

Abstract
The analysis of morphological changes that occur in the nervous system during normal aging could provide insight into cognitive decline and neurodegenerative disease. Previous studies have suggested that the nervous system of Caenorhabditis elegans maintains its structural integrity with age despite the deterioration of surrounding tissues. Unexpectedly, we observed that neurons in aging animals frequently displayed ectopic branches and that the prevalence of these branches increased with time. Within age-matched populations, the branching of mechanosensory neurons correlated with decreased response to light touch and decreased mobility. The incidence of branching was influenced by two pathways that can affect the rate of aging, the Jun kinase pathway and the insulin/IGF-1 pathway. Loss of Jun kinase signaling, which slightly shortens lifespan, dramatically increased and accelerated the frequency of neurite branching. Conversely, inhibition of the daf-2 insulin/IGF-1-like signaling pathway, which extends lifespan, delayed and suppressed branching, and this delay required DAF-16/FOXO activity. Both JNK-1 and DAF-16 appeared to act within neurons in a cell-autonomous manner to influence branching, and, through their tissue-specific expression, it was possible to disconnect the rate at which branching occurred from the overall rate of aging of the animal. Old age has generally been associated with the decline and deterioration of different tissues, except in the case of tumor cell growth. To our knowledge, this is the first indication that aging can potentiate another form of growth, the growth of neurite branches, in normal animals.
Introduction
Neurons are remarkable for their ability to maintain structural and functional integrity for decades. With age, however, neurons can become susceptible to degenerative processes that eventually result in neuronal death, cognitive decline, and clinical dementia. Thus, a better understanding of how aging affects individual neurons could provide important insights into age-related neuronal disease. For example, in humans, recent studies suggest that age-related alterations in cell–cell interactions, gene expression, and plasticity, including subtle morphological changes, may be responsible for cognitive decline in the elderly (Lu et al., 2004; Burke and Barnes, 2006).
The nematode Caenorhabditis elegans provides an attractive opportunity to study neuronal aging. Whereas the human brain is immensely complex, C. elegans contain only 302 neurons. Moreover, the positions of these neuronal cell bodies and processes are highly stereotyped and have been mapped anatomically (Ward et al., 1975; White et al., 1986; Hall and Russell, 1991), facilitating the identification of potentially important age-dependent morphological changes in neurons if they exist. Furthermore, the lifespan of the worm is relatively short, averaging 19 d, which facilitates the analysis of age-dependent events. Finally, and most important, many cell biological processes identified in C. elegans have been found to be conserved evolutionarily.
Previous studies have suggested that the nervous system of C. elegans remains structurally intact and unchanged as the animal ages (Herndon et al., 2002), whereas surrounding tissues deteriorate with time (Garigan et al., 2002; Herndon et al., 2002). These findings were intriguing, because they suggested that neurons of C. elegans are somehow rendered “aging resistant.” However, these observations also seem counterintuitive, because neurodegenerative diseases are age associated, even in C. elegans disease models (Link, 1995; Satyal et al., 2000; Morley et al., 2002).
Surprisingly, when we examined neuronal process morphology in C. elegans more closely, we observed dramatic age-dependent changes in their structure. Specifically, the neurons of old animals frequently exhibited novel neurite branches that were not present in young adults, presumably reflecting new process outgrowth. This is the first time such an observation has been made for neurons in normal aged animals. The timing and extent of neurite branching was influenced by pathways that affect the animal's overall rate of aging, including c-Jun N-terminal kinase signaling, insulin/IGF-1-like signaling, and mitochondrial respiration. By modulating these pathways in a tissue-specific manner, neuronal branching rates could be uncoupled from the rate of aging of the rest of the animal, indicating that factors within neurons themselves can dictate the timing of this age-related process.
Materials and Methods
Strains
The C. elegans hermaphrodite strains used in this study were as follows: CF3458 (zdIs5[Pmec-4::GFP] I), CF3561 (zdIs5[Pmec-4::GFP] I; jnk-1(gk7) IV), CF3654 (zdIs5[Pmec-4::GFP] I; jkk-1(km2) X), CF3733 (zdIs5[Pmec-4::GFP] I; mek-1(ks54) X), EG1285 (oxIs12[Punc-47::GFP] X), CF3707 (jnk-1(gk7) IV; oxIs12[Punc-47::GFP] X), EG4529 (oxIs268[Punc-47::GFP] III), CF3678 (oxIs268[Punc-47::GFP] III; mek-1(ks54) X), MJB1014 (dlk-1(ju476) I; oxIs12[Punc-47:GFP, lin-15+] X), MJB1015 (oxIs268[Punc-47:GFP] III; mkk-4(ju91) X), MJB1013 (pmk-3(ok169) IV; oxIs12[Punc-47:GFP, lin-15+] X), MJB1026 (nsy-1(ok593) II; oxIs12[Punc-47:GFP, lin-15+] X), MJB1022 (oxIs268[Punc-47:GFP] III; sek-1(km4) X), MJB1021 (oxIs268[Punc-47:GFP] III; jkk-1(km2) X), MJB1029 (mlk-1(ok2471) V; oxIs12[Punc-47:GFP, lin-15+] X), MJB1023 (jnk-1(gk7) IV; oxIs12[Punc-47:GFP, lin-15+] X), CF3714 (zdIs5[Pmec-4::GFP] I; jnk-1(gk7) IV; muEx329[Pjnk-1::jnk-1::GFP + myo-3::RFP]), CF3710 (zdIs5[Pmec-4::GFP] I; daf-2(e1370) III), CF3704 (daf-16(mu86) zdIs5[Pmec-4::GFP] I), CF3734 (daf-16(mu86) zdIs5 [Pmec-4::GFP] I; daf-2(e1370) III), CF3703 (zdIs5[Pmec-4::GFP] I; daf-2(e1370) III; jnk-1(gk7) IV), CF3701 (zdIs5[Pmec-4::GFP] I; eat-2(ad1116) II), CF3702 (zdIs5[Pmec-4::GFP] I; clk-1(qm30) III), CF3750 (zdIs5 [Pmec-4::GFP] I; clk-1(qm30) III; hif-1(ia4) V), CF3658 (zdIs5 [Pmec-4::GFP] I; mev-1(kn1) III), CF3761 (zdIs5[Pmec-4::GFP] I; rol-6(su1006) II), CF3798 (daf-16(mu86) zdIs5[Pmec-4::GFP] I; rol-6(su1006) II; daf-2(e1370) III), and CF3799 (daf-16(mu86) zdIs5[Pmec-4::GFP] I; daf-2(e1370) III; muIs131[Punc-119::GFP::daf-16 + rol-6]). EG1285, EG4529, MJB1014, MJB1015, MJB1013, MJB1026, MJB1022, MJB1021, MJB1029, and MJB1023 were a kind gift from E. Jorgensen (University of Utah, Salt Lake City, UT).
Lifespan analysis
For lifespan assays, ∼100 L4 larvae were picked and transferred every other day to a new plate seeded with Escherichia coli OP50 bacteria until the end of the reproductive period. Animals that were missing or died as a result of extruded internal organs or from internally hatched progeny were censored and statistically incorporated into the lifespan analysis (Lawless, 1998). All lifespan experiments were performed at 20°C. We generated lifespan curves using STATA 10, and p values were determined using the Mantel–Cox log-rank test.
Fluorescence microscopy
In each experiment, 20 or more age-synchronized GFP-expressing worms were examined using fluorescence microscopy at each time point for the presence of extra neuronal processes. Experiments were done blind to the genotype of the worms and in triplicate (unless otherwise indicated in the figure legend). Error bars in all figures represent the SD from the mean. Appropriately aged animals were mounted in 20 μl of M9 on 2% agarose pads dried overnight to immobilize the worms and examined without the use of any anesthetic. The animals were discarded after visualization. In analysis of the touch neurons, zdIs5[Pmec-4::GFP]-expressing animals scored positive for the presence of extra neuronal processes when a visible GFP-labeled branch was observed emanating from any one of the six touch neurons visualized using a 10× objective. Some day 1 animals (meaning day 1 of adulthood) displayed a misplaced posterior lateral microtubule (PLM) synaptic branch. This synaptic branch was located closer to the PLM cell body and has been observed previously in the zdIs5 transgenic strain (Wu et al., 2007). This more proximal misplaced synaptic branch was not scored as positive. Single processes emanating from the cell body opposite the canonical process were also not scored as extra neurite branches. For GABAergic neurons, oxIs12 or oxIs268[Punc-47::GFP] animals were scored positive for extra neuronal processes when a visible branch emanated from a commissure or when a burst of branches (a network) emanated from the nerve cord anywhere in the animal. These branches were visualized using a 25× oil objective. Two different Punc-47::GFP transgenes were used because they are integrated on different chromosomes and thus were differently suited for different strain constructions. For RNAi treatment, daf-16–RNAi bacteria, daf-2–RNAi bacteria, or empty-vector control bacteria (L4440) were seeded on NG plates containing carbenicillin and isopropyl-β-d-thiogalactopyranoside, and worms were grown from eggs until visualization. Pmec-4::GFP and Punc-47::GFP images were captured using a Retiga EXi Fast1394 CCD digital camera (QImaging) attached to a Carl Zeiss Axioplan 2 compound microscope. Openlab 4.0.2 software (Improvision) was used for image acquisition. The χ2 statistical test was used to compare all genotypes in which the total number of worms examined for each group was >40.
Response to light touch
A population of day 10 adults expressing Pmec-4::GFP was evaluated for the ability to respond to light touch using an eyelash glued to the end of a pipette tip (Chalfie and Sulston, 1981). Animals were classified into two different responsiveness categories according to their ability to respond appropriately to 10 touches. If the animal responded by either moving forward or backward after light touch in the posterior (between the tail and vulva) or in the anterior (between the head and vulva), respectively, then the animal received a score of “perfect response.” If the animal moved appropriately only after some touches but not all or could not move normally forward or backward, then the animal received a score of “imperfect response.” The touch neurons within these animals were then visualized as described above in “fluorescence microscopy.”
Reactive oxygen species treatment
Hydrogen peroxide.
Fifty day 2 adult Pmec-4::GFP worms were treated with 5 mm hydrogen peroxide on an unseeded agar plate for 15 min at room temperature. Worms were removed and allowed to recover at 20°C on an OP50-seeded plate containing fluorodeoxyuridine (FUDR) (18 mm) to prevent additional progeny production and bagging. Viability was scored the next day. An average of 23% of the treated population had died by this time. No animals on the untreated plates died. After 6 or 8 d of recovery, animals were visualized by fluorescence microscopy for the presence of ectopic neurite branches.
Paraquat.
Populations of 50 day 1 Pmec-4::GFP adults were placed on OP50-seeded, FUDR-treated plates containing various concentrations of paraquat (3, 4, and 6 mm). These concentrations of paraquat are known to shorten lifespan modestly (Lee et al., 2010). Only 44 and 59% of the population subjected to 3 and 4 mm paraquat, respectively, were still alive on day 15 compared with 78% that remained alive on the untreated plate. Animals were visualized by fluorescence microscopy at day 10 of adulthood for the presence of ectopic neurite branches.
Results
C. elegans neurons elaborate extra branches during aging
Using fluorescent reporters to visualize neuronal processes, we examined the morphology of neurons of C. elegans as the animals aged. We first observed the six mechanosensory neurons (the “touch neurons”) labeled with a Pmec-4::GFP reporter. In young animals, the structure of these neurons was simple and consistent from animal to animal (Fig. 1A), as predicted from EM reconstructions (White et al., 1986). Remarkably, we found that, with age, many of these neurons began to exhibit additional extensions emanating from their main processes (Fig. 1B,D). These branches could extend from anywhere along the axon or from the cell body itself, and they adopted a myriad of unique, often curving, trajectories (Fig. 2A). Consistent with a previous report (Herndon et al., 2002), we did observe a minority of older animals that maintained a morphology typical of the neurons in young animals (Fig. 1C), indicating that the phenotype is not obligatory in aged wild-type animals.
Figure 1.

Aged worms develop new neurite branches. A, A representative fluorescent image of a day 1 adult Pmec-4::GFP animal with structurally intact touch neurons. In all images, the tail of the animal is to the right. B, A representative fluorescent image of a day 15 adult Pmec-4::GFP animal with extra neurite branches. Bottom, Higher magnification of boxed area; the arrow highlights the ectopic neurite branches. C, A representative fluorescent image of a day 15 adult Pmec-4::GFP animal exhibiting morphologically intact neurons. D, Neurite branching increases during aging. Pmec-4::GFP animals were evaluated at each time point for the presence of extra neuronal processes in any one of the six touch neurons. The error bars represent the SD. n ≥ 3 experiments (χ2 test compared with day 1, *p < 0.01, ***p < 0.0001). E, A representative fluorescent image of GABAergic neurons in a day 1 adult Punc-47::GFP animal with normal commissures. F, A representative fluorescent image of a day 15 adult Punc-47::GFP animal with an extra neurite branch emanating from the commissure (arrow). G, A representative fluorescent image of a day 15 adult Punc-47::GFP animal exhibiting normal commissures (arrowheads). H, Neurite branches increase in GABAergic neurons during aging. Punc-47::GFP animals were evaluated at each time point for the presence of extra neuronal processes in commissures. n ≥ 2 experiments. (χ2 test compared with day 1, **p < 0.001, ***p < 0.0001).
Figure 2.
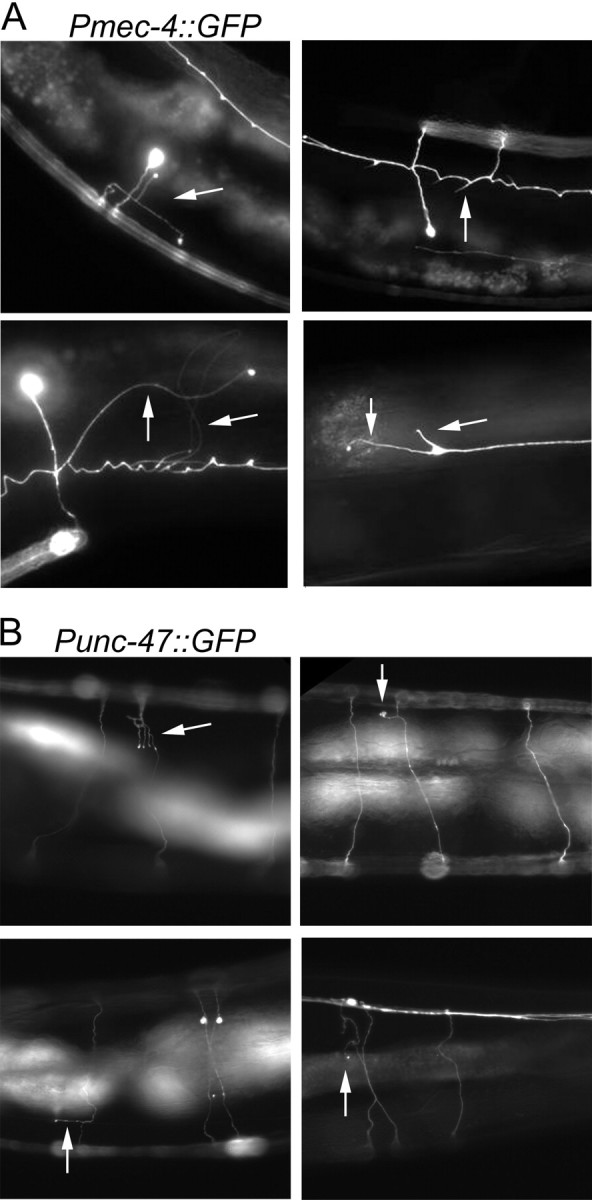
Representative images of age-associated extra neurite branches. Pmec-4::GFP-expressing animals (A) and Punc-47::GFP expressing animals (B) displaying extra neurite branches (arrows).
We next asked whether age-related branching was confined to the mechanosensory neurons. Using a Punc-47::GFP reporter, we also observed the GABAergic motor neurons. In addition to being functionally distinct from the touch neurons, these neurons also have circumferential in addition to longitudinal processes. Again, we observed an increasingly frequent number of ectopic neurite branches and projections emanating from the circumferential processes of these neurons as the animals aged (Figs. 1E–H, 2B). These findings indicate that more than one neuronal cell type can generate ectopic processes with age. In addition, they indicate that this phenomenon is unlikely to be attributable to a transgene-specific effect of Pmec-4::GFP, especially because Punc-47::GFP is integrated on a different chromosome. Instead, these findings suggest that the outgrowth of neurite branches could be a general neuronal phenomenon that accompanies the aging of C. elegans.
Not all worms age at the same rate (Garigan et al., 2002; Herndon et al., 2002). For example, as worms age, their mobility declines, but this decline occurs at different rates in different individuals. Those with the most rapid decline in mobility tend to die earlier (Herndon et al., 2002). Therefore, we wanted to know whether animals that were “physiologically older,” by the criteria of impaired movement, might exhibit more branches. To address this question, we subdivided a population of day 13 adult animals into two separate categories based on their ability to move and then looked for extra neurite branches in each animal. We observed a slight but significant correlation between decreased mobility and extra branches (percentage of animals with extra neuronal processes in animals with normal movement, 52 ± 5.7%; defective movement, 77 ± 14.6%; χ2 test, p = 0.04). We also performed the experiment using day 10 animals and again observed a significant correlation between a decline in movement and increased branching. In this same group of day 10 animals, we examined the response to gentle touch (with an eyelash) and found that an inability to respond appropriately also correlated with increased branching (percentage of animals with extra neuronal processes in animals with a perfect response to touch, 46 ± 5.1% vs an imperfect response to touch, 84 ± 12.2%; χ2 test, p = 0.007). Thus, the degree of branching correlated with both increased chronological and physiological age.
A specific MAP kinase pathway regulates neurite branching with age
Because the neurite branching phenotype was age associated, we asked whether genes that affect the rate of aging could delay or accelerate the appearance of branches. Overexpression of a neuronal c-jun N-terminal kinase, jnk-1, extends the lifespan of C. elegans; conversely, loss of jnk-1 shortens lifespan (Oh et al., 2005). When we examined the neurons of jnk-1(−) mutants, we discovered a striking acceleration of the branching phenotype (Fig. 3A–C). Loss of jnk-1 promoted neurite branching in both touch neurons (Fig. 3B) and GABAergic neurons (Fig. 3C). As in wild-type animals, the extent of branching increased with age in jnk-1 mutants.
Figure 3.

Loss of jnk-1 enhances neurite branching with age. A, A representative fluorescent image of a day 1 adult Pmec-4::GFP; jnk-1(gk7) mutant animal (top). A representative fluorescent image of a day 15 adult Pmec-4::GFP; jnk-1(gk7) mutant animal exhibiting extra neurite branches (bottom). Bottom, Magnification of boxed area. Arrows indicate extra neurite branches. B, jnk-1 mutant worms display more neurite branches in touch neurons than do control animals. Control (Pmec-4::GFP) and Pmec-4::GFP; jnk-1(gk7) mutant animals were evaluated at each time point for the presence of extra neurite branches. The error bars represent the SD (χ2 test, *p < 0.01, ***p < 0.0001). C, jnk-1(−) mutant worms exhibit more neurite branches in GABAergic neurons than do control animals. Control (Punc-47::GFP) and Punc-47::GFP; jnk-1(gk7) mutant animals were evaluated at each time point for the presence of extra neurite branches. n ≥ 2 experiments. (χ2 test, *p < 0.01, ***p < 0.0001). D, Touch neurons in day 10 adult control (Pmec-4::GFP), jnk-1(gk7), jkk-1(km2), and mek-1(ks54) animals were analyzed for the presence of extra neurite branches. (χ2 test, ***p < 0.0001). E, GABAergic neurons in day 5 adult control (Punc-47::GFP), jnk-1(gk7), dlk-1(ju476), mkk-4(ju91), pmk-3(ok169), mlk-1(ok2471), nsy-1(ok593), and sek-1(km4) were analyzed for the presence of extra neurite branches. Because of the use of two different Punc-47::GFP alleles, the data were normalized to control (ANOVA, ***p < 0.0001 compared with control). This analysis was performed at day 5 of adulthood because the jnk-1(−) mutant already exhibited a dramatic increase in branching compared with control.
To confirm that the enhanced branching phenotype we observed in the jnk-1 mutant was caused by loss of jnk-1, we introduced an extrachromosomal array encoding a functional JNK-1::GFP protein fusion expressed using 5 kb of the endogenous jnk-1 promoter into jnk-1(−) mutant animals. As predicted, this array suppressed the neurite branching phenotype [percentage of animals with extra neurite branching at day 10 in the jnk-1(+) rescued strain, 42.7 ± 10.3%; in jnk-1(−) strain, 95.9 ± 5.7%; χ2 test, p < 0.0001). jnk-1 mutants exhibit a locomotion defect that is easily visualized by the trails that they make in the bacterial lawn (Villanueva et al., 2001). The expression of this jnk-1(+) transgene in the jnk-1 mutant animals also eliminated this movement defect (data not shown), verifying the functional replacement of jnk-1 by the array. These findings demonstrate that JNK-1, a protein that influences aging in C. elegans, influences the rate of neurite branching. They also demonstrate that jnk-1 acts within neurons to influence branching, because the JNK-1::GFP signal was observed only within neurons in the rescued transgenic animals [consistent with previous reports (Kawasaki et al., 1999)].
JNK-1, a member of the MAP kinase superfamily, is a stress-response protein kinase that is activated by the upstream kinases JKK-1 and MEK-1 (Villanueva et al., 2001). We next asked whether either of these upstream kinases influenced age-related neurite branching. We observed the touch neurons by expressing Pmec-4::GFP in jkk-1(−) and mek-1(−) mutant animals. Like the jnk-1(−) mutation, loss of jkk-1 accelerated the branching phenotype; however, loss of mek-1 did not affect branching significantly (Fig. 3D). These results suggest that, in the wild type, JNK-1 and JKK-1 but not MEK-1 activities suppress age-related neurite branching.
Neuronal processes in developing C. elegans are capable of regeneration after laser axotomy (Yanik et al., 2004; Ghosh-Roy and Chisholm, 2010). Members of a specific MAP kinase pathway, containing dlk-1, mkk-4, and pmk-3, are completely required for axon regeneration in response to laser axotomy (Hammarlund et al., 2009). Could the neurite branching seen in aged animals be attributable to age-related activation of signaling pathways involved in axonal regeneration? To answer this question, we examined neurite branching in dlk-1, mkk-4, and pmk-3 mutant animals (Fig. 3E). All of these mutants exhibited wild-type levels of age-dependent branching. These results indicate that the pathways involved in age-related neurite branching and regeneration of injured axons are distinct from one another.
We also tested whether other MAP kinase signaling genes, specifically mlk-1, nsy-1, and sek-1, influenced neurite branching in aged worms. Loss-of-function mutations in nsy-1 and sek-1 did not affect neurite branching, yet loss-of-function mutations in mlk-1 (a MAP kinase kinase kinase thought to activate the p38 MAP kinase pmk-1) did affect neuronal branching. It is unclear how mlk-1 might interact with the JNK pathway to affect neuronal branching; however, we note that the loss of mlk-1 had a smaller affect than did loss of jnk-1. Overall, age-dependent branching seems to be most dramatically regulated by an MAP kinase pathway involving jkk-1 and jnk-1.
Insulin/IGF-1 signaling and DAF-16/FOXO activity also affect branching
The insulin-like growth factor receptor DAF-2 (Kimura et al., 1997) is a major lifespan determinant in C. elegans (Kenyon et al., 1993), and the effect of the insulin/IGF-1 signaling pathway on lifespan is highly conserved in evolution. In normal animals, the DAF-2 receptor activates a kinase cascade that ultimately phosphorylates and inhibits the DAF-16/FOXO transcription factor, which in turn limits longevity (Kenyon et al., 1993; Lin et al., 1997, 2001; Ogg et al., 1997; Henderson and Johnson, 2001; Lee et al., 2001). In daf-2(−) mutants, DAF-16/FOXO protein accumulates in the nucleus and alters the transcription of sets of metabolic, stress protective, antimicrobial and other genes that, in combination with one another, increase lifespan (Lee et al., 2003a; McElwee et al., 2003; Murphy et al., 2003; Curran et al., 2009). The lifespan extension produced by daf-2(−) mutations is dependent on daf-16, because daf-16; daf-2 double mutants are not long lived (Kenyon et al., 1993; Larsen et al., 1995). Instead, like daf-16 single mutants, they have a slightly shorter lifespan than wild-type animals (Larsen et al., 1995; Lin et al., 2001; Garigan et al., 2002).
We examined GFP-labeled touch neurons in animals carrying a reduction-of-function daf-2(e1370) mutation on day 10 of adulthood and found that they had far fewer branches than did wild-type animals (Fig. 4A). As expected, these same daf-2(−) mutants lived almost twice as long as normal. Because daf-16 is necessary for the extended lifespan of daf-2 mutants, we next asked whether daf-16 was necessary for the suppression of neurite branching in daf-2 mutants. In daf-16 single mutants, the degree of neurite branching was not significantly different from that of wild-type control animals. However, eliminating daf-16 activity completely blocked the inhibition of neurite branching by daf-2(−) mutations: daf-16(−); daf-2(−) double mutants had a branching phenotype that closely resembled that of wild-type worms (Fig. 4A). Thus, the suppression of neurite branching in daf-2(−) mutants requires daf-16.
Figure 4.

Insulin/IGF-1 signaling promotes extra neurite branching. A, The extra neurite branches were suppressed by loss of daf-2 in a daf-16-dependent manner. The touch neurons were analyzed in day 10 adult control (Pmec-4::GFP), daf-2(e1370) mutants, daf-16(mu86) mutants, and daf-16(mu86); daf-2(e1370) double mutants (χ2 test, **p < 0.001, ***p < 0.0001). B, daf-16 must be depleted in neurons to relieve daf-2-dependent suppression of branching. Touch neurons of daf-2(e1370) animals were analyzed at day 10 after growth on control (vector-only) RNAi bacteria and bacteria expressing dsRNA against daf-16. As predicted, daf-16; daf-2 double mutants fed daf-16--RNAi bacteria exhibited wild-type levels of neurite branching (χ2 test, **p < 0.001, ***p < 0.0001). C, Neuronal expression of daf-16 suppresses branching in daf-16(−); daf-2(−) mutant animals. The touch neurons were observed on day 10 of adulthood in control (Pmec-4::GFP), daf-16(mu86); daf-2(e1370), and daf-16(mu68); daf-2(e1370); Punc-119::GFP::daf-16 animals [χ2 test, ***p < 0.001 compared with control or daf-16(−); daf-2(−)]. D, daf-2 must be depleted from neurons to suppress branching. Pmec-4::GFP animals were analyzed on day 15 of adulthood after growth on control (vector-only) RNAi bacteria or bacteria expressing dsRNA against daf-2. E, daf-2 and jnk-1 mutations may act in different pathways to influence neurite branches. The touch neurons of daf-2(e1370) and daf-2(e1370); jnk-1(gk7) were evaluated on days 10, 15, 20, and 40 for the presence of neurite branches. Loss of jnk-1 enhanced the percentage of animals displaying branches in daf-2 mutant animals but not to the same extent as loss of jnk-1 in otherwise wild-type animals. n ≥ 2 experiments (χ2 test, **p < 0.001, ***p < 0.0001). F, Loss of jnk-1 did not shorten the daf-2 mutant lifespan. Lifespan analysis was performed on Pmec-4::GFP; daf-2(e1370) and Pmec-4::GFP; daf-2(e1370); jnk-1(gk7) animals. Mean lifespan for daf-2(−): 35.2 d. Mean lifespan for daf-2(−); jnk-1(−): 37.6 d. These lifespans were not significantly different.
Does daf-16, like jnk-1, act within neurons to influence branching? Like jnk-1, daf-16 is expressed in neurons, but it is expressed in other tissues as well (Henderson and Johnson, 2001; Lee et al., 2001; Lin et al., 2001). To determine whether daf-16 exerts its influence on neurite branching in a cell-autonomous manner, we took advantage of the fact that, in C. elegans, neurons are relatively resistant to RNAi. Previously, we showed that one can assay DAF-16 activity in vivo by using a broadly expressed DAF-16-regulated transcriptional reporter, Psod-3::GFP. Using this reporter, we found that feeding daf-2(−) mutants bacteria expressing daf-16 dsRNA completely eliminated Psod-3::GFP expression in non-neuronal cells but had no noticeable effect on neurons (Libina et al., 2003). In a similar manner, we now fed bacteria expressing daf-16 dsRNA to daf-2(−) mutant animals carrying the touch neuron-specific reporter Pmec-4::GFP. Surprisingly, and in stark contrast to daf-16; daf-2 double mutants, the RNAi-mediated knockdown of daf-16 had no measurable effect on the suppression of neurite branching caused by the daf-2(−) mutation (Fig. 4B). Although they had very low branching frequencies, their mean lifespan was normal [20 d compared with 47 d for daf-2(−) animals treated with vector RNAi-bacteria]. These observations imply that daf-16(+) function in the neurons of daf-2(−) mutants is sufficient to prevent neurite branching. To investigate the tissue specificity of daf-16 in another way, we evaluated daf-16(−); daf-2(−) double mutants containing a transgene that expressed daf-16 only in neurons. The touch neurons in this strain showed significantly fewer branches than did those in daf-16(−); daf-2(−) mutant animals (Fig. 4C). Conversely, we also asked whether daf-2-RNAi treatment of wild-type animals, which affects daf-2 activity in all tissues except neurons (Libina et al., 2003), affected the rate of neurite branching with age. Day 15 animals grown on control or daf-2-RNAi bacteria were found to have equivalent (i.e., wild-type) levels of neurite branching (Fig. 4D), despite the fact that the daf-2 RNAi-treated animals lived twice as long as control animals. Together, these results indicate that the insulin/IGF-1 pathway acts specifically within neurons to influence their ability to extend new branches with age.
Genetic dissociation of branching and lifespan
One interesting aspect of these experiments was that they uncoupled the rate of neurite branching from the animal's overall rate of aging. As predicted from previous studies (Dillin et al., 2002; Libina et al., 2003), daf-16 RNAi completely prevented the daf-2 mutation from extending lifespan. Thus, in this case, overall, the animals aged at a rate similar to that of wild type, but their neurons retained their youthful, unbranched, appearance (Fig. 4B,C). Likewise, animals fed daf-2 dsRNA-expressing bacteria were long lived, but their neurons elaborated a branching phenotype similar to that of wild type (Fig. 4D). In each case, perturbing the insulin/IGF-1 signaling pathway in neurons was necessary to influence neurite branching.
Because daf-2(−) and jnk-1(−) mutations have opposing effects on neurite branching in C. elegans (Figs. 3B, 4A), we wondered what the branching pattern would look like in animals carrying both mutations. We analyzed the double daf-2; jnk-1 mutant animals and found that they exhibited an intermediate branching phenotype (Fig. 4E). Whereas the entire population of day 15 jnk-1 single mutants had branches (Fig. 3B), only half of the daf-2; jnk-1 double mutants exhibited neurite outgrowths at this age (Fig. 4E). This finding is consistent with the interpretation that Jun kinase signaling and insulin/IGF-1 signaling act independently of one another to influence branching; however, because the daf-2 mutations are not null alleles—these prevent growth to adulthood—this interpretation remains tentative. Interestingly, despite the acceleration in branching caused by jnk-1(−) mutations in daf-2 mutant animals, the lifespan of the daf-2; jnk-1 double mutant was as long as that of the daf-2(−) single mutant (Fig. 4F), providing additional evidence that the branching phenotype can be regulated independently of organismal lifespan.
Caloric restriction can extend lifespan and delay age-related diseases in many organisms, so it was interesting to ask whether caloric restriction would delay the emergence of the neurite branching phenotype. C. elegans eat-2 mutants have impaired pharyngeal pumping and reduced food intake (Raizen et al., 1995). These mutants are long lived, presumably because of caloric restriction, and their longevity is independent of daf-16 (Lakowski and Hekimi, 1998). We found that, despite their increased lifespans, the temporal emergence of the branching phenotype in eat-2 mutants was similar to that of wild type (Fig. 5A). Thus again, the rates of aging and neurite branching could be uncoupled from one another (Fig. 5A,B). Together, these observations indicate that the neurite branching phenotype is strongly influenced by pathways that govern the overall rate of aging and lifespan; however, these pathways can affect neurite branching and other aspects of aging independently of one another. Furthermore, it indicates that branching itself is not a sufficient condition to age or kill the animal.
Figure 5.

Not all longevity mutations suppress neurite branching. A, Long-lived eat-2 mutants have similar wild-type levels of neurite branching, yet long-lived clk-1 mutants have suppressed branching. The touch neurons of day 15 control (Pmec-4::GFP), clk-1(qm30), and eat-2(ad1116) mutant animals were evaluated for the presence of extra neurite branches (χ2 test compared with control, ***p < 0.0001). B, A lifespan curve of control (Pmec-4::GFP), Pmec-4::GFP; clk-1(qm30), and Pmec-4::GFP; eat-2(ad1116) mutant animals, performed in parallel with the branching analysis in A. The mean lifespans were as follows: control at 18.5 d, clk-1(−) at 25.4 d, and eat-2(−) at 27.4 d (Mantel–Cox log-rank test, p < 0.0001).
Respiration can affect neurite branching
In yeast, worms, flies, and mice, a modest inhibition of respiration increases lifespan (Kayser et al., 2004; Kenyon, 2005; Dell'agnello et al., 2007; Lapointe and Hekimi, 2008; Copeland et al., 2009). This effect does not require daf-16 and is therefore mediated through a pathway separate from the insulin/IGF-1 signaling pathway (Lakowski and Hekimi, 1996; Dillin et al., 2002; Lee et al., 2003b). In contrast, mev-1(kn1) respiratory-chain mutations (Ishii et al., 1990) or RNAi treatments that severely inhibit respiration (Rea et al., 2007) shorten lifespan and, at least in some cases, appear to accelerate aging (Hosokawa et al., 1994; Adachi et al., 1998). Could activity of the respiratory chain affect neurite branching? To address this question, we examined the touch neurons in worms carrying a mutation in clk-1(qm30). Mutations that decrease the activity of the worm or mouse clk-1 genes, which encode ubiquinone biosynthetic enzymes, reduce respiration and extend lifespan (Lakowski and Hekimi, 1996; Braeckman et al., 1999; Kayser et al., 2004; Liu et al., 2005). The lifespan extension of clk-1(−) mutants was shown recently to be dependent on the hypoxia inducible factor (hif-1) transcription factor (Lee et al., 2010). We found that clk-1(qm30) significantly prolonged lifespan and delayed neurite branching (Fig. 5A,B). Interestingly, although loss of hif-1 reduced the lifespan of clk-1(−) mutant animals, neurite branching was not altered [percentage of animals with neurite branches at day 15 for clk-1(−); hif-1(+), 9.5%; clk-1(−); hif-1(−), 20%; χ2 test, p = 0.07]. Mutations in mev-1 had the opposite effect: mev-1 mutants displayed a shortened lifespan and exhibited accelerated neurite branching [the percentage of neurite branching at day 10 for mev-1(−), 86%; control, 37%]. These findings indicate that a perturbation that inhibits respiration affects neurite branching in C. elegans.
Exogenous reactive oxygen species do not cause neurite branching
Damage caused by reactive oxygen species (ROS) accumulates with age (Muller et al., 2007); thus, we wondered whether ROS could contribute to the neurite branching observed in older animals. Whereas high levels of agents that generate ROS, such as juglone or paraquat, are deleterious and shorten lifespan, low levels can induce cell-protective stress-response pathways that extend lifespan (Heidler et al., 2010; Lee et al., 2010). Likewise, both mev-1 (Senoo-Matsuda et al., 2001) and clk-1 (Lee et al., 2010; Yang and Hekimi, 2010) mutations increase ROS levels, but it has been proposed that the relatively high levels of ROS present in mev-1 mutants may shorten lifespan (Senoo-Matsuda et al., 2001), whereas the lower levels of ROS present in clk-1 mutants appear to extend lifespan (Lee et al., 2010; Yang and Hekimi, 2010). As described above, we found that mev-1 mutations accelerated neurite branching, whereas clk-1 mutations delayed branching. These observations made it particularly interesting to more directly assess the effects of ROS on neurite branching. To do this, first, we treated day 2 adults with 5 mm hydrogen peroxide. This dose killed 22% of the population by the next day; however, we failed to observe a significant change in the frequency of neurite branches in the animals that were still alive 6–8 d after treatment (∼50 animals were treated). To determine whether this result was specific for hydrogen peroxide, we also treated worms with paraquat, which stimulates the production of superoxide radicals within mitochondria. We supplemented the growth media with 3, 4, or 6 mm paraquat and observed touch neurons in animals expressing the Pmec-4::GFP reporter at day 10 of adulthood. The extent of neurite branching was not significantly different from that of control animals at any of these doses. These results suggest that ROS have little effect on the neurite branching phenotype; however, we cannot rule out the possibility that other types or levels of ROS are capable of influencing branching.
Discussion
In this study, we have described a new and unexpected age-related phenotype in C. elegans, neurite branching. Most of the neuronal processes in C. elegans are unbranched, although single branches are generated in a stereotyped manner, in certain neurons, including the anterior ventral microtubule (AVM) and PLM touch neurons that we examined in this study. However, we found that, although neurons in C. elegans do not exhibit the dramatic age-associated morphological deterioration seen in other tissues (Garigan et al., 2002; Herndon et al., 2002), they can elaborate a myriad of short and long, straight and curved branches in a variable manner. Our study of two different types of neurons, the touch neurons and the GABAergic motor neurons, allow us to conclude that (1) branches occur in mechanosensory neurons and in motor neurons, (2) branches occur in neurons that normally exhibit stereotypical branching during development and also in neurons that do not, and (3) branches can emanate from processes that run along the anteroposterior or dorsoventral axis.
We do not know whether the branches produced by old neurons are functional or whether they might be helpful or harmful to the animal. However, the finding that neurites in C. elegans sprout new branches with age raised several other questions, some of which were addressed in this study. Why is the branching frequency so greatly increased in older animals? What pathways affect the frequency of branching? Do the same mechanisms that govern the regeneration of severed axons in young animals influence branching in aged animals? What molecular mechanisms trigger branching in aged animals?
Age dependence of neurite branching
The finding that branching frequency increases with age implies that one or more factors that influence branching changes with age. Although we do not know what this factor is, our findings allow us to draw some conclusions about its nature. First, the factor can be dissociated from the overall rate of aging of the animal and from its lifespan. For example, in eat-2 mutants, lifespan is extended but the appearance of age-dependent neurite branches is similar to that of wild type. In daf-16(−); daf-2(−) mutants expressing daf-16(+) only in neurons, neurons remain unbranched despite the fact that, overall, the animals age rapidly (Fig. 4B,C). Conversely, in animals containing daf-2(+) activity solely within neurons, lifespan is doubled but neurite branching occurs at the same frequency as it does in wild type (Fig. 4D). There are two ways of explaining these data. First, it is possible that the age-associated factor that affects branching does not cause aging in general. Second, it is possible that the age-associated factor that affects branching does cause aging but specifically the aging of neurons. This latter possibility is consistent with the fact that branching frequency correlated with the genotype of neurons in our daf-2 and daf-16 mosaic animals. This model is difficult to test because neuron show very few morphological signs of aging even in normal old animals.
Two pathways that affect the frequency of neurite branch formation
We have identified two signaling pathways that play opposing roles in the regulation of branching. The first of these, a Jun kinase pathway, acts in wild-type C. elegans to suppress branching, because loss of this pathway dramatically accelerates and enhances branching. jnk-1 is expressed only in neurons (Kawasaki et al., 1999), indicating that Jun kinase-dependent events that occur within neurons affect the rate of branching.
JNK signaling is critical for the normal development and maintenance of the vertebrate nervous system (Waetzig et al., 2006; Haeusgen et al., 2009). It promotes neuronal development during embryogenesis, and it can stimulate neuronal regeneration or cell death after injury. Jun kinases have many targets in neurons, including transcription factors and cytoplasmic proteins (Bogoyevitch and Kobe, 2006). Thus, JNK signaling could potentially act in several different ways to suppress branching in aged C. elegans. One attractive model is that JNK signaling affects branching by influencing microtubule function. Interestingly, a phenotype at least superficially similar to the one we describe here has been observed in mouse cerebellar and motor-cortex neurons lacking JNK1. Loss of JNK signaling in these neurons increases dendritic arbor number, and it does so by reducing the level of phosphorylated MAP-2, which would otherwise bind to and stabilize microtubules (Brugg and Matus, 1991; Quinlan and Halpain, 1996; Sánchez et al., 1996, 2000; Björkblom et al., 2005). Disruptions in JNK signaling within C. elegans may elicit the neurite branching that we observed through similar mechanisms.
An additional signaling cascade, the insulin/IGF-1 pathway, also affects neurite branching in C. elegans. In wild-type animals, insulin/IGF-1 signaling promotes branching, at least in part, by inhibiting the activity of the DAF-16/FOXO transcription factor. Loss of daf-16 in otherwise wild-type animals has no effect on branching. In contrast, in the relatively branch-free daf-2 insulin/IGF-1-receptor mutants, which are known to have high levels of daf-16 activity, loss of daf-16 restores the level of branching back to that of wild type. Like Jun kinase, daf-16 acts within neurons to influence branching. Several hundred DAF-16-regulated genes have been identified (Lee et al., 2003a; McElwee et al., 2003; Murphy et al., 2003), and it will be interesting to ask which of these downstream genes may be responsible for the suppression of neurite branching in daf-2(−) mutants.
Neuronal regeneration and age-dependent branch formation
When neurons in young C. elegans are cut, they can regenerate (Yanik et al., 2004). Thus, it was interesting to ask whether the branching that occurs with age is actually a form of regeneration, particularly because one might expect old processes to be fragile and prone to breaking. Three arguments suggest that the answer is no. First, we saw no evidence of severed axons in aged animals, although it remains possible that tiny rips or fissures that are invisible to our eye stimulate branching. Second, we vortexed aged worms in liquid to cause physical stress, yet we observed no greater degree of ectopic neurite branching (data not shown). Third, the regeneration that occurs in young animals is dependent on dlk-1, mkk-4, and pmk-3 (Hammarlund et al., 2009), and we found that these genes do not alter age-dependent branching (Fig. 3E). Therefore, if the neurite branching that we observed in aged animals is a form of regeneration, it has a distinct underlying mechanism.
Interestingly, in young animals, axonal regeneration is enhanced by jnk-1 mutations (Hammarlund et al., 2009). Because jnk-1 mutations also stimulate age-dependent branching, it is possible that the substrates on which jnk signaling acts to influence branching are the same in the severed processes of young animals and the normal processes of old animals. In this model, the dlk-1/mkk-4/pmk-3 pathway would be required to generate these substrates in young neurons after axotomy but not in normal old neurons.
Could Jun kinase inhibition promote axon regeneration in older animals? Wild-type animals lose the ability to regenerate their axons when they are still middle aged (day 5) (Hammarlund et al., 2009), but our results suggest that inhibition of JNK-1 might potentiate neurite outgrowth in these animals as well.
Mitochondrial respiration and neurite branching
In addition to Jun kinase and insulin/IGF-1 signaling, we found that mutations affecting mitochondrial function could accelerate or delay neurite branching. Specifically, mev-1(kn-1) mutations, which shorten lifespan, accelerate the occurrence of branches, whereas clk-1(qm30) mutations, which extend lifespan, delay the occurrence of branches. The mechanism by which respiration affects neurite branching is not clear, although we can say that, in clk-1(−) mutants, HIF-1, which is required for longevity, is not required to suppress branching. We did not observe any effects of hydrogen peroxide or paraquat on branching, arguing against the possibility that ROS causes branching. However, we cannot rule out the possibility that the specific types or levels of ROS present in mev-1 and clk-1 mutants accelerate or delay branching.
Events that might stimulate neurite branching in elderly worms
In general, aging is associated with cognitive decline and neuronal degeneration, so the finding that neurons actively sprout branches in aged worms was unexpected and surprising. What is happening in the old worm to stimulate neurite growth? There are several possibilities. An inhibitor of neurite branching, or the ability of the cell to respond to such an inhibitor, might be lost during aging. Axon-regeneration inhibitors, such as Nogo or myelin-associated glycoprotein, prevent growth after CNS injury in mammals (Yang and Schnaar, 2008), and similar molecules could be downregulated during aging in C. elegans, allowing aberrant neurite growth. Neurite branching could also be a compensatory response to an age-dependent loss of neuronal cell function. Alternatively, a general inhibitor of cell growth might dissipate with age, or a factor that promotes cell growth could accumulate over time. A well-known type of growth often seen in the elderly is neoplasia. Both tumor growth and neurite branching are suppressed in C. elegans by daf-2 mutations in a daf-16-dependent manner (Pinkston et al., 2006; present study), raising the possibility of mechanistic overlap between the two phenomena. Neoplastic spread and invasion of neighboring tissues share essential features with embryogenesis, suggesting that normal developmental programs can be activated in malignancy. Likewise, perhaps reactivation of programs driving neuronal development also stimulates the neurite branching that we observed in aged animals.
Although many human neurodegenerative diseases are characterized by atrophy and neuronal loss, neurite sprouting has been observed in diseased brains (Masliah et al., 1993; Hashimoto and Masliah, 2003; Gordon et al., 2004). How neurite branching might be related to the onset, symptoms, or progression of neurodegenerative disease in humans remains unclear. Neurite branching might be an epiphenomenon, or a compensatory growth response, that occurs in degenerating or dysfunctional neurons. Alternatively, unaffected neurons could branch as a response to the loss of neighboring neurons or those within the same circuit. Thus, understanding the cellular mechanisms responsible for age-related neurite branching could potentially lead to new approaches to treat CNS injury or other forms of neuronal damage.
Footnotes
E.M.H.T is funded by a postdoctoral fellowship from the A. P. Giannini foundation. This work was supported by NIH Grant R01 AG11816 (C.K.), who is the director of the University of California, San Francisco Hillblom Center for the Biology of Aging and an American Cancer Society Research Professor. We thank members of the Kenyon laboratory for helpful discussions, comments on experiments, data analysis, and careful reading of this manuscript, with special thanks to H.-Y. Wu and A. E. Roux. We also thank S. Barmada for critical reading of this manuscript. We thank E. Jorgensen and the Caenorhabditis elegans Genetics Center (funded by NIH) for providing strains and K. Matsumoto for the plasmid Pjnk-1::jnk-1::GFP.
References
- Adachi H, Fujiwara Y, Ishii N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. J Gerontol A Biol Sci Med Sci. 1998;53:B240–B244. doi: 10.1093/gerona/53a.4.b240. [DOI] [PubMed] [Google Scholar]
- Björkblom B, Ostman N, Hongisto V, Komarovski V, Filén JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckman BP, Houthoofd K, De Vreese A, Vanfleteren JR. Apparent uncoupling of energy production and consumption in long-lived Clk mutants of Caenorhabditis elegans. Curr Biol. 1999;9:493–496. doi: 10.1016/s0960-9822(99)80216-4. [DOI] [PubMed] [Google Scholar]
- Brugg B, Matus A. Phosphorylation determines the binding of microtubule-associated protein 2 (MAP2) to microtubules in living cells. J Cell Biol. 1991;114:735–743. doi: 10.1083/jcb.114.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Curran SP, Wu X, Riedel CG, Ruvkun G. A soma-to-germline transformation in long-lived Caenorhabditis elegans mutants. Nature. 2009;459:1079–1084. doi: 10.1038/nature08106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161:1101–1112. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Chisholm AD. Caenorhabditis elegans: a new model organism for studies of axon regeneration. Dev Dyn. 2010;239:1460–1464. doi: 10.1002/dvdy.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, Hegedus J, Tam SL. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease. Neurol Res. 2004;26:174–185. doi: 10.1179/016164104225013806. [DOI] [PubMed] [Google Scholar]
- Haeusgen W, Boehm R, Zhao Y, Herdegen T, Waetzig V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience. 2009;161:951–959. doi: 10.1016/j.neuroscience.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Hall DH, Russell RL. The posterior nervous system of the nematode Caenorhabditis elegans: serial reconstruction of identified neurons and complete pattern of synaptic interactions. J Neurosci. 1991;11:1–22. doi: 10.1523/JNEUROSCI.11-01-00001.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Masliah E. Cycles of aberrant synaptic sprouting and neurodegeneration in Alzheimer's and dementia with Lewy bodies. Neurochem Res. 2003;28:1743–1756. doi: 10.1023/a:1026073324672. [DOI] [PubMed] [Google Scholar]
- Heidler T, Hartwig K, Daniel H, Wenzel U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010;11:183–195. doi: 10.1007/s10522-009-9239-x. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Hosokawa H, Ishii N, Ishida H, Ichimori K, Nakazawa H, Suzuki K. Rapid accumulation of fluorescent material with aging in an oxygen-sensitive mutant mev-1 of Caenorhabditis elegans. Mech Ageing Dev. 1994;74:161–170. doi: 10.1016/0047-6374(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Ishii N, Takahashi K, Tomita S, Keino T, Honda S, Yoshino K, Suzuki K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat Res. 1990;237:165–171. doi: 10.1016/0921-8734(90)90022-j. [DOI] [PubMed] [Google Scholar]
- Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J, Matsumoto K. A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J. 1999;18:3604–3615. doi: 10.1093/emboj/18.13.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser EB, Sedensky MM, Morgan PG, Hoppel CL. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J Biol Chem. 2004;279:54479–54486. doi: 10.1074/jbc.M403066200. [DOI] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. Determination of life-span in Caenorhabditis elegans by four clock genes. Science. 1996;272:1010–1013. doi: 10.1126/science.272.5264.1010. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J Biol Chem. 2008;283:26217–26227. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawless JF. Models and methods for lifetime data. New York: Wiley; 1998. [Google Scholar]
- Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003a;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003b;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, DeTeresa R, Alford M, Hansen L. Differing patterns of aberrant neuronal sprouting in Alzheimer's disease with and without Lewy bodies. Brain Res. 1993;617:258–266. doi: 10.1016/0006-8993(93)91093-8. [DOI] [PubMed] [Google Scholar]
- McElwee J, Bubb K, Thomas JH. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell. 2003;2:111–121. doi: 10.1046/j.1474-9728.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A. 2005;102:4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006;313:971–975. doi: 10.1126/science.1121908. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Halpain S. Postsynaptic mechanisms for bidirectional control of MAP2 phosphorylation by glutamate receptors. Neuron. 1996;16:357–368. doi: 10.1016/s0896-6273(00)80053-7. [DOI] [PubMed] [Google Scholar]
- Raizen DM, Lee RY, Avery L. Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics. 1995;141:1365–1382. doi: 10.1093/genetics/141.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez C, Tompa P, Szücs K, Friedrich P, Avila J. Phosphorylation and dephosphorylation in the proline-rich C-terminal domain of microtubule-associated protein 2. Eur J Biochem. 1996;241:765–771. doi: 10.1111/j.1432-1033.1996.00765.x. [DOI] [PubMed] [Google Scholar]
- Sánchez C, Díaz-Nido J, Avila J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Yasuda K, Tsuda M, Ohkubo T, Yoshimura S, Nakazawa H, Hartman PS, Ishii N. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J Biol Chem. 2001;276:41553–41558. doi: 10.1074/jbc.M104718200. [DOI] [PubMed] [Google Scholar]
- Villanueva A, Lozano J, Morales A, Lin X, Deng X, Hengartner MO, Kolesnick RN. jkk-1 and mek-1 regulate body movement coordination and response to heavy metals through jnk-1 in Caenorhabditis elegans. EMBO J. 2001;20:5114–5128. doi: 10.1093/emboj/20.18.5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waetzig V, Zhao Y, Herdegen T. The bright side of JNKs: multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol. 2006;80:84–97. doi: 10.1016/j.pneurobio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Ward S, Thomson N, White JG, Brenner S. Electron microscopical reconstruction of the anterior sensory anatomy of the nematode Caenorhabditis elegans.? 2UU. J Comp Neurol. 1975;160:313–337. doi: 10.1002/cne.901600305. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomas JH, Brenner FRS. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104:15132–15137. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LJ, Schnaar RL. Axon regeneration inhibitors. Neurol Res. 2008;30:1047–1052. doi: 10.1179/174313208X362523. [DOI] [PubMed] [Google Scholar]
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A. Neurosurgery: functional regeneration after laser axotomy. Nature. 2004;432:822. doi: 10.1038/432822a. [DOI] [PubMed] [Google Scholar]