

Abstract
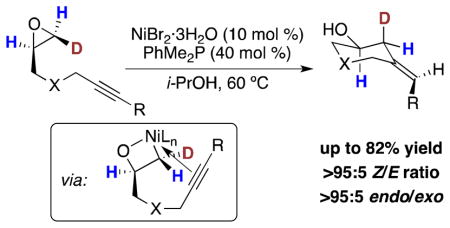
A Ni-catalyzed reductive coupling of alkynes and epoxides using Ni(II) salts and simple alcohol reducing agents is described. Whereas previously reported conditions relied on Ni(cod)2 and Et3B, this system has several advantages including the use of air-stable and inexpensive Ni(II) precatalysts (e.g., NiBr2·3H2O) as the source of Ni(0) and simple alcohols (e.g., 2-propanol) as the reducing agent. Deuterium-labeling experiments are consistent with oxidative addition of an epoxide C–O bond that occurs with inversion of configuration.
The development of methods for transition metal-catalyzed C–C bond formation using air-stable and inexpensive reagents continues to offer a challenge to synthetic chemists. Recent improvements in the scope and utility of Ni-catalyzed C–C bond forming reactions present a significant advance towards this goal.1,2 The advent of methods employing Ni(II) precatalysts, for example, has obviated the need to handle air-sensitive Ni(0) sources.3 In many cases, however, Ni-catalyzed coupling protocols are limited by the inclusion of air- and moisture-sensitive reducing agents. Herein, we describe a practical method for the Ni-catalyzed reductive coupling of alkynes and epoxides using air-stable and inexpensive Ni(II) salts as the precatalysts and 2-propanol (i-PrOH) as the reducing agent.4,5 Mechanistic analysis of the reductive coupling process via deuterium-labeling studies provides evidence that oxidative addition of the epoxide C–O bond occurs with inversion of configuration.
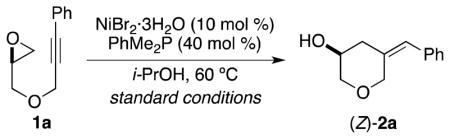
Our laboratory has previously demonstrated that alkynes and epoxides undergo reductive coupling in the presence of Ni(cod)2, Bu3P, and Et3B.6,7,8 For example, Ni-catalyzed reductive coupling of epoxide 1 provided homoallylic alcohol 2 in 50% yield (Scheme 1). The combination of Ni(cod)2 and Bu3P, both of which are exceedingly prone to air oxidation, was found to provide optimal results. In addition, the inclusion of Et3B, a highly pyrophoric reducing agent, was critical for the desired transformation to proceed. We hypothesize that Et3B may serve as a Lewis acid to facilitate oxidative addition of the epoxide, in addition to its standard role as a reducing agent.
Scheme 1.

Previously reported conditions for the Ni-catalyzed reductive couping of alkynes and epoxides6
In light of our recent studies involving regioselective epoxide-opening cascades promoted by hydroxylic solvents,9 we envisioned that alcohol solvents might promote the Ni-catalyzed reductive coupling of alkynes and epoxides. In this setting, the role of the alcohol solvent would be two-fold. First, oxidative addition of the epoxide C–O bond could be facilitated by hydrogen-bonding interactions between the epoxide oxygen and the alcohol solvent.9b,10 Second, the appropriate alcohol could serve as a reducing agent to provide the desired homoallylic alcohol product (2) and regenerate the active Ni(0) catalyst (vide infra, Scheme 2).4,5,11 In this manner, simple alcohols (e.g., i-PrOH) could serve as mild, air-stable, and inexpensive alternatives to Et3B.
Scheme 2.

Deuterium-Labeling Experiment and Proposed Mechanism for Ni-Catalyzed Reductive Coupling
Our investigations revealed that i-PrOH was an effective reducing agent for the Ni-catalyzed reductive coupling of alkynes and epoxides. For example, employing i-PrOH as the solvent in the presence of PhMe2P and either NiBr2·3H2O (Table 1, entry 1) or Ni(cod)2 (entry 2) provided high yields of the desired homoallylic alcohol 2a.12,13,14 The use of air-stable Ni(II) precatalysts and reducing agents (i.e., NiBr2·3H2O and i-PrOH, as opposed to previous conditions employing Ni(cod)2 and Et3B)6 allowed us to set-up and perform the coupling reactions outside of a glovebox. Furthermore, all reactions listed in Table 1 could be performed without drying of the reagents or glassware. Of note, reactions employing inexpensive NiX2·hydrate salts (entries 1 and 4) were of comparable efficiency to reactions performed with anhydrous NiBr2·diglyme (entry 3). Decreasing the catalyst loading to 5 mol % was tolerated and provided the desired homoallylic alcohol 2a in only slightly lower yields (entry 5). No product formation was observed in the absence of i-PrOH, the nickel catalyst, or phosphine ligand (entries 7, 8, 11).
Table 1.
Influence of Reaction Parameters on the Ni-Catalyzed Reductive Coupling of an Alkyne and Epoxide
 | |||
|---|---|---|---|
| entry | variation from standard conditions | yield (%)a | Z/Eb |
| 1 | nonec | 82 | 90:10 |
| 2 | Ni(cod)2 instead of NiBr233H2Od | 82 | 95:5 |
| 3 | NiBr23diglyme instead of NiBr233H2O | 76 | 88:12 |
| 4 | NiCl236H2O instead of NiBr233H2O | 74 | 89:11 |
| 5 | 5 mol % NiBr233H2O and 20 mol % | 75 | 95:5 |
| PhMe2P instead of standard catalyst/phosphine loading | |||
| 7 | THF instead of i-PrOH | <5e | --- |
| 8 | no NiBr233H2O | <5e | --- |
| 9 | 30 mol % PhMe2P instead of 40 mol % | 76 | 90:10 |
| 10 | 20 mol % PhMe2P instead of 40 mol % | 52 | 91:9 |
| 11 | no PhMe2P | <5e | --- |
Isolated yield of the mixture of olefin isomers.
Determined by 1H NMR spectroscopy of the crude reaction mixture.
Complete conversion of starting material observed within 3 h.
Phosphine loading was 20 mol % instead of 40 mol %.
Determined by 1H NMR spectroscopy relative to mesitylene as an internal standard.
Increased phosphine loadings were found to provide the highest yields of homoallylic alcohol 2a for reactions in which NiX2 salts were employed as the catalyst. Previously, a 2:1 (R3P:Ni) ratio was determined to be the optimal loading for reductive coupling reactions of epoxides and alkynes using Ni(cod)2 (Table 1, entry 2).6 When NiBr2·3H2O was employed as the catalyst, however, a phosphine loading of 20 mol % did not result in complete conversion of starting material (entry 10). Increasing the phosphine loading to 30 mol % led to complete consumption of the starting material and increased yields (entry 9). Finally, a phosphine loading of 40 mol % was found to provide the highest yield of homoallylic alcohol 2a (entry 1). We attribute the requirement for excess phosphine ligand to its involvement in the reduction of the Ni(II) precatalyst to the active Ni(0) species, which has been observed previously for both Ni(II)15 and Pd(II)16 precatalysts. Alternatively, excess phosphine may be necessary to suppress the formation of catalytically-inactive Ni(Oi-Pr)2 species.17
In all cases examined, reductive coupling of epoxide 1a proceeded to provide the desired tetrahydropyran product (2a) as a single regioisomer with respect to both the epoxide and the alkyne. Stereospecific cis addition to the alkyne occurred to provide the trisubstituted olefin (Z-2a);6 however, isomerization of the olefin geometry was observed to a limited extent under the standard conditions for reductive coupling. The amount of undesired isomerization product (E-2a) could be minimized by judicious choice of catalyst identity and loading (entries 1,2 and 5).
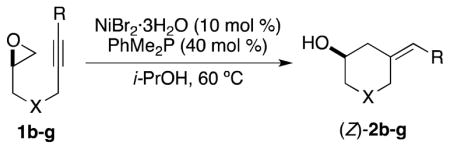
The mild conditions for Ni-catalyzed reductive coupling were found to effect product formation for substrates containing variation within the epoxide–alkyne tether. Both substituted cyclohexane (Table 2, entries 1 and 2) and piperidine (entry 3) derivatives could be prepared in this manner.12
Table 2.
Investigation of Reaction Scope
 | |||||
|---|---|---|---|---|---|
| entry | X | R | product | yield (%)a | Z/Eb |
| 1 | CH2 | Ph | 2b | 70 | 88:12 |
| 2 | C(CO2Me)2 | Ph | 2c | 76 | >95:5 |
| 3 | NBn | Ph | 2d | 74 | 88:12 |
| 4 | O | n-C5H11 | 2e | 75 | >95:5 |
| 5 | O | CO2Me | 2f | <5c | --- |
| 6 | O | H | 2g | 55d | --- |
Isolated yield of the mixture of olefin isomers.
Determined by 1H NMR spectroscopy of the crude reaction mixture.
Determined by 1H NMR spectroscopy relative to mesitylene as an internal standard.
Dropwise addition of substrate via syringe pump.
Varying the substituent on the alkyne had a more significant impact on the efficiency of reductive coupling. Alkyl-substitution was tolerated and provided the desired homoallylic alcohol (2e) in yields comparable to those observed for phenyl-substituted alkynes (Table 2, entries 1–4). Conversely, subjecting alkynoate 1f to the standard conditions for reductive coupling did not result in formation of the desired product (entry 5).18 Terminal alkynes are a particularly difficult class of substrates in Ni(0)-catalyzed coupling reactions, as this moiety often succumbs to deleterious cyclotrimerization.19 Under these conditions for reductive coupling, however, terminal alkyne 1g underwent reductive coupling in 55% yield (entry 6).
The proposed mechanism for Ni-catalyzed reductive coupling of epoxides and alkynes involves initial oxidative addition of the epoxide C–O bond to form nickella(II)oxetane 5 (Scheme 2).6 Previous reports have demonstrated that oxidative addition of epoxides with group 10 metals occurs with either inversion (Pd and Pt)20,21 or scrambling (Ni)22 of configuration, corresponding to SN2 ring opening or homolytic C–O bond cleavage, respectively. To the best of our knowledge, there is no experimental evidence describing oxidative addition of an epoxide C–O bond with Ni(0) that occurs with inversion of configuration.23
To gain insight into the oxidative-addition process, we prepared monodeuterated epoxide 3 and observed the relative configuration of the deuterated carbon center after subjection to the standard conditions for Ni-catalyzed reductive coupling (Scheme 2). Monodeuterated epoxide 3 was prepared with 83% deuterium incorporation at the terminal epoxide site bearing a cis relationship to the alkyne tether.12 Subjecting epoxide 3 to the standard conditions for reductive coupling provided homoallylic alcohol 4 with complete inversion of configuration at the deuterated carbon center.24 This result is consistent with inversion of configuration in the oxidative addition step to afford the nickella(II)oxetane intermediate (5), followed by migratory insertion that occurs with retention of configuration.25 We propose that inversion of configuration in the oxidative addition step occurs via SN2 attack by the nucleophilic Ni(0) species facilitated by hydrogen-bond activation of the epoxide.10 Importantly, identical results were obtained when epoxide 3 was subjected to the previously described conditions for reductive coupling (Scheme 1, PhMe2P instead of Bu3P) indicating that the active Ni(0) catalyst is responsible for the inversion process.
Following C–C bond formation, i-PrOH serves as a mild reducing agent to complete the catalytic cycle. Protonolysis of the Ni–O bond of strained bicyclic oxanickellacycle 6 results in formation of the LnNi(Oi-Pr) species (7) necessary for catalytic turnover. Subsequent β-H elimination occurs to liberate acetone, which is inert under the reaction conditions, followed by reductive elimination to afford homoallylic alcohol 4 and regenerate the active Ni(0) catalyst.
In summary, we have developed novel conditions for the Ni-catalyzed reductive coupling of alkynes and epoxides that employ air-stable and inexpensive reagents. These reaction conditions are significantly milder and more convenient than those previously used for similar reductive coupling reactions in which a combination of Ni(cod)2 and Et3B was necessary to effect C–C bond formation. Further investigation of the electrophile/π-nucleophile scope is warranted in an effort to improve the efficiency of previously developed methods and for the invention of novel reductive coupling procedures.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health, National Institute of General Medical Sciences (GM72566 and GM63755). M.G.B. is grateful to the National Institutes of Health for a postdoctoral fellowship (F32GM095014). We thank Dr. Christopher J. Morten (MIT) for helpful discussions. We thank Dr. Jeff Simpson (MIT) for assistance with NMR spectroscopy and Li Li (MIT) for obtaining mass spectrometric data.
Footnotes
Supporting Information Available. Complete experimental procedures and characterization data of novel compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For reviews, see: Montgomery J. Angew Chem, Int Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634.Montgomery J, Sormunen GJ. Top Curr Chem. 2007;279:1–23.Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun. 2007:4441–4449. doi: 10.1039/b707737h.Kimura M, Tamaru Y. Top Curr Chem. 2007;279:173–207.Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t.
- 2.For selected examples of recent advances in Ni-catalyzed C–C bond forming reactions, see: Malik HA, Sormunen GJ, Montgomery J. J Am Chem Soc. 2010;132:6304–6305. doi: 10.1021/ja102262v.Matsubara R, Jamison TF. J Am Chem Soc. 2010;132:6880–6881. doi: 10.1021/ja101186p.Cho HY, Morken JP. J Am Chem Soc. 2010;132:7576–7577. doi: 10.1021/ja101513d.Zhou CY, Zhu SF, Wang LX, Zhou QL. J Am Chem Soc. 2010;132:10955–10957. doi: 10.1021/ja104505t.Li W, Chen N, Montgomery J. Angew Chem, Int Ed. 2010;49:8712–8716. doi: 10.1002/anie.201004740.Ho CY, He L. Angew Chem, Int Ed. 2010;49:9182–9186. doi: 10.1002/anie.201001849.Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J Am Chem Soc. 2011;133:389–391. doi: 10.1021/ja108547u.Hoshimoto Y, Ohashi M, Ogoshi S. J Am Chem Soc. 2011;133:4668–4671. doi: 10.1021/ja109908x.
- 3.For selected examples of C3 C bond forming methods employing Ni(II) precatalysts, see: Watson MP, Jacobsen EN. J Am Chem Soc. 2008;130:12594–12595. doi: 10.1021/ja805094j.Quasdorf KW, Tian X, Garg NK. J Am Chem Soc. 2008;130:14422–14423. doi: 10.1021/ja806244b.Williams CM, Johnson JB, Rovis T. J Am Chem Soc. 2008;130:14936–14937. doi: 10.1021/ja8062925.Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920–921. doi: 10.1021/ja9093956.Lundin PM, Fu GC. J Am Chem Soc. 2010;132:11027–11029. doi: 10.1021/ja105148g.Owston NA, Fu GC. J Am Chem Soc. 2010;132:11908–11909. doi: 10.1021/ja105924f.
- 4.Alcohols bearing β-hydrogen atoms have proven to be competent reducing agents in Ni-catalyzed reductive coupling reactions: Herath A, Li W, Montgomery J. J Am Chem Soc. 2008;130:469–471. doi: 10.1021/ja0781846.Li W, Herath A, Montgomery J. J Am Chem Soc. 2009;131:17024–17029. doi: 10.1021/ja9083607.Phillips JH, Montgomery J. Org Lett. 2010;12:4556–4559. doi: 10.1021/ol101852w.
- 5.For additional examples of i-PrOH-mediated reductive C–C bond-forming reactions see: Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134–15135. doi: 10.1021/ja077389b.Bower JF, Patman RL, Krische MJ. Org Lett. 2008;10:1033–1035. doi: 10.1021/ol800159w.Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338–6339. doi: 10.1021/ja801213x.Patman RL, Chaulagain MR, Williams VM, Krische MJ. J Am Chem Soc. 2009;131:2066–2067. doi: 10.1021/ja809456u.
- 6.Molinaro C, Jamison TF. J Am Chem Soc. 2003;125:8076–8077. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]
- 7.For examples of the utility of Ni-catalyzed alkyneepoxide reductive3 coupling in the context of natural product synthesis, see: O’Brien KC, Colby EA, Jamison TF. Tetrahedron. 2005;61:6243–6248.Woodin KS, Jamison TF. J Org Chem. 2007;72:7451–7454. doi: 10.1021/jo071132e.Sparling BA, Simpson GL, Jamison TF. Tetrahedron. 2009;65:3270–3280. doi: 10.1016/j.tet.2008.11.086.Trenkle JD, Jamison TF. Angew Chem, Int Ed. 2009;48:5366–5368. doi: 10.1002/anie.200902079.
- 8.Reductive opening of epoxides has also been observed under titanocene catalysis: Gansäuer A, Pierobon M, Bluhm H. Angew Chem, Int Ed. 1998;37:101–103.Gansäuer A, Bluhm H, Pierobon M. J Am Chem Soc. 1998;120:12849–12859.
- 9.See, for example: Vilotijevic I, Jamison TF. Science. 2007;317:1189–1192. doi: 10.1126/science.1146421.Byers JA, Jamison TF. J Am Chem Soc. 2009;131:6383–6385. doi: 10.1021/ja9004909.Morten CJ, Byers JA, Van Dyke AR, Vilotijevic I, Jamison TF. Chem Soc Rev. 2009;38:3175–3192. doi: 10.1039/b816697h.
- 10.a) Hine J, Linden SM, Kanagasabapathy VM. J Am Chem Soc. 1985;107:1082–1083. [Google Scholar]; b) Hine J, Linden SM, Kanagasabapathy VM. J Org Chem. 1985;50:5096–5099. [Google Scholar]; c) Yamada T, Morisseau C, Maxwell JE, Argiriadi MA, Christianson DW, Hammock BD. J Biol Chem. 2000;275:23082–23088. doi: 10.1074/jbc.M001464200. [DOI] [PubMed] [Google Scholar]; d) Omoto K, Fujimoto H. J Org Chem. 2000;65:2464–2471. doi: 10.1021/jo9916333. [DOI] [PubMed] [Google Scholar]; e) Fleming EM, Quigley C, Rozas I, Connon SJ. J Org Chem. 2008;73:948–956. doi: 10.1021/jo702154m. [DOI] [PubMed] [Google Scholar]; e) Wurst JM, Liu G, Tan DS. J Am Chem Soc. 2011;133:7916–7925. doi: 10.1021/ja201249c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For selected reviews describing transition-metal catalyzed transfer hydrogenation, see: Zassinovich G, Mestroni G, Gladiali S. Chem Rev. 1992;92:1051–1069.Noyori R, Hashiguchi S. Acc Chem Res. 1997;30:97–102.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem, Int Ed. 2009;48:34–46. doi: 10.1002/anie.200802938.
- 12.Details of substrate synthesis are provided as Supporting Information.
- 13.Subjecting enantioenriched epoxide 1a (88% ee) to the standard conditions for Ni-catalyzed reductive coupling provided homoallylic alcohol 2a (88% ee) with complete preservation of optical purity. See Supporting Information for details.
- 14.Preliminary investigation of the reaction conditions indicated that i-PrOH was the optimal alcohol reducing agent. See Supporting Information for additional details of reaction optimization including alcohol reducing agent, Ni(II) precatalyst and temperature.
- 15.Ananikov VP, Gayduk KA, Starikova ZA, Beletskaya IP. Organometallics. 2010;29:5098–5102. [Google Scholar]
- 16.a) Amatore C, Jutand A, M’Barki MA. Organometallics. 1992;11:3009–3013. [Google Scholar]; b) Ozawa F, Kubo A, Hayashi T. Chem Lett. 1992:2177–2180. [Google Scholar]; c) Fors BP, Krattiger P, Strieter E, Buchwald SL. Org Lett. 2008;10:3505–3508. doi: 10.1021/ol801285g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Sacco A, Mastrorilli P. J Chem Soc, Dalton Trans. 1994:2761–2764. [Google Scholar]; b) Hubert-Pfalzgraf LG, Kessler VG, Vaisserman J. Polyhedron. 1997;16:4197–4203. [Google Scholar]
- 18.Phosphine-catalyzed conjugate addition of i-PrOH to the alkynoate was observed, which has been reported previously: Inanaga J, Baba Y, Hanamoto T. Chem Lett. 1993:241–244.Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035–1050.
- 19.Saito S, Yamamoto Y. Chem Rev. 2000;100:2901–2915. doi: 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]
- 20.a) Aye KT, Gelmini L, Payne NC, Vittal JJ, Puddephatt RJ. J Am Chem Soc. 1990;112:2464–2465. [Google Scholar]; b) Bäckvall JE, Bökman F, Blomberg MRA. J Am Chem Soc. 1992;114:534–538. [Google Scholar]; c) Kulasegaram S, Kulawiec RJ. Tetrahedron. 1998;54:1361–1374. [Google Scholar]
- 21.Oxidative addition of epoxides with [Lewis acid][Co(CO4)] complexes has also been observed to occur with inversion of configuration: Church TL, Getzler YDYL, Coates GW. J Am Chem Soc. 2006;128:10125–10133. doi: 10.1021/ja061503t.
- 22.Bäckvall J-E, Karlsson O, Ljunggren SO. Tetrahedron Lett. 1980;21:4985–4988. [Google Scholar]
- 23.Oxidative addition of tosyl-aziridines with Ni(0) has been observed to occur with inversion of configuration: Lin BL, Clough CR, Hillhouse GL. J Am Chem Soc. 2002;124:2890–2891. doi: 10.1021/ja017652n.
- 24.The relative stereochemistry of monodeuterated tetrahydropyran 4 was confirmed by synthesis of the tert-butyldiphenyl silyl ether (TBDPS-4) and comparison to the protio-congener (TBDPS-2a). See Supporting Information for complete experimental details and stereochemical analysis.
- 25.The mechanism of migratory insertion is generally accepted to occur with retention of configuration at the metal-bonded carbon. For the seminal study, see: Bock PL, Boschetto DJ, Rasmussen JR, Demers JP, Whitesides GM. J Am Chem Soc. 1974;96:2814–2825.For further discussion of migratory insertion as it pertains to the stereochemical outcome of transition-metal mediated processes, see: Lau KSY, Wong PK, Stille JK. J Am Chem Soc. 1976;98:5832–5840.Spencer J, Pfeffer M. Tetrahedron: Asymmetry. 1995;6:419–426.Malinakova HC. Chem–Eur J. 2004;10:2636–2646. doi: 10.1002/chem.200305667.Lu G, Malinakova HC. J Org Chem. 2004;69:4701–4715. doi: 10.1021/jo040148r.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.