

First documented over a century ago, conjugate additions are among the most utilized organic reactions. In carbon-carbon bond-forming variants, the nucleophile is typically organometallic in nature. Earlier technology employed enolate, organolithium, Grignard, or organocopper reagents, and more recently organozinc and organoboron compounds have enhanced this transformation significantly.[1,2] Despite increased functional group tolerance, an organometallic or organometalloid is nonetheless required in these powerful methods. Herein we describe a novel conjugate addition reaction in which a simple, unactivated alkene (ethylene, an alpha olefin, or styrene) takes the place of the organometal (eq 1). In other words, although an alkene is not an alkenylmetal reagent per se, it functions as one in this C–C bond-forming process.
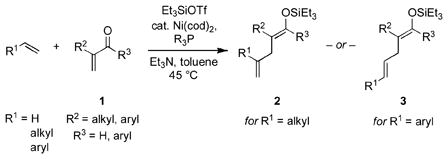 |
(1) |
Catalyzed polymerization of alkenes is one of the most important industrial processes,[3] and Ni-catalyzed two-alkene coupling reactions have also received significant attention, including hydrovinylation.[4] Montgomery has found that nickel complexes catalyze a wide variety of conjugate addition reactions,[5] but the closest precedent to the transformation reported here (catalytic 1,4-addition of simple alkene to unsaturated carbonyl) appears to be Lewis-acid promoted conjugate addition of electron-rich alkenes.[6,7] However, in these cases migration of the double bond of the alkene nucleophile occurs, in contrast to the Ni-catalyzed reactions described below.
Ogoshi reported that stoichiometric amounts of Ni(cod)2 and Me3SiOTf effected intramolecular coupling of an alkene and an aldehyde, and shortly thereafter, we reported that alpha olefins are excellent nucleophiles in intermolecular carbonyl addition reactions catalyzed by complex derived from Ni(cod)2 and a phosphine or an N-heterocyclic carbene.[8] Depending on the nature of the ligand, addition at either the terminus or the 2-position of the alkene occurs. The latter provides direct access to allylic alcohol derivatives, and the former yields products of a carbonyl-ene-like reaction. With the aim of broadening the scope of alkenes as nucleophiles in carbon-carbon bond-forming reactions, we turned our attention to electrophiles containing unsaturated carbonyl functional groups.
In order to focus on issues of alkene reactivity in initial studies, we selected ethylene as the coupling partner and decided to address issues of regioselectivity in subsequent experiments. As shown in Table 1, Et3SiOTf and catalytic amounts of Ni(cod)2 and Bu3P afford good to excellent yields of the conjugate addition product, isolated as the enolsilane (entries 1–4). Moreover, the stereoselectivity with respect to formation of the enolsilane is at least 92:8. Unsaturated ketones are also effective electrophiles (entries 5–11) but proceed with lower selectivity in some cases.
Table 1.
Ni-Catalyzed Conjugate Addition Reactions of Alkenes.[a]
| entry | R1 | R2 | R3 | Major product | Yield (%)[b] | E:Z (2)[b] |
|---|---|---|---|---|---|---|
| 1 | H | Me | H | 2a | 52 | 95:5 |
| 2 | n-hexyl | 2b | 76 58[c] 64[d] |
95:5 95:5[c] 95:5[d] |
||
| 3 | PhCH2 | 2c | 83 | 8:92 | ||
| 4[e,f] |
|
2d | 97 | 7:93 | ||
| 5 | Me | Ph | 2e | 90 | 95:5 | |
| 6 | n-Pr | 2f | 94 86[d] |
90:10 90:10[d] |
||
| 7 | i-Pr | 2g | 78 | 13:87 | ||
| 8[g] | Ph | 2h | 70 | n.d. | ||
| 9[h] | Me | p-anisyl | 2i | 94 | 91:9 | |
| 10 | n-Bu | 2-furyl | 2j | 95 | 75:25 | |
| 11 | Et | 2-thienyl | 2k | 95 | 75:25 | |
| 12[f] | n-hexyl | n-hexyl | H | 2l | 67[i] | 95:5 |
| 13[f] | Ph | n-hexyl | H | 3a | 70 | 81:19 |
| 14[f] | Me | p-anisyl | 3b | 55 | 91:9 |
See Supporting Information and eq 1. Standard conditions (entries 1–11): To a solution of Ni(cod)2 (0.075 mmol) and Bu3P (0.15 mmol) in toluene (1.5 mL) at 23 °C under ethylene (1 atm) were added Et3N (1.5 mmol) and the enal or enone (0.25 mmol). Triethylsilyl triflate (0.44 mmol) was added dropwise at 0 °C. The mixture was stirred 48 h at 45 °C and purified by chromatography (SiO2). In some cases CyPPh2 (entry 12) or tricyclopentylphosphine (entries 13–14) was used in place of Bu3P.
Determined by 1H NMR.
Ethylene pressure 2 atm.
Fourfold larger scale (1 mmol enal used).
Compound 1d added over 48 h.
Reaction time 72 h; a lower yield was obtained with 24 h reaction time.
A dihydropyran from hetero-Diels–Alder reaction of 2 equiv of 1h was isolated (13%).
Reaction time 24 h.
Combined yield of 2l and 2l′ (79:21; compound 2l′ is the result of addition to the 1-position of 1-octene). See text and Supporting Information.
As demonstrated in entry 9, electron-rich enones are superior electrophiles, and certain heterocycles are also tolerated (entries 10–11). Despite reduced selectivity, reactions with furan- and thiophene-containing enones proceed in high chemical yield. Overall, most of the above cases are highly selective, and thus the transformation represents a direct and stereoselective assembly of tetrasubstituted siloxyalkenes.[9,10]
Several observations regarding the optimum reaction conditions are noteworthy. Increasing either the ethylene pressure from 1 atm to 2 atm or the scale of the reaction fourfold resulted in only a marginal reduction in yield (entries 2 and 6). Out of 25 additives investigated (see Supporting Information), Bu3P was by far the most effective for coupling reactions of ethylene. Toluene is the superior solvent; for example, ethereal solvents such as Et2O, THF, and 1,4-dioxane completely suppress the coupling reaction.
Significant effort was expended to reduce the rather high catalyst loading (30 mol%); however, small decreases in the amount of Ni(cod)2 resulted in significantly reduced yield. For example, 2b was afforded in 49% yield when 15 mol% Ni(cod)2 was used (76% yield under standard conditions). Similarly, a 63% yield of 2f was obtained at 20% loading, down from 94% yield at 30% loading.
Other critical variables are the amounts of Et3N and Et3SiOTf employed. Decreasing or increasing the former lowered the yield or completely suppressed the reaction, and reducing the amount of silyl triflate from 1.75 to 1.25 equiv decreased the yield of 2b from 76% to 47% under otherwise identical conditions. It should also be noted that Me3SiOTf can be used in place of Et3SiOTf, but this substitution tends to diminish the product yield.
Unactivated monosubstituted olefins are also good coupling partners in this reaction. For example, 1-octene and 2-hexylacrolein are united in 67% yield, and with very high enolsilane E/Z selectivity (entry 12). Coupling occurs with approximately 4:1 regioselectivity, favoring coupling at the 2-position of the alkene. Since there are comparatively a greater number of general methods for the preparation of 1-alkenyl organometallics (e.g., hydrometalation of terminal alkynes), the fact that 1-octene functions as a 2-alkenyl organometallic reagent highlights a particularly useful aspect of this reaction.
Aryl alkenes, on the other hand, afford the opposite alkene regioselectivity (entries 13–14). Coupling at the 2-position of styrene is not observed; carbon-carbon bond formation at 1-position occurs exclusively, whether the electrophile is an enal or an enone.
These trends and observations noted above suggest a basic mechanistic framework (Scheme 1). The proposed sequence of events is based largely on a crystal structure of a complex derived from Ni(cod)2, Cy3P, a 1,3-diene, and PhCHO reported recently by Ogoshi.[11] We believe that the alkene (ethylene shown) and the electrophile (enal or enone, 1) afford an oxa-π-allyl nickel complex (A) during the formation of the carbon-carbon bond. The silyl triflate reacts with this species, giving an enolsilane and a Ni(II) complex (B) that undergoes rapid β-H elimination. Product (2) release and Et3N abstraction of TfOH from complex C affords a Ni(0) species (not shown), completing the catalytic cycle.
Scheme 1.
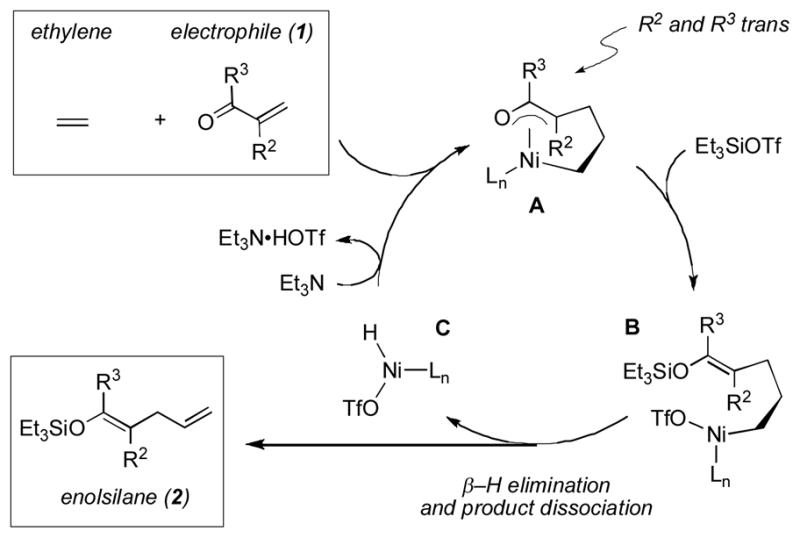
Proposed Mechanistic Framework
The E/Z selectivity thus appears to be dictated by two factors that in most cases reinforce each other. The placement of R2 and R3 away from each other and the chair-like chelation of Ni in complex A are consistent with the observed sense of alkene geometry. The superior performance of electron-rich enals and enones is consistent with the fact that reaction with silyl triflate is a critical step in the cycle. Mackenzie has reported Ni-catalyzed conjugate addition reactions between alkenyltributyltin reagents and α,β-unsaturated aldehydes that are assisted by chlorotrialkylsilanes and likely proceed via 1-((trialkylsilyl)oxy)allyl]nickel(II) intermediates.[7] In this vein, it is possible that the silyl triflate and enal (or enone) first combine, and that the resulting species then undergoes coupling with the alkene. Morken has proposed a similar sequence of events in Ni-catalyzed coupling reactions between allylboron reagents and enones.[12]
With the caveat that different ligands are used in coupling reactions of alpha olefins (CyPPh2) and styrene (P(cyclopentyl)3), our working hypothesis for the complementary regioselectivity in these two cases is as follows: It is possible that the regioselectivity observed for styrene (coupling at the alkene 1-position) is due primarily to an electronic consideration, specifically, the formation of a benzylic Ni species. On the other hand, the sense of selectivity in alpha olefin cases is that resulting from avoidance of steric repulsion between the Ni-ligand complex and the alkene substituent. We have proposed an explanation similar to the latter for the behavior of alpha olefins in other Ni-catalyzed coupling reactions that we have developed.[8b–f]
Several aspects of this transformation are noteworthy. First, it is a rare example of selective conjugate addition of an alkenyl equivalent to an unsaturated aldehyde. Typically in such reactions 1,2-addition is favored, or complex mixtures are afforded.[1] The high E/Z selectivity in most cases also merits further comment. Enolsilanes are starting materials in a wide range of enantioselective transformations leading to carbonyl compounds with quaternary stereogenic centers in the α-position, in many cases with very high enantioselectivity.[13,14] The double bond configuration is generally critical for high facial selectivity, and thus the nickel-catalyzed conjugate addition reaction provides rapid access to important tri-and tetrasubstituted enolsilanes that would otherwise be difficult to prepare with high selectivity via enolization of an aldehyde or ketone[9] (cf. Table 1, entry 2, (allyl vs. n-hexyl). Finally, the products derived from ethylene possess a monosubstituted alkene that is an excellent substrate for catalytic olefin cross-metathesis.[15] This combination therefore affords products that are regiocomplementary to those of the nickel-catalyzed conjugate addition reaction with aliphatic, monosubstituted alkenes (e.g., 1-octene).
Our current efforts expanding the scope and utility of the conjugate addition of monosubstituted alkenes to unsaturated carbonyl compounds. More broadly, we continue to explore catalytic reactions that utilize simple, widely available chemical feedstocks, including alpha olefins, and provide important synthetic intermediates in a single operation.
Supplementary Material
Footnotes
Support for this work was provided by the National Institute of General Medical Sciences (GM-063755). C.-Y. H. and H. O. thank the Croucher Foundation and the JSPS, respectively, for postdoctoral fellowships. We are grateful to Dr. Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Supporting Information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Chun-Yu Ho, Center of Novel Functional Molecules, The Chinese University of Hong Kong, Shatin, NT, Hong Kong SAR, PR China (current address)
Dr. Hirohisa Ohmiya, Department of Chemistry Massachusetts Institute of Technology Cambridge, MA 02139 (USA)
Prof. Dr Timothy F. Jamison, Department of Chemistry Massachusetts Institute of Technology Cambridge, MA 02139 (USA).
References
- 1.Posner GH. An Introduction to Synthesis Using Organocopper Reagents. Wiley-Interscience; New York: 1980. Perlmutter P. Conjugate Addition Reactions in Organic Synthesis. Pergamon Press; Oxford, New York: 1992. Catalyzed conjugate addition: Lopez F, Minnaard AJ, Feringa BL. Acc Chem Res. 2007;40:179. doi: 10.1021/ar0501976.Christoffers J, Koripelly G, Rosiak A, Rössle M. Synthesis. 2007;9:1279.Tsogoeva SB. Eur J Org Chem. 2007;9:1701.
- 2.For pioneering work in chlorotrimethylsilane-modified dialkylcuprate conjugate addition reactions, see: Corey EJ, Hannon FJ, Boaz NW. Tetrahedron. 1989;45:545.Horiguchi Y, Komatsu M, Kuwajima I. Tetrahedron Lett. 1989;30:7087.
- 3.a) Lappin GR, Sauer JD, editors. Alpha Olefins Applications Handbook. Marcel Dekker; New York: 1989. [Google Scholar]; b) Gladysz JA, Guest, editors. Chem Rev. 4. Vol. 100. 2000. Frontiers in Metal-Catalyzed Polymerization. [DOI] [PubMed] [Google Scholar]; c) Yeston JS. Science. 2005;309:2139. [Google Scholar]
- 4.Review: RajanBabu TV. Chem Rev. 2003;103:2845. doi: 10.1021/cr020040g.See also: b) Ru-catalyzed hydrovinylation of 2,4-dienoate esters (not conjugate addition: vinyl group and H add to 4- and 5-position, respectively): He Z, Yi CS, Donaldson WA. Synlett. 2004:1312.Catalyzed hydrovinylation of enoates and/or enones with double bond migration: c) Ni: Muller G, Ordinas JI. J Mol Cat A: Chem. 1997;125:97.Ru: Yi CS, He Z, Lee DW. Organometallics. 2001;20:802.
- 5.Review: Montgomery J. Angew Chem. 2004;116:3980.Angew Chem Int Ed. 2004;43:3890.Herath A, Thompson BB, Montgomery J. J Am Chem Soc. 2007;129:8712. doi: 10.1021/ja073300q.
- 6.Thermal: Albisetti CJ, Fisher NG, Hogsed MJ, Joyce RM. J Am Chem Soc. 1956;78:2637.Lewis acid-promoted: Buchi G, Koller E, Perry CW. J Am Chem Soc. 1964;86:5646.Snider BB, Deutsch EA. J Org Chem. 1983;48:1822.
- 7.Enal- and enone-derived coupling reactions of allylnickel complexes (stoichiometric in Ni, sunlamp irradiation): Johnson JR, Tully PS, Mackenzie PB, Sabat M. J Am Chem Soc. 1991;113:6172.
- 8.Ogoshi S, Oka M, Kurosawa H. J Am Chem Soc. 2004;126:11802. doi: 10.1021/ja0460716.Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194. doi: 10.1021/ja055363j.Ho CY, Ng SS, Jamison TF. J Am Chem Soc. 2006;128:5362. doi: 10.1021/ja061471+.Ng SS, Ho CY, Jamison TF. J Am Chem Soc. 2006;128:11513. doi: 10.1021/ja062866w.Ho CY, Jamison TF. Angew Chem. 2007;119:796. doi: 10.1002/anie.200603907.Angew Chem Int Ed. 2007;46:782.Isocyanates as electrophiles: Schleicher KD, Jamison TF. Org Lett. 2007;9:875. doi: 10.1021/ol063111x.
- 9.Preparation of geometrically defined silyl enol ethers from aldehydes and ketones: House HO, Czuba LJ, Gall M, Olmstead HD. J Org Chem. 1969;34:2324.Heathcock CH, Buse CT, Kleschick WA, Pirrung MC, Sohn JE, Lampe J. J Org Chem. 1980;45:1066.Corey EJ, Gross AW. Tetrahedron Lett. 1984;25:495.Hall PL, Gilchrist JH, Collum DB. J Am Chem Soc. 1991;113:9571.Denmark SE, Pham SM. J Org Chem. 2003;68:5045. doi: 10.1021/jo034092x.
- 10.(E)-2-methyl cinnamaldehyde also undergoes coupling with ethylene; however, a significant amount of 1,2-addition occurs together with the usual 1,4-addition. See Supporting Information for details.
- 11.Ogoshi S, Tonomori K-i, Oka M-a, Kurosawa H. J Am Chem Soc. 2006;128:7077. doi: 10.1021/ja060580l. [DOI] [PubMed] [Google Scholar]
- 12.Sieber JD, Liu S, Morken JP. J Am Chem Soc. 2007;129:2214. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]
- 13.General reviews: Brownbridge P. Synthesis. 1983;2:85.Kuwajima I, Nakamura E. Acc Chem Res. 1985;18:181.Berrisford DJ. Angew Chem. 1995;107:192.Angew Chem, Int Ed Eng. 1995;34:178.
- 14.Protonation: Ishihara K, Nakashima D, Hiraiwa Y, Yamamoto H. J Am Chem Soc. 2003;125:24. doi: 10.1021/ja021000x.Alpha-chlorination: Zhang YH, Shibatomi K, Yamamoto H. J Am Chem Soc. 2004;126:15038. doi: 10.1021/ja0454485.Fluorination: Cahard D, Audouard C, Plaquevent JC, Roques N. Org Lett. 2000;2:3699. doi: 10.1021/ol006610l.Epoxidation: Davis FA, Sheppard AC, Chen BC, Haque MS. J Am Chem Soc. 1990;112:6679.Ishii A, Kojima J, Mikami K. Org Lett. 1999;1:2013.Dihydroxylation: Morikawa K, Park J, Andersson PG, Hashiyama T, Sharpless KB. J Am Chem Soc. 1993;115:8463.Aldol: Evans DA, Masse CE, Wu J. Org Lett. 2002;4:3375. doi: 10.1021/ol026488l.Evans DA, Wu J, Masse CE, MacMillan DWC. Org Lett. 2002;4:3379. doi: 10.1021/ol026489d.
- 15.Reviews: Grubbs RH. Tetrahedron. 2004;60:7117.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem. 2005;117:4516. doi: 10.1002/anie.200500368.Angew Chem Int Ed. 2005;44:4490.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.