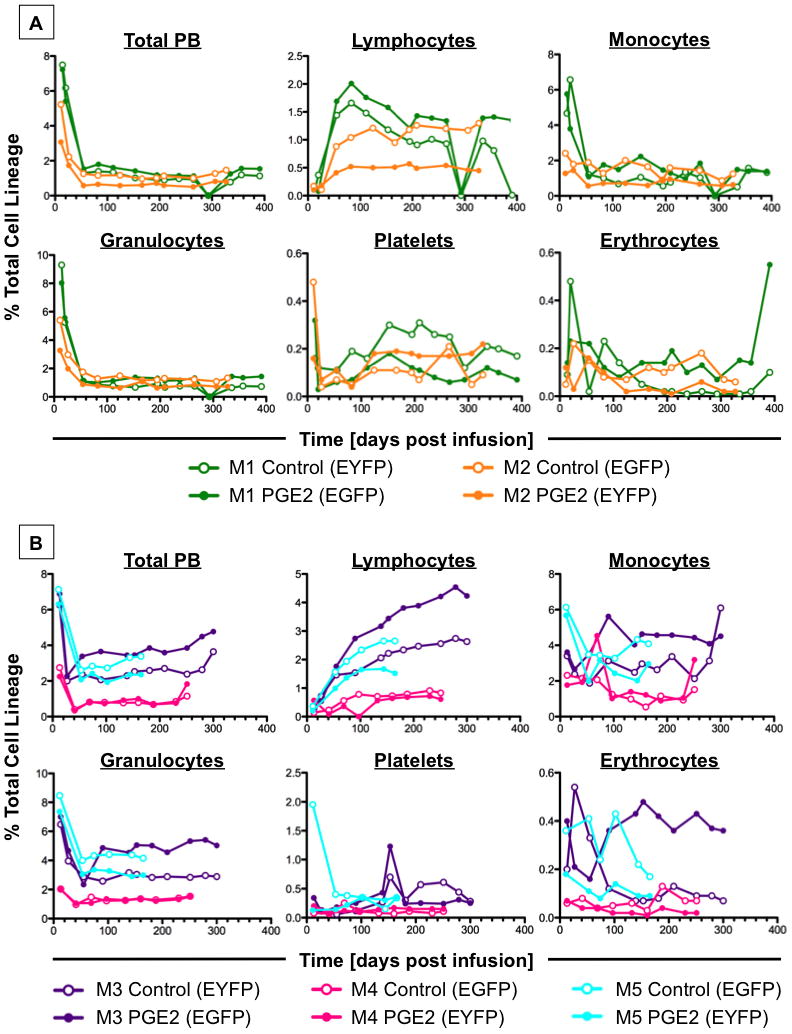
A-Whole PB and multilineage longitudinal FACS analysis of primates treated with dmPGE2 prior to transduction (n=2). Equivalent long-term function of fluorescently-marked control (open circles) and dmPGE2-treated MPBSCs (solid circles) was shown.
B-Whole PB and multilineage longitudinal FACS analysis of primates treated with dmPGE2 post transduction (n=3). Long-term survival and differentiation capacity is maintained irrespective of compound exposure (as indicated above).
Comparative genomic analysis of human and non-human primate CD34+ MPBSCs exposed to increasing doses of dmPGE2 shows high conservation of gene expression
In order to identify the potential mechanism of action of dmPGE2 on gene expression and HSC function, and to possibly explain the muted response of rhesus CD34+ MPBSCs to dmPGE2 exposure, microarray gene expression analysis was conducted. Human and rhesus CD34+ MPBSCs were exposed to DMSO or dmPGE2 (10 and 50μM dose), and processed for microarray analysis at 2, 6 and 12hrs post treatment (Supplemental Figure 7). 10μM dmPGE2 had minimal impact on gene expression in rhMPBSCs compared to treatment at 50μM, while significant changes were observed at each dose using hMPBSCs (Figure 5A–C). The greatest relative alterations in gene expression were observed for all treatments at 6hrs post exposure, so that time point was utilized for further comparative analysis. 110 genes were commonly regulated between human and rhesus MPBSCs, indicating an evolutionarily conserved response to PGE2 stimulation (Figure 5D; Supplemental Table 3A); among this set were genes representative of PGE2/cAMP signaling including: CREM, CREB, PDE4B, and PTGER2; several chemokines and cytokines including: CXCL1, CXCL3, CXCL10, IL1R1, IL18R1, IL23A and ANG; and many genes known to be specific to cell cycle regulation or hematopoietic cell differentiation. 262 genes were similarly regulated in human cells at both dmPGE2 doses (Figure 5D; Supplemental Table 3B), but not significantly altered in rhesus samples; among these were many major regulators of HSC development and maturation such as CXCR4, HHEX, HOXA9, JUNB, LCK, LMO2, LY6A, RUNX1 and TF. Considerable and exclusive overlap was also found between each of the PGE2 doses in human cells and that of the high dose in rhesus macaques (Figure 5D; Supplemental Table 3C,D); in particular, 10μM hMPBSC/50μM rhMPBSC show coordinated up regulation of genes like FLT3, JAK1, CCR1 and CD8a, perhaps indicative of enhanced proliferative and differentiation potential in the transplantation assays, while 50μM hMPBSC/50μM rhMPBSC displayed common regulation among genes influencing hematopoietic quiescence such as ANGPT1, ATXN1, FOXO1, HIF1A, STAT3 and VAV1. Together these data suggest that rhMPBSC may require a higher level of PGE2-stimulation for optimal HSC-related gene regulation than hMPBSCs, correlating with receptor distribution and transplant results.
Figure 5. Genome-wide expression analysis reveals commonly regulated genes in human and rhesus MPBSC (see also Figure S7 and Table S3).
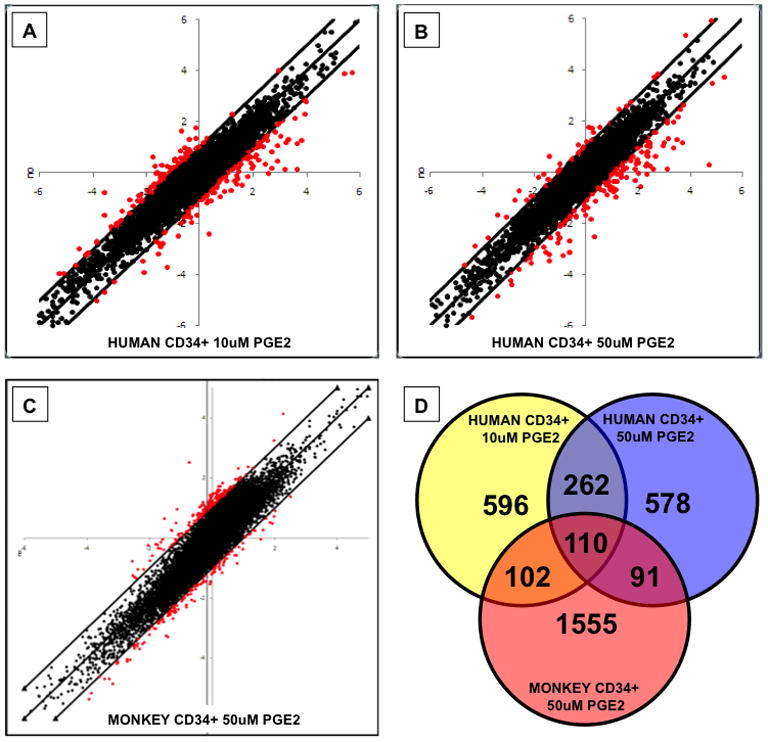
A–C-Scatter plots show genes significantly up- and down-regulated in response to dmPGE2 (x-axis) compared to controls (y-axis). Genes with >2-fold changes are highlighted in red.
D-Schematic display of genes regulated in response to dmPGE2 treatment, showing overlap between treatment groups of MPBSCs.
PGE2 regulates common signaling pathways in human and rhesus CD34+ MPBSC
To futher delineate the cellular signals mediated by dmPGE2, the microarray data were subjected to Ingenuity Pathway Analysis, which revealed a number of commonly up- (24) and down-regulated (12) pathways across all treatment groups, and in pairwise comparisons (Figure 6A,B, Supplemental Table 4A,B). Strikingly, among the up-regulated gene sets, 3 of the top 5 are directly influenced by cAMP-mediated signaling cascades (Figure 6C), suggesting high conservation of immediate early responses to PGE2 stimulation across vertebrate species (Goessling et al., 2009); chemokine/cytokine-related pathways were likewise represented by multiple gene families. The significance of the downregulated pathways is less clear, although several gene sets appear to be related to alternative differentiation potential, including that of endothelial cells (Figure 6D). A roughly equal number of pathways were commonly up-(17) and down-regulated (18) in hMPBSCs treated at varying dmPGE2 concentrations (Supplemental Table 5A,B). Through Ingenuity analysis, we again find that while many pathways are coordinately regulated between the 50 μM human MPBSC and rhesus treatment groups (33 up, 7 down), a number of pathways were conversely common only to the 10 μM PGE2 dose of hMPBSC and 50uM PGE2 in rhesus macaques (11up, 8 down) (Supplemental Tables 6A,B and 7A,B), indicating that PGE2-stimulation potential of rhesus CD34+ MPBSCs may be several fold lower than that of human CD34+ cells.
Figure 6. Ingenuity Pathway Analysis reveals conserved response patterns between human and rhesus MPBSCs (see also Tables S4-7).

Microarray data were subjected to Ingenuity Pathway Analysis.
A,B-Venn diagram illustrations of the overlap of significantly up- and down-regulated signaling pathways between humans and rhesus macaques MPBSCs.
C,D-List of significantly up- and down-regulated pathways common between human and rhesus dmPGE2 treated MPBSCs; the ratio indicates the fraction of affected genes of the number of total genes included in each pathway.
hCB exposed to dmPGE2 shows gene expression alterations in cell cycle, PGE2 signaling, and stem cell behavior which correlate to the MPBSC microarray analysis
To validate the gene expression signatures observed in the MPBSC analysis and to show conservation of the potential mechanism of action of dmPGE2 in hCB samples, we examined relative changes in gene expression following 10uM dmPGE2 treatment at fixed intervals post exposure (1, 3, 6 and 9hrs) by qPCR. To confirm that dmPGE2 was mediating effects on hCB cell proliferation and apoptosis, we examined a panel of 10 cell cycle related genes (Figure 7A). As previously shown for murine HSCs, expression of the classical WNT-pathway target and pro-proliferative cell cycle regulator, CYCLIN D1, increased following PGE2 treatment; CYCLIN E1 showed a similar response. Expression of the anti-apoptotic genes BCLXL and BCL2 were enhanced by PGE2, while that of the pro-apoptotic gene BAX decreased at 3 and 6 hrs. These data suggest one consequence of dmPGE2 treatment may be to preserve hCB cells in a viable state, capable of homing, engrafting and proliferating to repopulate the BM niche and reconstitute PB lineages.
Figure 7. dmPGE2 affects expression of cell cycle, PGE2-related and HSC genes.
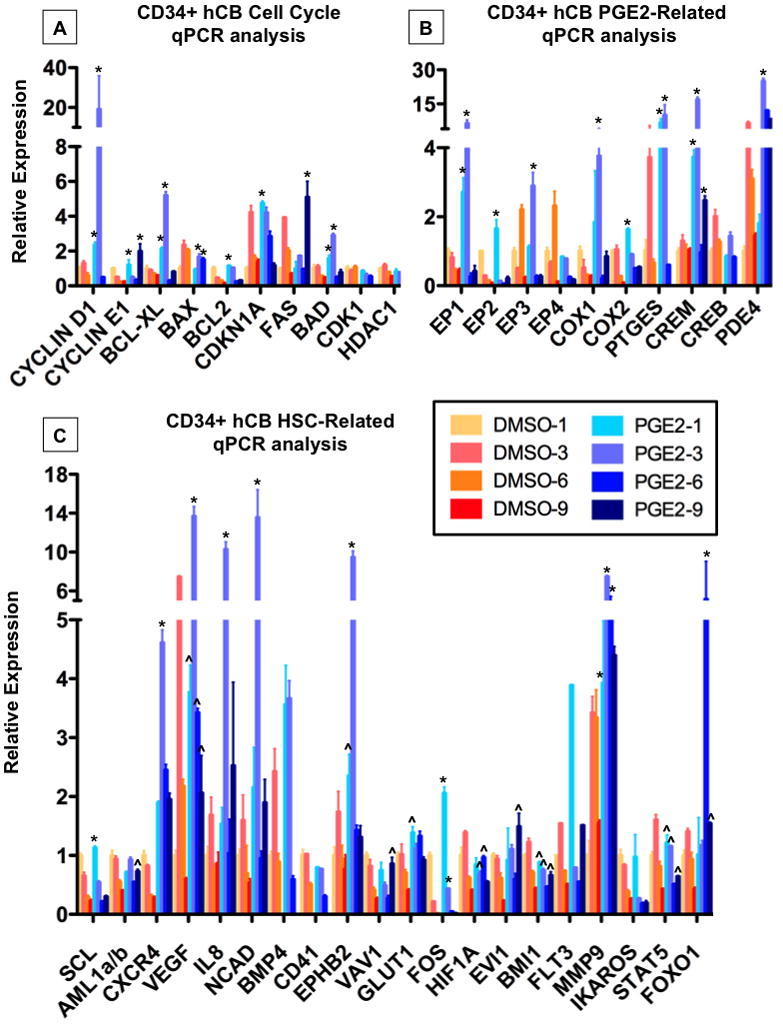
CD34+ hCB was incubated with 10μM dmPGE2 over a range of 1–9hrs; results of qPCR analysis are shown. n=3–5; ANOVA, *p<0.001 vs. all untreated controls. ^p<0.001 vs. control at corresponding time point.
A-dmPGE2 exposure altered expression of genes related to cell cycle and apoptosis.
B-dmPGE2 induced expression of PGE2 pathway related genes.
C-dmPGE2 affected genes associated with hematopoietic stem cell function.
We have previously shown that PGE2 not only enhances HSC formation and function, but is required for the normal kinetics of hematopoietic recovery (North et al., 2007). Clinical and murine studies have demonstrated elevated levels of PGE2 following hematopoietic transplantation (Ballinger et al., 2006; Cayeux et al., 1993). Additionally, PGE2 it is a well-characterized mediator of inflammatory responses to injury, where we have recently shown it can stimulate wnt activity and regeneration (Goessling et al., 2009). While PGE2 is capable of binding its four cognate receptors with high affinity, to rapidly elicit changes in cAMP-mediated signaling, it has an extremely short serum half-life. Here, following stimulation of CD34+ hCB cells with dmPGE2, we find that several components related to PGE2 production and signaling are specifically regulated (Figure 7B): dmPGE2 treatment induces expression of prostaglandin synthesis enzymes COX1 and 2, as well as the PGE specific endoperoxidase, PTGES, which could thereby enable a perpetuation of PGE2-mediated signaling. Conversely, the PGE2 receptors EP1 and EP3, generally considered to be negative response elements, were also upregulated in response to dmPGE2. These seemingly opposing responses could serve as a means to “fine tune” the extent and targets of PGE2-mediated signaling; similar counter-regulatory responses have been observed for pathways that partner with PGE2, such as WNT (Dichtel-Danjoy et al., 2009). In support of this concept, correlative responses were also observed for pathway components downstream of ligand binding: the cAMP response element (CREM), as well as phosphodiesterase 4D (PDE4D), which regulates the breakdown of cAMP, were 13- and 3.8-fold upregulated, respectively.
To assess which factors critical to HSC formation and function in a vascular niche, such as the umbilical cord, may be influenced by dmPGE2 to positively impact CD34+ hCB cells, we conducted qPCR expression analysis of matched samples following 10μM dmPGE2 treatment for a panel of 20 HSC-related genes identified in the literature and/or in the microarray analysis. CXCR4 expression was highly responsive to dmPGE2 exposure (Figure 7C), consistent with previous reports (Hoggatt et al., 2009). Vascular endothelial growth factor (VEGF) is a potent stimulator of endothelial and HSC proliferation, which regulates the maturation of the vascular endothelial niche, thereby enabling HSC induction from hemogenic endothelium (Gerber et al., 2002); VEGF expression was robustly increased, 3.7-fold, by dmPGE2. IL8 (CXCL8) is one of the inflammatory cytokines involved in proliferation and migration of endothelial progenitors and HSCs (Laterveer et al., 1995); IL8 expression increased up to 6-fold in response to dmPGE2. N-cadherin, which has a controversial role in adult HSC function (Li and Zon, 2010), but is expressed in the arterial HSC niche, was elevated up to 8.5-fold post dmPGE2 treatment. Expression of Bone-Morphogenic Protein-4 (BMP4), required to demarcate and specify the hemogenic endothelial population in the arterial niche (Durand et al., 2007), and that of Ephrin B2 (EPHB2) required for arterial niche identify (Lawson et al., 2001) were 3.5-fold and >9-fold upregulated, respectively. Expression of traditional markers of immature HSCs such as RUNX1 (AML1) and SCL decreased over time in untreated cells, consistent with the observation that HSCs progressively de-differentiate in culture; dmPGE2 limited the loss of RUNX1 expression and increased the expression of regulators of quiescence such as FOXO1 (Tothova et al., 2007), suggesting it may enable the hCB HSCs to maintain their identity or bias HSCs toward self-renewal.
Discussion
The data presented here suggest that ex vivo stimulation of human HSCs with dmPGE2 prior to transplantation may positively impact gene expression, engraftment capacity and HSC function in the clinical setting. While advantageous in both its availability and immunologic flexibility, hCB contains significantly fewer HSCs than BM or mobilized PB. The development of a rapid and safe means to enhance hCB HSC number and/or function has been an area of significant research. The data presented here, and previously (Goessling et al., 2009; Hoggatt et al., 2009; North et al., 2007), suggest that short ex vivo dmPGE2 treatment can enhance several factors that should improve human HSC function and multilineage hematopoietic recovery including homing, apoptosis, and proliferation. Interestingly, gene programs indicative of HSC production from a vascular niche (VEGF, BMP4) are also significantly upregulated or maintained (RUNX1) in response to dmPGE2 stimulation of hCB. As the CD34 antigen is expressed on both HSCs and hemogenic endothelium, this data could imply that a portion of the effect of PGE2 in hCB is mediated by endothelial cells, which have recently been shown to provide signals that retain HSCs in an undifferentiated state (Butler et al., 2010); dmPGE2 exposure could alternatively promote the “budding” or transition of immature HSCs from hemogenic endothelial precursors, similar to the original HSC screen in zebrafish (North et al., 2007). More work is needed to examine these intriguing alternatives, however, the maintenance of a significant number of PGE2-regulated gene programs across species suggests dmPGE2 will elicit beneficial regulatory responses over multiple hematopoietic contexts.
As recently discussed (McDermott et al., 2010), despite years of intensive investigation, the xenotransplantation model remains an approximation of human engraftment potential. As part of the preclinical evaluation of the safety of dmPGE2 for ex vivo treatment of hCB, we made several selective choices in the treatment scheme to optimize the potential to elucidate any effects-positive or negative- of dmPGE2 on the hCB sample. In particular, we chose to treat and transplant whole cord blood samples, in addition to purified CD34+ cells; this was done to mimic current clinical protocols, which use whole hCB units, and elucidate any unanticipated or unwanted effects on non-HSC cell-types contained in the cord. We observed no significant differences in negative outcomes between recipients of dmPGE2-treated versus control-treated cords, with or without CD34-enrichment. Despite the positive effect of dmPGE2 on both in vitro and in vivo models of hCB HSC function, these assays still cannot directly account for the variety of factors that influence the ultimate ability of a given hCB sample to engraft. The immunological consequences of HSC transplantation alone, notwithstanding the complexities added by the use of two independent immune systems in the setting of double cord transplantation, are still difficult to model; the standard isogenic murine competitive transplantation schemes do not account for immunological reactions, while cross-species transplantation introduces rejection and engraftment scenarios that do not exist in the clinical setting. Meaningful xenotransplantation of two unmatched hCB units to fully illustrate advantageous treatment protocols is not possible at this time as no specific criteria exists to reliably predict engraftment success of each individual cord unit prior to use in transplant assays.
The competitive autologous transplantation protocol in the non-human primate model enables the comparison of dual-treated, retrovirally marked HSC populations in vivo without the confounding influences of immunorejection. While these experiments are resource-intensive, prohibiting in vivo dose-finding studies to demonstrate a statistically significant benefit for dmPGE2, they allow for longitudinal evaluation of individual primates to assess durability, functionality and overall safety of the treated-HSC samples. Here, we show that cells treated with PGE2 prior to re-infusion are capable of contributing to stable multilineage repopulation at time points more than 1 year post injection, with an absence of any lineage skewing or compromise of HSC self-renewal potential. Long-term non-human primate transplantation experiments were initiated using 10μM dmPGE2 based on available safety and engraftment data from murine (North et al., 2007) and xenotransplantation models. While this treatment was not able to elicit a significant enhancement in repopulation potential compared to matched controls in the majority of rhesus macaques subjected to the protocol utilized, the result most likely reflects sub-optimal compound dosing rather than the inability of dmPGE2 to elicit functional responses in HSCs. Receptor distribution analysis obtained while the primate studies were ongoing revealed a significant difference in EP receptor expression between human and non-human primate CD34+ HSCs. Further, while 10μM dmPGE2 could elicit changes in cAMP activity, higher doses caused more dramatic effects, and statistically meaningful differences in gene expression were only observed at 50μM dmPGE2. We anticipate that use of 50μM dmPGE2 in the transplant protocol may cause a more discernable difference in repopulation potential in the autologous transplantation assay, but strong conservation of genetic regulation across species, and clear evidence of safety and efficacious results in alternate vertebrate models preclude the justification of repeating these studies in the non-human primate model to confirm these expectations.
Through our investigations, we identified EP4 as a novel marker highly enriched in the hCB CD34+ and CD133+ population. Additional studies are needed to define whether EP4 could be used alone or in combination to further refine the isolation of cells with in vivo hCB HSC function. In the setting of clinical hematopoietic transplantation, the entire hCB sample will be treated with dmPGE2 ex vivo prior to transplant; the selective distribution of EP4 indicates that despite the presence of multiple cell types in the whole hCB sample, the main effect of treatment is likely focused on the stem and progenitor cell populations. The fact that similar results were obtained for whole hCB and CD34+ cell xenotransplantation into NOD/SCID mice, further supports a targeted effect on non lineage-specified cell types. The selective distribution of the EP4 receptor to the CD34+ population in hCB samples will hopefully serve to minimize any potential negative side effects while mediating the full benefits of dmPGE2 stimulation on hematopoietic recovery in the clinical setting.
Many signaling pathways have been identified that regulate aspects of both HSC production and adult homeostasis, however, it is unclear if HSCs from a developmental vascular niche, such as CD34+ hCB, will respond predictably to stimulation from BM-derived factors in the clinic. No prospective study to date has examined whether the simple process of stem cell isolation prior to in vitro culture and transplantation may deprive HSCs of factors normally produced by supporting cell-types that influence their long-term function and maintenance in the patient. Similarly, a theoretical risk of any in vivo treatment approach is that it may not only affect the transplanted cells but also cells of the host, particularly surviving cancer cells, which could be more receptive to stimulation. One advantage of the brief ex vivo whole hCB unit incubation method is that no long-term manipulation or cell isolation is required, and there is limited potential of in vivo stimulation of persisting cancer cells in the transplant recipient. Based on the data presented herein, the FDA approved an investigational new drug (IND) application for dmPGE2 in April 2009; this represents the first compound discovered in Zebrafish to be used in patients. An independently conducted phase 1 clinical trial evaluating dmPGE2 in hCB transplantation is in progress (http://clinicaltrials.gov/ct2/show/NCT00890500). Further investigation will be needed to refine the ex vivo treatment approach for optimal clinical use in multicenter trials; furthermore, combinatorial strategies with other bourgeoning therapies may prove to elicit the largest benefit for the transplant patient while minimizing complications.
Experimental Procedures
16,16-dimethyl Prostaglandin E2 (dmPGE2)
dmPGE2, purchased from Cayman Chemicals, was resuspended in DMSO, and diluted to working concentrations of 1μM to 1mM as indicated; ex vivo dmPGE2 exposure duration was as indicated, followed by substrate removal and washing prior to analysis or use.
hCB Samples
Xenotransplant studies utilized fresh whole hCB samples discarded from clinical use (due to low cell number), obtained through the Center for Human Cell Therapy (Dana-Farber Cancer Institute, Boston, MA) and the Carolinas Cord Blood Bank (Duke University Medical Center, Durham, NC). Following red cell depletion (50–60%) and FACS-based viability assessment, samples were divided for parallel treatment. In vitro viability, proliferation, cell culture and gene expression analyses utilized frozen purified (CD34+, CD133+, MNC or HUVEC) hCB samples from pooled donors (Stem Cell Technologies). Cells were prepared to manufacturers directions, assessed for viability and split for use.
Apoptosis and Proliferation Analysis
5 million frozen CD34+ hCB cells were thawed, split and treated with 1μM dmPGE2 or DMSO in 2% FCS in IMDM for 3, 6 or 9 hrs at 37°C. Apoptosis was assessed by Annexin V-FITC (R&D Systems) and 7-AAD (BD Pharmingen) FACS. Proliferating cells were identified using the Click-iT EdU Flow Cytometry Assay Kit (Invitrogen).
Colony Forming Units-Culture
CD34+ CB cells were treated with DMSO or 1μM dmPGE2 for 15min, 30min, 1, 3, or 6hrs in 2% FCS-supplemented IMDM at 37°C. Cell suspensions were mixed with MethoCult H4434 (Stem Cell Technologies), plated in triplicate at limiting dilutions: 2000, 800, or 320 cells per 33 mm plate/1.5 ml H4434, and counted on day 13.
NOD/SCID Xenotransplantation
Individual whole or CD34-enriched hCB samples were split and exposed to dmPGE2 (10μM) or DMSO vehicle control ex vivo for 1 hr in dextran/albumin. Following sublethal irradiation (6.5Gy), 20 million whole or 2500 CD34+ hCB cells were introduced by retroorbital injection into NOD/SCID (Charles River) recipients. PB was obtained at 1 and 3 months, and BM isolated at >3 months post-transplant. Following red cell lysis, hCD45 (Becton-Dickenson) expression was examined by FACS; 1% PB and 0.2% BM hCB chimerism were utilized to indentify positively engrafted recipients.
Quantitative PCR (see also Supplemental Experimental Procedures 1)
CD34+ CB cells treated with DMSO or 1μM dmPGE2 for 1hr in IMDM with 2% FCS at 37°C were washed and incubated for a fixed time (+2, 5, or 8hrs) prior to RNA extraction (Trizol, Invitrogen). cDNA was synthesized with the SuperScript III first-strand synthesis kit (Invitrogen). Quantitative RT-PCR (58°C annealing) was performed in triplicate utilizing SybrGreen (Invitrogen) on the iQ5 Multicolor RTPCR Detection System (BioRad); primer sequences are listed in Supplemental Experimental Procedures 1. Data were normalized to β-actin and shown relative to DMSO at 1hr post-exposure.
Statistical Analysis
SigmaStat software was used for statistical analysis. Differences between treatments for all in vitro and qPCR analyses were calculated by t-test and one-way ANOVA. hCB engraftment in NOD/SCID mice was analyzed by Fisher’s Exact test (one-tailed).
Non-human Primate Cell Collection
Rhesus macaques (Macaca mulata) were housed and handled in accordance with the guidelines set by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (DHHS publication No. NIH 85-23). PB HSCs were mobilized with granulocyte colony-stimulating factor (G-CSF) and stem cell factor (gift of Amgen Inc.), harvested by leukapheresis, and enriched for CD34+ cells as previously described (Donahue et al., 2005).
Human CD34+ cell collection
hMPBSCs were collected following G-CSF mobilization, and enriched by CD34+ immunoselection according to IRB approved protocols (02-H-0160, 96-H-0049).
Microarray analysis
Human and non-human primate MPBSCS were incubated with either 10 or 50μM dmPGE2 in X-Vivo media (Lonza) for 1hr. Cells were collected at 6, 12 and 24hrs post incubation, and total RNA (Trizol extraction) was co-hybridized to custom-made 17.5K cDNA (UniGene cluster) microarrays. The complete list of genes in the Hs-CCDTV-17.5k-1px printing is available at: http://nciarray.nci.nih.gov/galfiles. Hierarchical cluster analysis and TreeView software were used for visualization. Gene clustering analysis was performed using Ingenuity Pathway Analysis software.
Autologous competitive transplant study (see also Supplemental Experimental Procedures 2)
Rhesus macaques (n=5) were mobilized daily with 10μg/kg G-CSF and 200μg/kg stem cell factor (SCF) for 5 days. On day 5, CD34+ enriched MPBSC were isolated from leukapheresed blood as described (Donahue et al., 2005). CD34+ cells from each donor were split and transduced with a chimeric HIV vector to express EGFP or EYFP (Uchida et al., 2009). Rhesus CD34+ cells were treated with 10μM dmPGE2 for 1hr at 37°C either prior to or post viral transduction (Supplemental Experimental Procedures 2). Cells were re-infused on day 3 following leukapheresis, after 2 consecutive days of 500 Gy total body irradiation.
Supplementary Material
Highlights.
Human cord blood and rhesus macaque CD34+ HSCs respond to dmPGE2 stimulation
dmPGE2 enhances colony number and xenotransplants of unfractionated and CD34+ hCB
dmPGE2-treated non-human primate HSCs show long-term multilineage engraftment
Preclinical safety evidence on dmPGE2 supports initiating a phase 1 clinical trial
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams GB, Martin RP, Alley IR, Chabner KT, Cohen KS, Calvi LM, Kronenberg HM, Scadden DT. Therapeutic targeting of a stem cell niche. Nat Biotechnol. 2007;25:238–243. doi: 10.1038/nbt1281. [DOI] [PubMed] [Google Scholar]
- Ballen KK, Hicks J, Dharan B, Ambruso D, Anderson K, Bianco C, Bemiller L, Dickey W, Lottenberg R, O’Neill M, et al. Racial and ethnic composition of volunteer cord blood donors: comparison with volunteer unrelated marrow donors. Transfusion. 2002;42:1279–1284. doi: 10.1046/j.1537-2995.2002.00191.x. [DOI] [PubMed] [Google Scholar]
- Ballen KK, Shpall EJ, Avigan D, Yeap BY, Fisher DC, McDermott K, Dey BR, Attar E, McAfee S, Konopleva M, et al. Phase I trial of parathyroid hormone to facilitate stem cell mobilization. Biol Blood Marrow Transplant. 2007a;13:838–843. doi: 10.1016/j.bbmt.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Ballen KK, Spitzer TR, Yeap BY, McAfee S, Dey BR, Attar E, Haspel R, Kao G, Liney D, Alyea E, et al. Double unrelated reduced-intensity umbilical cord blood transplantation in adults. Biol Blood Marrow Transplant. 2007b;13:82–89. doi: 10.1016/j.bbmt.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger MN, Aronoff DM, McMillan TR, Cooke KR, Olkiewicz K, Toews GB, Peters-Golden M, Moore BB. Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J Immunol. 2006;177:5499–5508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE, Cooper S, Hass DM, Hathaway JK, Stehman FB, Hangoc G. Experimental basis of cord blood transplantation. Bone Marrow Transplant. 2009;44:627–633. doi: 10.1038/bmt.2009.285. [DOI] [PubMed] [Google Scholar]
- Broxmeyer HE, Douglas GW, Hangoc G, Cooper S, Bard J, English D, Arny M, Thomas L, Boyse EA. Human umbilical cord blood as a potential source of transplantable hematopoietic stem/progenitor cells. Proc Natl Acad Sci U S A. 1989;86:3828–3832. doi: 10.1073/pnas.86.10.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, Seandel M, Shido K, White IA, Kobayashi M, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TB, Hangoc G, Liu Y, Pollok K, Broxmeyer HE. Inhibition of CD26 in human cord blood CD34+ cells enhances their engraftment of nonobese diabetic/severe combined immunodeficiency mice. Stem Cells Dev. 2007;16:347–354. doi: 10.1089/scd.2007.9995. [DOI] [PubMed] [Google Scholar]
- Cayeux SJ, Beverley PC, Schulz R, Dorken B. Elevated plasma prostaglandin E2 levels found in 14 patients undergoing autologous bone marrow or stem cell transplantation. Bone Marrow Transplant. 1993;12:603–608. [PubMed] [Google Scholar]
- Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med. 2010;16:232–236. doi: 10.1038/nm.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtel-Danjoy ML, Caldeira J, Casares F. SoxF is part of a novel negative-feedback loop in the wingless pathway that controls proliferation in the Drosophila wing disc. Development. 2009;136:761–769. doi: 10.1242/dev.032854. [DOI] [PubMed] [Google Scholar]
- Donahue RE, Kuramoto K, Dunbar CE. Large animal models for stem and progenitor cell analysis. Curr Protoc Immunol. 2005;Chapter 22(Unit 22A):21. doi: 10.1002/0471142735.im22a01s69. [DOI] [PubMed] [Google Scholar]
- Durand C, Robin C, Bollerot K, Baron MH, Ottersbach K, Dzierzak E. Embryonic stromal clones reveal developmental regulators of definitive hematopoietic stem cells. Proc Natl Acad Sci U S A. 2007;104:20838–20843. doi: 10.1073/pnas.0706923105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch BJ, Porter RL, Gigliotti BJ, Olm-Shipman AJ, Weber JM, O’Keefe RJ, Jordan CT, Calvi LM. In vivo prostaglandin E2 treatment alters the bone marrow microenvironment and preferentially expands short-term hematopoietic stem cells. Blood. 2009;114:4054–4063. doi: 10.1182/blood-2009-03-205823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002;417:954–958. doi: 10.1038/nature00821. [DOI] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood. 2009;113:5444–5455. doi: 10.1182/blood-2009-01-201335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laterveer L, Lindley IJ, Hamilton MS, Willemze R, Fibbe WE. Interleukin-8 induces rapid mobilization of hematopoietic stem cells with radioprotective capacity and long-term myelolymphoid repopulating ability. Blood. 1995;85:2269–2275. [PubMed] [Google Scholar]
- Laver JH, Hulsey TC, Jones JP, Gautreaux M, Barredo JC, Abboud MR. Assessment of barriers to bone marrow donation by unrelated African-American potential donors. Biol Blood Marrow Transplant. 2001;7:45–48. doi: 10.1053/bbmt.2001.v7.pm11215698. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- Li P, Zon LI. Resolving the controversy about N-cadherin and hematopoietic stem cells. Cell Stem Cell. 2010;6:199–202. doi: 10.1016/j.stem.2010.02.007. [DOI] [PubMed] [Google Scholar]
- McDermott SP, Eppert K, Lechman E, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116:193–200. doi: 10.1182/blood-2010-02-271841. [DOI] [PubMed] [Google Scholar]
- North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46:213–220. [PubMed] [Google Scholar]
- Rocha V, Broxmeyer HE. New approaches for improving engraftment after cord blood transplantation. Biol Blood Marrow Transplant. 2010;16:S126–132. doi: 10.1016/j.bbmt.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Trobridge GD, Kiem HP. Large animal models of hematopoietic stem cell gene therapy. Gene Ther. 2010;17:939–948. doi: 10.1038/gt.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med. 2006;12:89–98. doi: 10.1038/nm1339. [DOI] [PubMed] [Google Scholar]
- Uchida N, Washington KN, Hayakawa J, Hsieh MM, Bonifacino AC, Krouse AE, Metzger ME, Donahue RE, Tisdale JF. Development of a human immunodeficiency virus type 1-based lentiviral vector that allows efficient transduction of both human and rhesus blood cells. J Virol. 2009;83:9854–9862. doi: 10.1128/JVI.00357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CC, Kaba M, Iizuka S, Huynh H, Lodish HF. Angiopoietin-like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood. 2008;111:3415–3423. doi: 10.1182/blood-2007-11-122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.