

Abstract
Förster (fluorescence) resonance energy transfer (FRET) and fluorescence polarization (FP) are widely used technologies for monitoring bimolecular interactions and have been extensively used in high-throughput screening (HTS) for probe and drug discovery. Despite their popularity in HTS, it has been recognized that different assay technologies may generate different hit lists for the same biochemical interaction. Due to the high cost of large-scale HTS campaigns, one has to make a critical choice to employee one assay platform for a particular HTS. Here we report the design and development of a dual-readout HTS assay that combines two assay technologies into one system using the Mcl-1 and Noxa BH3 peptide interaction as a model system. In this system, both FP and FRET signals were simultaneously monitored from one reaction, which is termed “Dual-Readout F2 assay” with F2 for FP and FRET. This dual-readout technology has been optimized in a 1,536-well ultra-HTS format for the discovery of Mcl-1 protein inhibitors and achieved a robust performance. This F2 assay was further validated by screening a library of 102,255 compounds. As two assay platforms are utilized for the same target simultaneously, hit information is enriched without increasing the screening cost. This strategy can be generally extended to other FP-based assays and is expected to enrich primary HTS information and enhance the hit quality of HTS campaigns.
Introduction
Protein–protein interactions are involved in the control of diverse physiological and pathological processes in living organisms such as cell apoptosis and proliferation, which represent an emerging class of molecular targets for novel drug discovery.1 To monitor molecular interactions, a number of assay technologies have been developed, such as time-resolved Förster (or fluorescence) resonance energy transfer (TR-FRET), fluorescence polarization (FP), and surface plasmon resonance.2–5 These assay technologies, particularly in homogenous format, have been extensively used in high-throughput screening (HTS) campaigns for the identification of new chemical entities in the drug discovery field and new molecular probes for chemical biology studies.5 However, the application of different assay technologies often gives rise to different hit lists even when monitoring the same biochemical interaction. Because of the high cost of screening large chemical libraries, HTS campaigns are often conducted in a single-point format and investigators are forced to choose a single-assay technology. To enhance the efficiency of HTS campaigns, we have designed and developed a novel HTS technology that allows the generation of two HTS readouts from one reaction by combining FRET and FP technologies into one platform. This technology is termed dual-readout F2 assay, where F2 standing for FRET and FP. We have further miniaturized the F2 assay to a 1,536-well ultra-HTS (uHTS) format. To provide a proof of concept, this F2 uHTS assay technology was used to monitor the interaction of Mcl-1 and Noxa for the eventual goal of discovering the next generation of small molecule modulators of apoptosis.
Apoptosis, or programmed cell death, is a critical process in both development and homeostasis of multicellular organisms.6 Alterations in apoptotic pathways can disrupt the delicate balance between cell proliferation and cell death and lead to a variety of diseases.6,7 Mcl-1 belongs to the prosurvival Bcl-2 subfamily along with Bcl-XL, Bcl-2, Bcl-w, and A1.7–9 Mcl-1 is overexpressed in many human cancers and its overexpression contributes to chemoresistance and disease relapse.10–12 Recently, a number of groups have reported the discovery of small-molecules known as BH3 mimetics, which induce apoptosis by inhibiting antiapoptotic Bcl-2 family members.13–23 This family of molecules demonstrates a wide range of both potency and selectivity for different antiapoptotic Bcl-2 proteins. However, there is still a need for developing BH3 mimetics that can efficiently and selectively target Mcl-1 protein.
One of the essential elements in discovering and identifying small-molecule Mcl-1 inhibitors is the development of a robust, quantitative, and high-throughput assay for evaluation of the binding affinities of potential small molecule inhibitors. In vitro binding studies have demonstrated that BH3 peptides from pro-apoptotic proteins exhibit preferences in binding to anti-apoptotic proteins (Bcl-2, Bcl-xL, and Mcl-1).24 Noxa BH3 peptide is highly selective for Mcl-1 and Bcl-2A1 proteins (within the nM range) but does not bind detectably to the other members of this family (>100 μM).24 Recently published structures of Mcl-1 in complex with the Noxa and Puma BH3 domains demonstrate that Noxa specifically targets Mcl-1 and exploits a basic patch unique to the Mcl-1 sequence.25 These interactions between Mcl-1 and the Noxa BH3 peptide form the basis for the design of the dual-readout F2 assay, which can be used to screen for small molecule inhibitors that selectively disrupt the interaction of Mcl-1 protein and Noxa.
Materials and Methods
Peptides
All the peptides were synthesized at Emory Microchemistry and Proteomic Facility. The 26-mer Noxa peptide (residues 18–43: PAELEVECATQLRRFGDKLNFRQKLL-NH2) used in this study was synthesized and labeled with 5/6-carboxytetramethyl-rhodamine (TMR). The nonlabeled Noxa peptide and 21-residue Bid-BH3 peptide (residues 79–99; QEDIIRNIARHLAQVGDSMDR-NH2) were synthesized and used as peptide antagonists.
Expression and Purification of Recombinant Mcl-1 Protein
Human Mcl-1 cDNA was purchased from Origene. The Mcl-1 fragment, amino acid residues 171–327, was cloned into the pHis-TEV vector (a modified pET vector) through BamHI and EcoRI sites, using the oligonucleotides: 5′-CGGGATCCGAGGACGAGTTGTA CCG-GCAG-3′ and 5′-GGAATTCCTAGCCAC-CTTCTAGGTCCTC TAC-3′. Mcl-1 protein with an N-terminal 6xHis tag was produced in Escherichia coli BL21 (DE3) cells. Cells were grown at 37°C in 2xYT containing antibiotics to an OD600 of 0.6. Protein expression was induced by 0.4 mM isopropyl β-D-1-thiogalactopyranoside at 37°C for 4 h. Cells were lysed in 50 mM Tris pH 8.0 buffer containing 500 mM NaCl, 0.1% bME, and 40 μL of Leupeptin/Aprotin. Mcl-1 protein was purified from the soluble fraction using Ni-NTA resin (Qiagen), following the manufacturer's instructions. The protein was further purified on a Source Q15 column (resin and column are from Amersham Biosciences) in 25 mM Tris pH 8.0 buffer, with NaCl gradient. The purified recombinant Mcl-1 protein was stored at −80°C in following buffer: 25 mM Tris pH 8.0, 100 mM NaCl, 5 mM DTT, and 25% glycerol.
Dual FRET/FP Measurements
TR-FRET and FP measurements were performed using black 1,536-well plates (Corning Costar, Cat# 3724) in an Envision Multilabel plate reader (Perkin Elmer Life Sciences). To each well, a mixture of TMR-Noxa peptide, Mcl-1 protein, and terbium-anti-His antibody (His-Tb) were added to a final volume of 5 μL in the assay buffer (20 mM Tris buffer, pH 7.5, 50 mM NaCl, and 0.01% NP40). The TR-FRET and FP signals were measured from the same well either in TR-FRET or FP module with an Envision Multilabel plate reader.
In the TR-FRET readout, Mcl-1 protein with a 6xHis tag was indirectly labeled with the terbium through a terbium-conjugated anti-6xHis antibody (LanthaScreen™; Invitrogen). Terbium and rhodamine comprise a fluorescence energy transfer pair. Interaction of Mcl-1 protein with TMR-Noxa peptide brings two conjugated fluorophores into proximity, leading to an energy transfer from terbium to rhodamine and the generation of FRET signals (Fig. 1). FRET signal is detected in an Envision Multilabel plate reader with a laser excitation at 337 nm, and emission at 545–7 and 572–7.5 nm to measure the fluorescence signal from terbium and rhodamine, with a dual-dichroic mirror at 400/555 nm. The delay time is set at 100 μs. The TR-FRET signal is expressed as TR-FRET signal ratio: F572/F545 nm × 104, where F572 and F545 nm are fluorescence counts at 572 and 545 nm for rhodamine and terbium, respectively. The TR-FRET signal window was calculated as the difference between the TR-FRET signal values for bound TMR-Noxa (TMR-Noxa peptide with Mcl-1) and the TR-FRET signal values for the free TMR-Noxa (TMR-Noxa peptide only without Mcl-1).
Fig. 1.

Schematic diagram of the design for the dual-readout F2 assay technology for monitoring bimolecular interactions with the Mcl-1 protein and Noxa peptide interaction as a model system. Mcl-1 protein is labeled with a FRET donor terbium, whereas its binding partner, Noxa peptide, is labeled with TMR. Binding of Mcl-1 protein to TMR-Noxa peptide brings donor (terbium) and acceptor (TMR) fluorophores together, leading to the energy transfer from terbium to TMR upon excitation at 337 nm and generating TR-FRET signal (A). Meanwhile, binding of Mcl-1 protein to TMR-Noxa peptide slows down the movement of the TMR-Noxa peptide in the same reaction, leading to the generation of FP signal (B). FP, fluorescence polarization; FRET, fluorescence resonance energy transfer; TMR, 5/6-carboxytetramethyl-rhodamine; TR-FRET, time-resolved fluorescence resonance energy transfer.
In the FP readout, the same reaction mixtures for TR-FRET readout were used to measure the FP signals using the FP module (Fig. 1). The binding of the Mcl-1 protein to rhodamine-labeled Noxa-peptide slows down the rotation of the TMR-Noxa peptide and generates the FP signal upon excitation by a polarized light.26 The assay wells for FP readout were the same as for TR-FRET experiment. For TMR-Noxa peptide, an excitation filter at 531–25 nm and dual-emission filters (p and s) at 595–60 nm were used with a dual-dichroic mirror at 555/595 nm. All FP signals were recorded and expressed as millipolarization (mP) units. The FP assay window was calculated by subtracting the mP values recorded for free TMR-Noxa peptide from mP values recorded for bounded TMR-Noxa peptide in the presence of Mcl-1 protein. The FP data were corrected with G factor as optimized for the Envision Multilabel plate reader to avoid instrument artifacts as recommended.27,28
Development and Optimization of the Dual-Readout F2 Assay for Mcl-1 Protein
The key point for the dual F2 assay development was to determine the concentrations of the binding partners at the optimal conditions for both readouts. We first determined the optimal concentrations of the TMR-Noxa peptide. Increasing concentrations of TMR-Noxa peptide with terbium-anti-His antibody (2 nM) were mixed with buffer in the presence or absence of Mcl-1 protein (50 nM). A 5 μL of the mixture was dispensed to each well of 1,536-well plates. The dual TR-FRET/FP measurements were then performed as described above.
To determine the equilibrium binding of TMR-Noxa peptide and Mcl-1 protein using dual TR-FRET and FP measurements, increasing amounts of Mcl-1 protein were incubated with TMR-Noxa peptide (125 nM) terbium-anti-His antibody (2 nM) at room temperature for 1 h. The dual TR-FRET and FP measurements were performed as described above. All experimental data were analyzed using Prism 5.0 software (Graphpad Software) and the IC50s were determined by nonlinear curve fitting as the concentration of the Mcl-1 protein at which 50% of the ligand is bound.
Assay Validation with Unlabeled Peptide Antagonists in the Dual TR-FRET/FP Assay
To verify the specificity of the dual TR-FRET/FP Mcl-1 binding assay, two peptide antagonists, nonlabeled Noxa and Bid peptides, were used to test their ability to compete with the binding of TMR-Noxa peptide to Mcl-1 protein measured by the dual TR-FRET and FP. An increasing concentration of nonlabeled Noxa (0.5 μL) or Bid peptide diluted in dimethyl sulfoxide (DMSO) was added to 4.5 μL of the reaction containing Mcl-1 protein (62.5 nM), TMR-Noxa peptide (125 nM), and terbium-anti-His antibody (2 nM). After incubating at room temperature for 1 h, the TR-FRET and FP signals were measured. For each assay, background reaction (in the absence of Mcl-1) and control reaction (containing vehicle, DMSO, without competition peptide) were included in each assay plate. The competitive effect of the peptides on binding was expressed as percentage of control TR-FRET or FP signal as the following:
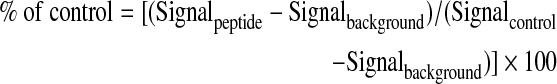 |
Data were plotted against log10 values of peptide concentrations and analyzed using Prism 5.0 software (Graphpad Software). IC50 values were determined by nonlinear curve fitting as the concentrations of the peptides at which 50% of control FRET or FP signal was inhibited.
High-Throughput Assay Performance
To evaluate the quality and suitability of the dual-readout F2 Mcl-1 binding assay for HTS, the Z′ factor was calculated for both TR-FRET and FP measurement based on the following equation: Z′ factor = 1–(3SDb + 3SDf)/(μb – μf), where SDb and SDf are the standard deviations for bound (b) and free (f) peptides without Mcl-1 protein, whereas μb and μf are the mean FRET or FP signals for bound and free peptides, respectively. The Z′ factor reflects the quality of the assay itself without intervention of test compounds. To monitor assay sensitivity, the signal-to-noise ratio (S/N) was calculated for FP measurement as follows: S/N = (μb – μf)/(SD2b + SD2f)0.5 and the signal-to-background ratio (S/B) was determined for TR-FRET assay using the following equation: S/B = μb/μf.
Validation of the Dual-Readout F2 Assay in 1,536-Well uHTS Format
For the validation of the dual-readout F2 assay in uHTS format, a library of 102,255 compounds from Molecular Library Screening Center Network was tested. About 4.5 μL of assay reaction buffer that contains Mcl-1 protein (62.5 nM), TMR-Noxa (125 nM), and terbium-anti-His (2 nM) was dispensed into 1,536-well assay plates using a MultiDrop Combi (Thermo-Fisher Scientific). Then, compounds (0.1 μL; 1 mM in 100% DMSO) in 384-well storage plates (Corning) were transferred to 1,536-well assay plates using a PinTool (VP Scientific) integrated with Beckman NX liquid handler (Beckman Coulter). The reactions were mixed thoroughly using the shaker integrated with BeckmanNX and incubated at room temperature for 1 h. The plates were delivered through integrated robotic systems to the Envision multimode reader as described above for recording TR-FRET and FP signals. Screening data were analyzed using the CambridgeSoft Software (CambridgeSoft Corporation).
Results and Discussions
Design of the Dual-Readout F2 Assay
FRET and FP are two most widely used fluorescence-based homogenous HTS technologies for monitoring bimolecular interactions. FRET is a nonradioactive, photophysical effect in which energy that is absorbed by a donor fluorophore is transferred to an acceptor fluorophore.29 Energy transfer from donor to acceptor fluorophores occurs when (1) the emission spectrum of the donor fluorophore significantly overlaps with the absorption spectrum of the acceptor fluorophore; (2) the donor and acceptor transition dipole orientations are approximately parallel; and (3) the donor and acceptor molecules are in close proximity (10–100 Å). By coupling the donor and acceptor fluorophores to two interacting molecules, the fluorophores may be brought into close proximity and induce a FRET signal. The attractive feature of TR-FRET is the time delay of the signal measurement, which reduces the prompt autofluorescence from tested library compounds. FP is a sensitive nonradioactive technique for the study of biomolecular interactions in solution. When fluorescent small molecules (such as tagged peptides) in solution are bound by large molecules (such as a protein), the movement of the resulting complex becomes slower. In this way, the binding of a fluorescently labeled peptide to a protein can be monitored by the change in emitted polarization signals.
TR-FRET or FP assay format is usually employed individually for a specific HTS campaign. However, it is well documented that different assay and detection technologies, such as FRET versus FP, can generate quite different hit lists for the same biochemical interaction.30–32 To enhance the quality of the hit list from primary screens, this study evaluated the feasibility of combining two assay formats, FRET and FP, into one HTS reaction, in which both FRET and FP signals were read from the same well. The Mcl-1 and Noxa interaction involved in apoptosis regulation was used as a model system. For the F2 assay design, epitope-tagged Mcl-1 protein is indirectly labeled with a TR-FRET donor, terbium, through terbium-conjugated anti-epitope-antibody (Fig. 1). Noxa peptide is directly labeled with a corresponding acceptor fluorophore, TMR, for terbium pairing in this case. For the TR-FRET readout, the binding of TMR-labeled Noxa peptide to Mcl-1 bring donor (terbium) and acceptor (TMR) into proximity, leading to the energy transfer from terbium to TMR upon excitation and the generation of the FRET signal, which can be detected at 572 nm. The same TMR-labeled Noxa peptide used as TR-FRET acceptor serves as fluorescence tracer in the FP readout. The TMR-Noxa-peptide is relatively small and rotates faster in the solution. Upon excitation with plane-polarized light, the emitted light is random and results in a lower FP signal. Whereas, binding of TMR-Noxa peptide to the large Mcl-1 protein slows down the movement of the TMR-Noxa peptide, leading to the emission of polarized light and an increased FP signal. Thus, both TR-FRET and FP signals can be obtained from the same reaction without the need of additional reagents. As expected, addition of antagonists or compounds that disrupt the Mcl-1/Noxa complex formation will result in lowered signals in both TR-FRET and FP readouts.
Dual-Readout F2 Assay Development
Analysis and optimization of TMR-labeled Noxa peptide in the dual-assay platform
The key point for the successful dual TR-FRET/FP assay is to choose the assay conditions optimal for both assays in the same reaction. We first evaluated the concentrations of TMR-Noxa-peptide optimal for the dual measurements. Increasing concentrations of TMR-Noxa-peptide were mixed with constant amount of terbium-anti-His antibody in the presence or absence of Mcl-1 protein (50 nM). Five microliters of the mixture was transferred to a 1,536-well plate, and TR-FRET signal, FP signal, and fluorescence intensity (FI) for TMR-labeled Noxa peptide were measured using the Envision plate reader. For FP measurement, the tracer's fluorescent quantum yield has to be significantly higher than that of the background (buffer) itself.33 As shown in Figure 2A, as low as 4 nM of TMR-Noxa-peptide alone, the FI signal from tracer is >15 times higher than background from buffer-only wells and reached about 1,100 times higher at 250 nM of the tracer. At around 500 nM of TMR-Noxa peptide, the detected FI signal was saturated and reached the maximum detection limitation using the current reader settings, which already generated a wide range of signal window between 1 and 1,100 times. As a control, the FI was not significantly changed in the presence of Mcl-1 protein. The addition of Mcl-1 protein (50 nM) to increasing concentrations of TMR-Noxa peptide resulted in an increased FP signal (Fig. 2B). The maximum FP assay window was 78.8 mp at 31.3 nM of TMR-Noxa peptide. Further increasing the TMR-Noxa peptide to 125 nM resulted in a decrease in the FP signal window. The estimated Kd for the binding of TMR-Noxa peptide to 50 nM of Mcl-1 protein is about 2.6 nM.
Fig. 2.

Evaluation of TMR-Noxa peptide in dual-readout F2 assay in 1,536-well format. Increasing concentrations of TMR-Noxa peptide were incubated with 50 nM Mcl-1 protein at room temperature for 1 h. The total reaction volume is 5 (L per well. (A) Total FI values were measured for each TMR-Noxa peptide concentration and compared to those of buffer alone in the presence or absence of Mcl-1 protein. (B) The polarization signal was recorded and expressed as FP signal windows after subtracting the mP values for tracer alone. The data shown are average values with SD from four replicates. (C) TR-FRET signal with increasing concentrations of Noxa-Rho peptide were measured and plotted against TMR-Noxa peptide concentrations. TR-FRET signal = A572nm/A545nm × 104; (D) the S/B values of TR-FRET (expressed as the calculated values × 2 to approximate similar scale) or S/N values of FP were obtained from data in (B) and (C) and plotted against TMR-Noxa peptide concentrations. (E) Z′ factors of the assay were calculated for both TR-FRET and FP measurements from the data in (B) and (C). FI, fluorescence intensity; mP, millipolarization; S/B, signal-to-background ratio; SD, standard deviation; S/N, signal-to-noise ratio.
We then measured the TR-FRET signal for the same reaction in the same wells used for FP measurement. As shown in Figure 2C, in the presence of 50 nM of Mcl-1 protein, increasing TMR-Noxa peptide led to a dose-dependent increase in TR-FRET signals. The maximum FRET signal window is about 2,300. TMR-Noxa peptide alone in the absence of Mcl-1 protein gave minimal FRET signal. The Kd for the binding of TMR-Noxa peptide to 50 nM of Mcl-1 in the TR-FRET measurement is ∼85.5 nM. This result is consistent with reported binding affinity of Noxa BH3 peptide,24 as well as with our results obtained with surface plasmon resonance direct binding assay (Kd of 72 nM), and FP-based competitive assay (Ki = 97 nM) (data not shown).
The goal of the designed dual-readout F2 assay is to provide a multiplexed assay platform that is simple for routine use as well as applicable for HTS or uHTS. Therefore, we evaluated the dual-readout assay performance through assessing its Z′ factor in both TR-FRET and FP measurement, S/B of the TR-FRET read, and S/N of the FP read. Binding of 50 nM Mcl-1 to increasing concentrations of TMR-Noxa peptide resulted in an increase in both S/B (TR-FRET) and S/N (FP) values (Fig. 2D). The S/B of TR-FRET signal at 125 nM of TMR-Noxa peptide was about 8.6 and achieved maximum (9.1) when 250 nM of TMR-Noxa peptide was tested. The S/N of the FP read was 20 at 62.5 nM of TMR-Noxa peptide and reached saturation at 62.5 nM or higher concentrations of TMR-Noxa peptide. Z′ factors were all above 0.5 for both TR-FRET and FP readouts when higher than 30 nM of TMR-Noxa peptide were tested (Fig. 2E), indicating a robust and high-quality assay for HTS.34
The Kds obtained by TR-FRET and FP measurements for the same reaction appear to be distinct. The Kd for the binding of TMR-Noxa peptide to 50 nM of Mcl-1 measured in the TR-FRET read (85.5 nM) is ∼32.9 times higher that the Kd obtained from FP read (2.6 nM). This may be due to the different signal strength measured by a given fluorophore-ligand concentration in each assay format. The FP assay format is amenable to using a lower fluorophore-ligand concentration than the TR-FRET assay format. To achieve high enough signal and remain within the dynamic range of the assay for both TR-FRET and FP measurements, the concentrations of TMR-Noxa peptide used in the dual-readout assays need to be carefully selected. For the FP assay format, when used at a lower concentration of the fluorophore, fluorescence contributions from library compound will have a more dramatic impact on the assay because the detection readout is a measure of the sum of all fluorescent species in the assay. As the fluorophore concentration increases in the assay, the contributions from fluorescent compounds are reduced. Therefore, the use of higher concentrations of fluorophore-labeled ligand would have the advantage to reduce the fluorescence interference from library compounds in the FP assay format.35 Thus, based on the titration curves of TMR-Noxa peptide in FP readout and TR-FRET readout, concentrations of 62.5 and 125 nM TMR-Noxa peptide were selected for the further evaluation in the dual-readout F2 assay.
Analysis and optimization of Mcl-1 protein in the dual-readout F2 assay platform
To determine the Mcl-1 protein concentrations optimal for the dual-readout F2 assay, we carried out titration experiments. A constant concentration of the TMR-Noxa peptide (62.5 or 125 nM) was titrated with increasing concentrations of the Mcl-1 protein (1–1,000 nM). The binding signals were measured in both TR-FRET and FP modes.
Increasing concentrations of Mcl-1 led to dose-dependent increase of the FRET signal with either 62.5 or 125 nM of TMR-Noxa peptide (Fig. 3A). The TR-FRET signal reached a maximum when 31.3 nM of Mcl-1 protein was mixed with 62.5 nM of TMR-Noxa, or when 62.5 nM of Mcl-1 was incubated with 125 nM of TMR-Noxa peptide, respectively. The estimated Kd of Mcl-1 was 6.2 and 20.6 nM, respectively, under these two conditions.
Fig. 3.

Optimization of Mcl-1 protein in dual-readout F2 assay. (A) Increasing concentrations of Mcl-1 protein were incubated with 62.5 or 125 nM of TMR-Noxa peptide and incubated at room temperature for 1 h. The TR-FRET signal and the FP signal were measured using Envision Multilabel plate reader. (A) The FRET signal window was obtained by subtracting peptide-only values from values in the presence of the amount of added Mcl-1 protein and plotted against Mcl-1 protein concentration. (B) The FP assay window was obtained by subtracting the mP values of tracer alone from the values in the presence of protein. Data were analyzed using a nonlinear regression method in Prism 5.0. (C) The comparison between dose–response curves of the binding of 125 nM TMR-Noxa peptide to Mcl-1 protein between TR-FRET and FP measurements. The values of FP assay window in mP were multiple by 17 to bring to the similar scale of TR-FRET signal window for comparison. The data shown are average with SD from four replicates. (D) The S/B values of TR-FRET (values × 2 to approximate same scale) and S/N values of FP were obtained from data in (A) and (B) for the binding of 125 nM TMR-Noxa peptide to Mcl-1. (E) Z′ values for TR-FRET and FP measurements were calculated from data in (A) and (B) for the binding of 125 nM TMR-Noxa peptide to increasing concentrations of Mcl-1 protein.
In the FP measurement, as the amount of Mcl-1 protein increased, progressively increased FP signal was observed until reaching saturation. A maximum assay window about 100 mP was achieved. The Kds for the binding to 62.5 and 125 nM of TMR-Noxa peptide in the FP read were 11.5 and 20.5 nM, respectively.
Both TR-FRET and FP assays from the same reaction gave rise to a consistent and robust assay performance. The Kd of the Mcl-1 binding to 125 nM of TMR-Noxa peptide was 20.6 nM in the TR-FRET read and 20.5 nM in the FP read (Fig. 3C). Mcl-1 protein at 62.5 nM generated the maximum S/B (10.6) in the TR-FRET read and S/N (25.1) in the FP read (Fig. 3D). Further increasing Mcl-1 protein concentration led to decreased signals in TR-FRET readout, although the FP readout remained constant (Fig. 3C, D). Z′ factors were above 0.5 for both TR-FRET and FP readouts when Mcl-1 protein concentrations were >7.8 nM. From these analyses, it appears that it is feasible to identify assay conditions that permit the optimal performance by both TR-FRET and FP assay types for the Mcl-1/Noxa interaction. Accordingly, an assay condition that utilizes 125 nM of TMR-Noxa peptide and 62.5 nM of Mcl-1 protein was selected and used for the following studies. With this assay condition, optimized performance regarding assay window and robustness can be achieved for both TR-FRET and FP measurement.
Validation of the Dual-Readout F2 Technology in a Competition Assay
To validate the dual-readout F2 assay format for HTS, we have developed a competition assay by using unlabeled Noxa peptide. The unlabeled Noxa peptide is expected to compete with the TMR-Noxa for Mcl-1 binding. Indeed, the addition of unlabeled Noxa-peptide dose-dependently decreased both TR-FRET and FP signals (Fig. 4). Importantly, both TR-FRET and FP readouts gave rise to similar IC50 values for the Noxa antagonist peptide with 93.4 nM for TR-FRET and 81.1 nM for FP, respectively. These data indicate that the dual-readout F2 assay can be used for measuring the inhibitory effect of potential antagonists and searching for Mcl-1 protein inhibitors.
Fig. 4.

Competition study with the dual-readout F2 assay. Competition of nonlabeled Noxa peptide for the binding of TMR-Noxa peptide to Mcl-1 protein. Increasing concentrations of nonlabeled Noxa peptide were added to the reaction buffer containing 125 nM of TMR-Noxa peptide and 62.5 nM of Mcl-1 protein in a final volume of 5 (L. Free (125 nM TMR-Noxa peptide alone) and bound (125 nM TMR-Noxa peptide with 62.5 nM Mcl-1 protein) tracer controls were included on each plate. The TR-FRET and FP signal were measured after a 10-h incubation at room temperature. The competition effects in TR-FRET and FP measurements were expressed as percentage of control and was calculated as described in the Materials and Methods section. The data were plotted against log10 nonlabeled Noxa peptide concentrations, and the EC50 were calculated using a nonlinear regression in Prism 5.0.
uHTS Format Development
To evaluate the suitability of the dual-readout F2 technology for the identification of Mcl-1 inhibitors in an uHTS format, DMSO tolerance and stability of the assay were examined. DMSO is a common solvent used in dissolving many natural and organic compounds as employed in generating small molecular libraries for HTS. To access the full applicability of dual-readout F2 for uHTS, it is necessary to test the effect of the DMSO on the assay performance before screening for Mcl-1 protein inhibitors. For this purpose, both TR-FRET and FP signals were recorded upon the addition of increasing concentrations of DMSO to wells in a 1,536-well plate. As shown in Figure 5, the FRET as well as FP readouts remained unchanged in the presence of up to 2.3% (v/v) DMSO. Further increase of DMSO slightly decreased the FP signal and significantly interfered with the TR-FRET signal. These results indicate that dual-readout F2-based Mcl-1 uHTS assay can be reliably performed in the presence of up to 2.3% DMSO.
Fig. 5.

Effect of DMSO on dual-readout F2 assay performance and the stability of the competition assay. The interaction of Mcl-1 (62.5 nM) with TMR-Noxa peptide (125 nM) was carried out in the presence of an increasing amount of DMSO (0%–9%) at room temperature for 2 h. (A) The TR-FRET signals were measured and plotted against the increasing amount of DMSO. (B) The FP signal was measured and plotted against an increasing amount of DMSO. (C, D) The stability of the inhibitory effect of the nonlabeled Noxa peptide on the binding of TMR-Noxa peptide (125 nM) to Mcl-1 protein (50 nM) measured by TR-FRET readout (C) or FP readout (D) after 2 or 10 h incubation at room temperature. DMSO, dimethyl sulfoxide.
Good temporal stability upon compound addition would be a highly attractive feature for the uHTS assay and particularly for the uHTS campaign. Thus, we examined the stability of the dual-readout F2-based Mcl-1 assay in a competition assay format with unlabeled Noxa peptide as a competitor. At two different time points (2 and 10 h) after adding increasing concentrations of unlabeled Noxa peptide to the reaction containing TMR-Noxa peptide (125 nM) and Mcl-1 (62.5 nM), FRET and FP signals in a 1,536-well plate were measured. The results showed that the inhibitory effect of the unlabeled Noxa peptide on the assay is indeed very stable as revealed by virtually unchanged IC50 values in the TR-FRET read (Fig. 5C) and the FP read (Fig. 5D) at both time points. Such assay stability in the presence of an antagonist allows for the large batch sizes typical of uHTS screens.
Validation of the Dual-Readout F2-Based Mcl-1 Assay in an uHTS Format
To be amenable for uHTS, the dual-readout F2 assay should exhibit minimal variation between well-to-well, plate-to-plate, and day-to-day operations. To test the utility of our optimized dual-readout assay in an uHTS format, we examined the precision and robustness of the assay in eighty 1,536-well microplates performed in a low volume on different days. About 4.5 μL of reaction buffer containing TMR-Noxa peptide (125 nM), Mcl-1 (62.5 nM), and terbium-anti-His antibody (2 nM) was dispensed to a 1,536-well black plate using a Multidrop Combi. On each 1,536-well plate, each of 32 wells contains a free TMR-Noxa peptide control (125 nM) and terbium-anti-His antibody (2 nM) without Mcl-1 protein, which gives a minimal TR-FRET and FP signal. Another 32 wells contain TMR-Noxa peptide, Mcl-1 protein (62.5 nM), and terbium-anti-His antibody (2 nM), which defines the assay window for both TR-FRET and FP readout. The reactions were incubated for 2 h. Both TR-FRET and FP analyses were performed in the TR-FRET mode and FP mode, respectively, with the EnVision microplate reader. Analysis across eighty 1,536-well plates demonstrated stable signals in the TR-FRET (Fig. 6A) and FP reads (Fig. 6B) with large assay windows. The average TR-FRET signal from 80 plates was 1,553.6 cps with an SD of 136.8. The average FP signal was 133.9 mP with an SD of 10.2. To further evaluate the applicability of the dual-readout F2 technology for the 1,536-well based uHTS, the assay performance parameter S/B (TR-FRET), S/N (FP), and Z′ factors were calculated. The S/B values for TR-FRET readout, as determined for each plate, were consistently higher than 5 and the average value was 6.2 from 80 plates with an SD of 0.8 (Fig. 6C). The S/N values for the FP read were constantly higher than 8 and the average was 8.8 with an SD of 0.5 (Fig. 6C). The Z′ factors were all above 0.5 for both TR-FRET read and FP read (Fig. 6D), demonstrating a robust and consistent assay with minimal variations. Thus, the dual-readout F2-based Mcl-1/Noxa binding assay in 1,536-well plate is of excellent quality for uHTS.
Fig. 6.

Evaluation of the dual-readout F2 assay performance in an uHTS format. Eighty 1,536-well plates each containing 32 free tracer control wells (125 nM TMR-Noxa peptide) and 32 bound tracer control wells (125 nM TMR-Noxa and 62.5 nM Mcl-1) in a total of 5 (L assay buffer per well were used to determine the assay performance for uHTS. (A) In the TR-FRET readout, the TR-FRET signal for each well was recorded; the average values from each plate were plotted against the corresponding plate number. (B) In the FP readout, the FP signal for each well was measured and the average FP signals from free and bound tracer were plotted against plate number. (C) The S/B values of TR-FRET readout and the S/N values of FP readout were calculated for each plate. (D) Z′ factors for each plate were calculated for both TR-FRET and FP readout. uHTS, ultra-high-throughput screening.
To test the utility of the dual-readout F2 assay for uHTS, we performed a screening using a 102,255-compound library from the Molecular Library Screening Center Network. Library compounds (0.1 μL of 1 mM stock in DMSO) were dispensed into 1,536-well plates containing 4.5 μL per well of reaction buffer (125 nM TMR-Noxa peptide, 62.5 nM Mcl-1, and 2 nM terbium-anti-His conjugate). The final compound concentration was 21.7 μM with a final DMSO concentration of 2.2%. Analysis of the TR-FRET-derived data revealed a total of 1,568 positive compounds that decreased the TR-FRET assay window by >50% with a hit rate of 1.5% (Fig. 7A). Compounds with intrinsic fluorescence or fluorescence-quenching properties were identified by the FI at 545 nm. Fold of control (FOC) was calculated by FI of 545 nm from compound wells over that from vehicle control wells. FI artifacts were defined by FOC ≤ 0.2 (80% quenching) or ≥ 2 (twofold fluorescence over the control). Among those 1,568 positives obtained from TR-FRET, 53 compounds were possible artifact compounds, which resulted in an artifact rate of 3.4%.
Fig. 7.

Validation of the dual-readout F2 assay in an uHTS format. The Mcl-1 F2 assay was validated for uHTS using a 102,255 compounds library from Molecular Library Screening Center Network as an example. Library compounds were added to the 1,536-well plates containing reaction buffer with TMR-Noxa peptide and Mcl-1 and incubated at room temperature for 2 h. The TR-FRET signal and the FP signal from each well containing a library compound were measured and plotted. (A) The percentage of inhibition from TR-FRET readout was plotted against the FI of the compound. FI of the compound was expressed as FOC and calculated by the fold increase of FI at 545 nm from compound wells over that from vehicle control wells for each plate. (B) The percentage of inhibition from FP readout was calculated and plotted against the FI of the compounds expressed as FOC. FOC was calculated by the fold increase of the p-channel FI counts from a compound well over that from vehicle control wells for each plate. Compounds that showed percentage of inhibition > 50 in both TR-FRET and FP readout were considered as potential hits. Compounds with FI ≤ 0.2, or FI ≥ 2 were defined as FI artifacts. (C) FI of the library compounds from FP readouts was plotted against that from TR-FRET readouts. (D) The percentage of inhibition from FP readout was plotted against that from TR-FRET readout. FOC, fold of control.
On the other hand, the FP readout gave rise to 325 positives when the hit cutoff was set at 50% inhibition of FP assay window compared to DMSO control wells (Fig. 7B). p-Channel FI of the FP read was used for reference to identify fluorescent or fluorescence-quenching compounds in FP readout. FI of the compound was expressed as FOC and calculated as p-channel counts from compound well over that from vehicle control wells. Fifty-seven compounds have been found to interfere with the assay with fluorescence (FI ≥ 2) or fluorescence quenching (FI ≤ 0.2). The artifact rate was 21.3%.
These data indicate that FP readout generates higher artifact rate (21.3%) than that from TR-FRET readout (3.4%), suggesting that FP assay might be more sensitive to fluorescence interference from screening compounds, which is consistent with previous reports.35 When we plotted the FI of FP from the compound against FI of TR-FRET, we found that some compounds interfered only with the FP readout or TR-FRET, whereas some fluorescent-quenching compounds interfered with both readouts.
Interestingly, most of FP-derived positives also showed up as positives in the TR-FRET assay format (Fig. 7C). Two hundred fifty-nine compounds showed % inhibition > 50 in both TR-FRET and FP readout with 52 fluorescence-interfering compounds, resulting in 207 top hits. Thus, by requiring that a hit be positive in both TR-FRET and FP readouts, the strategy using the dual readouts would expect to greatly reduce the number of false positives that are caused by compound fluorescence or quenching at the wavelengths employed in either assay. Therefore, the dual F2 assay technology allows us to better rank and prioritizes hits from HTS and enables the selection of high quality hits for the follow-up studies.
Interestingly, 1,268 compounds were identified as positives only in the TR-FRET assay and 60 positive compounds were exclusive to the FP readout (Fig. 7C). This suggests that different assay technologies may generate different hit list, which is consistent with previous reports.31 With the advances of the instrumentation and the precision of the modern liquid-handling technologies, the variation issue of HTS has been drastically reduced. Simply repeating the screening will not significantly change the hit list. While false-positive issues have been extensively addressed in the HTS field, false-negatives have not gained much attention primarily because that large-scale screening for one target using multiple platforms is not practical. The application of the F2 assay platform permits the recovery of missed hits from one assay, such as FP, by the other, such as TR-FRET, resulting in the increased quality of screening by reducing the number of false-negatives. Such an apparent advantage of the F2 assay requires further experimental validation.
Summary
We have developed a novel HTS technology termed dual-readout F2 assay through combining FRET and FP assays into one platform for monitoring bimolecular interactions. Using the interaction of Mcl-1 and Noxa as a model system, we have optimized the F2 assay in a 1,536-well format for the screening of Mcl-1 protein inhibitors. It should be noted that a main technical challenge of the dual-readout F2 approach is the opposing requirements of TR-FRET verses FP for optimal reagent concentrations. A donor assay component labeled with lanthanide (Tb-Mcl-1 in this study) defines sensitivity of the TR-FRET readout. Conversely, the fluorescent peptide (TMR-Noxa) provides the readout in FP assay. Optimizations of FP and TR-FRET readouts would require fixing peptide and protein concentration, respectively, and titrating with protein and peptide, correspondingly. To avoid potential pitfalls, titration of both assay components in the presence of a broad range of multiple concentrations of its counterpart, often around a reported Kd value, would be critical to achieve an optimized condition. Compilation of these data would help selecting the most appropriate ratio and concentrations of the two assay components, specifically advantageous for protein–peptide pairs with a priori unknown affinities.
Further validation of the dual-readout F2 assay by screening a 102,255-compound library revealed that the number of positives in dual readouts was less than that from a single-readout platform. These dual-positive hits after the exclusion of false-positives, such as fluorescent compounds or quenchers, as shown in Figure 7, should be selected as primary positives for secondary and biological validation assays. As two assay formats are utilized for the same target at the same time in the same reaction, the dual-readout F2 technology is likely to minimize the number of false-positives and false-negatives, enrich primary HTS information, and increase screening quality and efficiency.
Abbreviations
- DMSO
dimethyl sulfoxide
- FI
fluorescence intensity
- FOC
fold of control
- FP
fluorescence polarization
- HTS
high-throughput screening
- mP
millipolarization
- S/B
signal-to-background ratio
- SD
standard deviation
- S/N
signal-to-noise ratio
- TMR
5/6-carboxytetramethyl-rhodamine
- TR-FRET
time-resolved fluorescence resonance energy transfer
- uHTS
ultra high-throughput screening
Acknowledgments
This work was supported in part by the grants from the National Institutes of Health PHS 5U54 HG003918 (to R.D. and H.F.), NS056915-01 (to Z.N.-C.), P01 CA116676 (to H.F.), Emory University's SPORE in Head and Neck Cancer Career Development award (P50 CA128613; to Y.D.), Emory Faculty Distinction Fund, and Georgia Cancer Coalition and Georgia Research Alliance (to H.F.).
Disclosure Statement
No competing financial interests exist.
References
- 1.Wells JA. McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.Jameson DM. Croney JC. Fluorescence polarization: past, present and future. Comb Chem High Throughput Screen. 2003;6:167–173. doi: 10.2174/138620703106298347. [DOI] [PubMed] [Google Scholar]
- 3.Rich RL. Myszka DG. Advances in surface plasmon resonance biosensor analysis. Curr Opin Biotechnol. 2000;11:54–61. doi: 10.1016/s0958-1669(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 4.Selvin PR. The renaissance of fluorescence resonance energy transfer. Nat Struct Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 5.Inglese J. Johnson RL. Simeonov A, et al. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 6.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 7.Adams JM. Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 8.Chao DT. Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 9.Petros AM. Medek A. Nettesheim DG, et al. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci USA. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wuilleme-Toumi S. Robillard N. Gomez P, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19:1248–1252. doi: 10.1038/sj.leu.2403784. [DOI] [PubMed] [Google Scholar]
- 11.Kaufmann SH. Karp JE. Svingen PA, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91:991–1000. [PubMed] [Google Scholar]
- 12.Hussain SR. Cheney CM. Johnson AJ, et al. Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin Cancer Res. 2007;13:2144–2150. doi: 10.1158/1078-0432.CCR-06-2294. [DOI] [PubMed] [Google Scholar]
- 13.Degterev A. Lugovskoy A. Cardone M, et al. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 14.Tzung SP. Kim KM. Basanez G, et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol. 2001;3:183–191. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- 15.Oltersdorf T. Elmore SW. Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 16.Petros AM. Dinges J. Augeri DJ, et al. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J Med Chem. 2006;49:656–663. doi: 10.1021/jm0507532. [DOI] [PubMed] [Google Scholar]
- 17.Bruncko M. Oost TK. Belli BA, et al. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 18.Wang G. Nikolovska-Coleska Z. Yang CY, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006;49:6139–6142. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]
- 19.Tang G. Yang CY. Nikolovska-Coleska Z, et al. Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins. J Med Chem. 2007;50:1723–1726. doi: 10.1021/jm061400l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang G. Ding K. Nikolovska-Coleska Z, et al. Structure-based design of flavonoid compounds as a new class of small-molecule inhibitors of the anti-apoptotic Bcl-2 proteins. J Med Chem. 2007;50:3163–3166. doi: 10.1021/jm070383c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang G. Nikolovska-Coleska Z. Qiu S. Yang CY. Guo J. Wang S. Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins. J Med Chem. 2008;51:717–720. doi: 10.1021/jm701358v. [DOI] [PubMed] [Google Scholar]
- 22.Tse C. Shoemaker AR. Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 23.Chen S. Dai Y. Harada H. Dent P. Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 24.Chen L. Willis SN. Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 25.Day CL. Smits C. Fan FC. Lee EF. Fairlie WD. Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol. 2008;380:958–971. doi: 10.1016/j.jmb.2008.05.071. [DOI] [PubMed] [Google Scholar]
- 26.Du Y. Masters SC. Khuri FR. Fu H. Monitoring 14–3-3 protein interactions with a homogeneous fluorescence polarization assay. J Biomol Screen. 2006;11:269–276. doi: 10.1177/1087057105284862. [DOI] [PubMed] [Google Scholar]
- 27.Tetin SY. Hazlett TL. Optical spectroscopy in studies of antibody-hapten interactions. Methods. 2000;20:341–361. doi: 10.1006/meth.1999.0927. [DOI] [PubMed] [Google Scholar]
- 28.Rajkowski KM. Cittanova N. Corrected equations for the calculation of protein-ligand binding results from fluorescence polarization data. J Theor Biol. 1981;93:691–696. doi: 10.1016/0022-5193(81)90230-7. [DOI] [PubMed] [Google Scholar]
- 29.Ullman EF. Kirakossian H. Singh S, et al. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc Natl Acad Sci USA. 1994;91:5426–5430. doi: 10.1073/pnas.91.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X. Glickman JF. Bowen BR. Sills MA. Comparison of assay technologies for a nuclear receptor assay screen reveals differences in the sets of identified functional antagonists. J Biomol Screen. 2003;8:381–392. doi: 10.1177/1087057103256466. [DOI] [PubMed] [Google Scholar]
- 31.Sills MA. Weiss D. Pham Q. Schweitzer R. Wu X. Wu JJ. Comparison of assay technologies for a tyrosine kinase assay generates different results in high throughput screening. J Biomol Screen. 2002;7:191–214. doi: 10.1177/108705710200700304. [DOI] [PubMed] [Google Scholar]
- 32.Wu X. Sills MA. Zhang JH. Further comparison of primary hit identification by different assay technologies and effects of assay measurement variability. J Biomol Screen. 2005;10:581–589. doi: 10.1177/1087057105275628. [DOI] [PubMed] [Google Scholar]
- 33.Du Y. Moulick K. Rodina A, et al. High-throughput screening fluorescence polarization assay for tumor-specific Hsp90. J Biomol Screen. 2007;12:915–924. doi: 10.1177/1087057107306067. [DOI] [PubMed] [Google Scholar]
- 34.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 35.Turek-Etienne TC. Small EC. Soh SC, et al. Evaluation of fluorescent compound interference in 4 fluorescence polarization assays: 2 kinases, 1 protease, and 1 phosphatase. J Biomol Screen. 2003;8:176–184. doi: 10.1177/1087057103252304. [DOI] [PubMed] [Google Scholar]