

Abstract
Synthetic studies toward the total synthesis of (+)-acutiphycin (1) resulted in the discovery of additive-free, highly regioselective nickel-catalyzed reductive coupling reactions of aldehydes and 1,6-enynes and the construction of an advanced intermediate in studies directed toward the synthesis of 1. Ultimately, though not employing the nickel-catalyzed reaction, a highly convergent total synthesis of (+)-acutiphycin featuring an intermolecular SmI2-mediated Reformatsky coupling reaction and macrolactonization initiated by a retro-ene reaction of an alkoxyalkyne was achieved. The resulting synthesis was 18 steps in the longest linear sequence from either methyl acetoacetate or isobutyraldehyde.
Introduction
The complex macrolide (+)-acutiphycin (1) was isolated in 1984 by Moore and coworkers and possesses potent in vivo antineoplastic activity against murine Lewis lung carcinoma, as well as significant cytotoxicity against KB and NIH/3T3 cell lines.1 Since the natural source of acutiphycin (the blue-green alga Oscillatoria acutissima) no longer produces this metabolite, detailed investigations of its mechanism of action and therapeutic potential have been very limited and further studies must be fueled by chemical synthesis. Smith reported the first total synthesis of 1 in 1995,2 and a series of studies directed towards the total synthesis of 1 have also been described by Kiyooka.3 The strategies employed in both the Smith synthesis and the Kiyooka approach are linear in nature, whereas we recently reported the first convergent total synthesis of (+)-acutiphycin.4 Herein, we describe our initial strategy for the total synthesis of (+)-acutiphycin and the discoveries that resulted from this approach. A detailed description of the successful route to (+)-acutiphycin is also provided.
The nickel-catalyzed reductive coupling of alkynes and aldehydes5 has been shown to be a versatile tool in the synthesis of natural products.6 Although regioselectivity is optimal for aromatic alkynes7 (Scheme 1; eq 1) and 1,3-enynes (eq 2),8 good levels of regiocontrol have also been observed for alkynes containing two distinct alkyl substituents (eq 3).9 All of these transformations give exclusive syn addition to the alkyne, resulting in the formation of (E)-trisubstituted allylic alcohols, and allows for the possibility of catalyst and/or reagent control. In our initial approach to (+)-acutiphycin, we intended to form both of the (E)-trisubstituted olefins and to establish the configurations at C7 and C13 using these catalytic processes (Scheme 2). In addition, due to the challenges associated with macrolactonization en route to 1,2b we initially investigated an alternative C–C bond-forming strategy to close the macrocycle: nickel-catalyzed reductive macrocyclization. Although we considered both reductive coupling reactions to be challenging, the C14–C15 bond was targeted for the ring closing step since the range of oxidation states present along the C1–C7 backbone would make it difficult to reveal the C7 aldehyde. In contrast the remaining C–C bond would be formed via a Claisen condensation with acetate 6.
Scheme 1.

Nickel-catalyzed reductive coupling reactions of alkynes
Scheme 2.

Retrosynthetic analysis of (+)-acutiphycin
Results and Discussion
The synthesis of the C2–C7 fragment began with enantiomerically enriched 7,10 a well known intermediate available by alkylation of methyl acetoacetate and subsequent asymmetric reduction (Scheme 3).11,12 Protection of 7 as the silyl ether followed by reductive debenzylation and oxidation provided 3. Although hydroxyl groups have been shown to direct addition to aldehydes via chelation,13 we choose a non-chelating protective group, TBDPS, since chelation control via hydroxyl groups has not, to date, been demonstrated in nickel-catalyzed reductive coupling reactions of alkynes and aldehydes.14
Scheme 3.

Synthesis of aldehyde fragment 3
As shown in Scheme 4, enyne 5 (X = CH2) was selected rather than 4 (X = O) in order to avoid competitive reductive cyclization during the fragment coupling with 3, as well as other competing reactions in the Claisen condensation with 6. After the reductive coupling step, oxidative cleavage of the terminal olefin would reveal the necessary aldehyde functional group. Additionally, although C11 is in the ketone oxidation state in the natural product, the potential for epimerization2,3 at C10 and other complications suggested that the prudent choice would be to mask C11 as a protected hydroxyl group. The synthesis of 5 began with an indium-mediated addition of prenyl bromide to 8, a commonly used derivative of the Roche ester,15 to give 9 (Scheme 4).16,17 Protection of the secondary alcohol followed by selective deprotection of the primary alcohol and the Ley oxidation18 provided 10 in good yield over three steps. Treatment of 10 with the Seyferth-Gilbert reagent19 provided a terminal alkyne that was then methylated to yield 5. The third necessary fragment was available from racemic heptene oxide by way of Jacobsen’s hydrolytic kinetic resolution (Scheme 5).20 Addition of a lithium anion derived from propyne to 11 and subsequent conversion to the acetate ester provided 6.
Scheme 4.

Synthesis of enyne 5 via indium-mediated addition of prenyl bromide
Scheme 5.

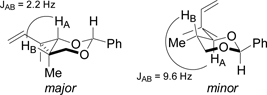
Synthesis of alkyne 6 via HKR
Studies of Nickel-Catalyzed Reductive Fragment Coupling Operations
Based on data obtained in early model studies,21 we reasoned that (+)-NMDPP would be an excellent candidate ligand for stereoselective reductive coupling of 3 and 5 (Scheme 6; Table 1, entry 1). Although the regioselectivity was much greater than expected,22 the yield in these reactions was disappointingly low. Moreover, the diastereoselectivity was largely invariant with respect to the ligand, as demonstrated by the fact that both the other enantiomer of NMDPP and an achiral ligand provided the same sense and essentially the same degree of diastereoselectivity (entries 2 and 3). The latter result was most unexpected and prompted us to test the reaction in the absence of a phosphine ligand. Not only was this reaction effective, but a significant increase in yield was observed, and the high degree of regio- and diastereocontrol were maintained (entry 4). The success of this coupling stood in stark contrast to all of our previous experience with this chemistry, in which we had never observed any coupling product in the absence of a phosphine ligand.
Scheme 6.

Nickel-catalyzed reductive coupling reactions of 1,6-enyne 5
Table 1.
Discovery of an Olefin-Directing Effect in 1,6-Enynesa
| entry | Phosphine | Yield | drb |
|---|---|---|---|
| 1 | (+)-NMDPP | 39% | 80 : 20 |
| 2 | (–)-NMDPP | 45% | 77 : 23 |
| 3 | P(o-anisyl)3 | 52% | 80 : 20 |
| 4 | none | 84% | 80 : 20 |
In all cases the reaction was run neat in 350 mol% Et3B using 10 mol% Ni(cod)2, and (if employed) 10 mol% phosphine.
Determined by 1H NMR.
Knochel had previously reported the favorable interaction of a distal alkene in nickel-catalyzed cross-coupling reactions of alkyl halides with dialkylzinc reagents.23 Based on our own results and this precedent, we proposed that the terminal olefin was coordinating to the nickel center, forcing the aldehyde to bind adjacent to carbon a (Scheme 7).24 This hypothesis was studied in more detail, and we have since determined that the high regioselectivity in phosphine-free nickel-catalyzed reductive coupling reactions is general for and specific to 1,6-enynes, while other enynes failed to react (Scheme 8, Table 2).25
Scheme 7.

Possible binding mode of 1,6-enyne 5
Scheme 8.

Directing effects of tethered alkenes
Table 2.
Directing Effects of Tethered Alkenes a
| entry | enyne | n | yield (%) | regioselectivity (A:B)b |
|---|---|---|---|---|
| 1 | 1,3- | 0 | <5 | n.d. |
| 2 | 1,4- | 1 | <5 | n.d. |
| 3 | 1,5- | 2 | <5 | n.d. |
| 4 | 1,6- | 3 | 53 | >95 : 5 |
| 5 | 1,7- | 4 | <5 | n.d. |
Standard procedure: The alkyne (0.50 mmol) was added to a 0 °C solution of Ni(cod)2 (0.05 mmol), i-PrCHO (1.00 mmol), and Et3B (1.00 mmol) in EtOAc (0.5 mL), and the solution was allowed to stir 15 h at room temperature.
Determined by 1H NMR and/or GC.
Unfortunately, the major diastereomer observed in the coupling of 3 and 5 was of the opposite configuration to that found in 1. As the use of a phosphine additive was detrimental to reaction yield and the possibility of achieving efficient reagent control was limited, we were left to consider the impact of the stereocenters of 3 and 5. As C11 is a ketone in (+)-acutiphycin we had the luxury of using epi-C(11)-5 (13). In order to probe the hypothesis that chiral centers on the ‘tether’ of a 1,6-enyne might influence the diastereoselectivity in nickel-catalyzed reductive coupling reactions of aldehydes and 1,6-enynes, we first synthesized model substrate 14 and investigated it in a reductive coupling with isobutyraldehyde (Scheme 9).26
Scheme 9.

Diastereselective reductive coupling reaction of 1,6-enyne 14
Since 15 was isolated as a single regioisomer and 95:5 diastereoselectivity was observed, the reductive coupling study of 14 clearly demonstrated the impact of chiral centers on the tether of a 1,6-enyne on the stereochemical outcome of coupling reactions. With this result in hand, we prepared 13, in which the C11 stereocenter on the tether of our 1,6-enyne fragment had been inverted, using Marshall coupling27 of aldehyde 1628 and propargylic mesylate 1729 (Scheme 10). Despite the steric bulk of aldehyde 16, this Marshall coupling proceeded with excellent yield and enantioselectivity to afford the desired anti product 18 as the only observable diastereomer. Protection and methylation then provided 13 in six linear steps from tiglic acid.
Scheme 10.

Synthesis of 1,6-enyne 13 via the Marshall coupling
Gratifyingly, 13 coupled with 3 in a manner analogous to 5, in this way providing the desired (S)-allylic alcohol as the major product (Scheme 11). The diastereomeric alcohols were then converted to their corresponding lactones with PPTS to enable their chromatographic separation and characterization.30 The strong dependence of diastereoselectivity on the configuration of the remote C11 stereocenter provides further evidence of olefin coordination to the metal center. It is also noteworthy that this coordination appears favorable despite the considerable steric bulk along the tether of the 1,6-enyne.
Scheme 11.

Phosphine free nickel-catalyzed reductive coupling reactions
Consequences of the 1,6-Enyne Approach to (+)-Acutiphycin
Allylic alcohol 20 was carried on to the Claisen condensation as a mixture of diastereomers (Scheme 12). Fortuitously, the undesired (minor) diastereomer failed to form the hemiketal and was easily separated from the desired (major) isomer by passing the crude material through a pad of silica. After methanolysis, compound 22 was obtained as a single diastereomer in 36% overall yield from 3 and 13. This route thus afforded the entire carbon skeleton of (+)-acutiphycin in three consecutive steps. Unfortunately, conversion of the terminal olefin of 22 to the necessary aldehyde proved extremely challenging, as reaction at the C8–C9 olefin was observed exclusively under ozonolysis or epoxidation conditions. Although dihydroxylation was selective for desired terminal olefin, conversion was very low (<10%) and failed to provide sufficient material to study subsequent steps.
Scheme 12.

Claisen condensation to macrocyclic precursor 22
Although the initial retrosynthetic plan for (+)-acutiphycin did not lead to the completion of the total synthesis, it revealed the general utility of 1,6-enynes as substrates for highly regioselective nickel-catalyzed reductive coupling reactions with aldehydes. Additionally, the phosphine-free nickel-catalyzed reductive coupling of 3 and 13 had successfully provided a challenging stereocenter and the E-trisubstituted olefin, while also serving as an effective fragment coupling.
Total Synthesis of (+)-Acutiphycin
The initial approach to (+)-acutiphycin allowed for efficient access to three complex fragments. In the revised approach, we sought to retain this convergence as much as possible. However, as the C13–C14 bond had proven to be a significant obstacle in our synthetic efforts, we decided to consider the C14–C15 olefin as an alternate disconnection (Scheme 13). The C7–C8 disconnection was retained, and both the C14–C15 bond and the ester linkage were considered candidates for ring closing. The C15–C22 and C3–C7 fragments were largely unchanged from our initial route; however, our 1,6-enyne now required a ketone functional group and two additional carbons. A silyl-enol ether (25) was targeted for its potential use in a Mukaiyama aldol strategy.
Scheme 13.

Revised retrosynthetic analysis of (+)-acutiphycin
Once again the Marshall coupling served as an excellent means to access homopropargylic alcohol 27 (Scheme 14). Although we were unaware of any precedent for performing the Marshall coupling in the presence of a ketone, this approach seemed viable since organozinc species react significantly more slowly with ketones than with aldehydes.31 Indeed, the β-keto-aldehyde 2632 proved a viable substrate for these conditions, providing 27 as the anti diastereomer in excellent yield and enantioselectivity.33 Protection and methylation afforded 25 in five linear steps from isobutyraldehyde.
Scheme 14.

Synthesis of silyl-enol ether 25 via the Marshall coupling
Ideally, the enol ether would act similarly to the terminal olefin of 13 in directing regioselectivity and diastereoselectivity of nickel-catalyzed reductive coupling reactions with 3. Unfortunately, it was discovered that trisubstituted enol ethers were not suitable directors in phosphine-free nickel-catalyzed reductive coupling reactions.34 However, both 25 and 2835 could be joined with 3 via the hydrozirconation-transmetallation chemistry of Wipf (Scheme 15).36 This sequence provided the E-trisubstituted allylic alcohols in excellent regioselectivity as easily separable mixtures of diastereomers, with the desired (S)-allylic alcohols (23, 29) being favored.37 A Claisen condensation with 3038 and subsequent methanolysis provided 31. Oxidation of the primary alcohol to the aldehyde was successful; however, the resultant β-acetoxy aldehyde was prone to elimination, liberating a carboxylic acid. This sensitivity, coupled with the stability of the silyl enol ether, prevented the use of a Mukaiyama aldol reaction to close the macrocycle.
Scheme 15.

Consecutive fragment coupling reactions of 25, 3 and 30
Unanticipated Macrodiolide Formation
The SmI2-promoted Reformatsky reaction was considered milder way to access the necessary enolate equivalent.39 Electrophilic bromination of 31 and subsequent oxidation of the primary alcohol afforded 32 (Scheme 16). Slow addition of 32 to a dilute solution of SmI2 in THF at –78 °C resulted in the formation of a new product originally thought to be the desired macrocycle. However, exposure to Martin sulfurane40 resulted in the formation of a product which contained both the characteristic signals of an enone and of a β-hydroxy ketone in the 1H NMR. A HRMS determined that the exact mass of this compound was 1711.0651, which corresponds to the sum of the exact masses of a monomeric macrocyclic enone (846.5286) and a monomeric macrocyclic β-hydroxy ketone (864.5392).41 Neither the mass of the monomeric enone nor the β-hydroxy ketone were observed in the mass spectrum. Consequently, it was concluded that the SmI2 Reformatsky reaction had produced the macrodiolide (32 membered ring) and the product obtained after the mono-dehydration was 33.42 This intermolecular-coupling macrocylization sequence was very unexpected since the preference for intramolecular addition in SmI2 mediated Reformatsky reactions is well documented.39,43
Scheme 16.

Unexpected formation of the macrodiolide
Macrolactonization Based Strategy
Our focus then shifted to formation of the C14–C15 olefin via an intermolecular strategy, with the intention of using macrolactonization to close the ring. Some of these strategies are briefly summarized by their respective fragments as shown in Scheme 17. The main obstacle in all these approaches was poor reactivity at the C14 center, due presumably to the steric bulk at C12. Originally it was anticipated that the pKa difference between an ester and a ketone would be sufficient to obtain selective enolate formation at C14. However, when the aldol reaction was explored with 35 and 36, the C4 protons proved to be more easily abstracted than those at C14, resulting either in the elimination of the TBDPSO group, or C–C bond formation between C4 and 24. Therefore, strategies such as the Mukaiyama aldol, cross-metathesis, and Zn-mediated Reformatsky reactions, which include a built-in bias towards reactivity at C14, were considered. However, these systems simply proved unreactive and failed to provide any of the desired C–C bond. The Horner-Wadsworth-Emmons (HWE) strategy was not fully tested because of our inability to form the necessary β-keto-phosphonate from 36, probably also due to the steric bulk at C12.
Scheme 17.

Summary of fragment coupling attempts at C14–C15
An unusual application of the Reformatsky reaction, however, provided an efficient and novel solution to this problem (Scheme 18). Electrophilic bromination of 23 provided the requisite α-bromoketone (36) in quantitative yield. While activated zinc had failed to generate the desired enolate,44 SmI2 did so, affording a β-hydroxy ketone derived from 36 and 2445 in excellent yield (90%, 1.0 mmol scale) as a mixture of diastereomers. Dehydration with the Martin sulfurane provided 39 in an overall yield of 72% over two steps.
Scheme 18.

SmI2-mediated fragment coupling reaction.
While SmI2 has been commonly employed in intramolecular Reformatsky reactions, its use in intermolecular cases has been extremely limited due to the numerous side reactions that can occur.46 We propose that the α-quaternary center of 37, which had proved the downfall of the previous methods, prevents oxidative dimerization of the samarium enolate and other competing SmI2-mediated pathways. When coupled with subsequent dehydration, this two-step sequence is complementary to the Horner-Wadsworth-Emmons strategies, and it may find use in other sterically hindered systems. Further studies to investigate the generality of this approach are currently underway.
Hydrofluoric acid selectively removed both Et3Si groups in the presence of the TBDPS group affording the β-hydroxyl group necessary for directed reduction (Scheme 19). Although syn-selective directed reductions of β-hydroxy ketones are well known, we were surprised to find that there were no prior examples with an all-carbon quaternary center between the directing alcohol and the carbonyl undergoing reduction, as is the case here.47 Indeed, one of the most common syn-selective techniques (Et2BOMe, NaBH4)48 was completely unsuccessful in this system; however, the technique developed by Evans utilizing catecholborane provided and efficient solution to this problem.49 The syn-stereochemistry of diol 40 and the C14-C15 olefin geometry was determined by nOe analysis of the acetonide derivative 41, the configuration at C13 was further supported by the 13C spectra of 41.50
Scheme 19.

Syn-selective reduction and determination of stereochemistry
Our early attempts at macrolactonization focused on a strategy similar to that of Smith (Scheme 20).2 Although the Yamaguchi protocol51 was successful in formation of the macrolactone, the mixed anhydride intermediate was very moisture sensitive and consequently the yield was variable and often very low. Moreover, elimination of methanol resulted in formation of 44 as the major product, which could be partially converted to 43 by refluxing in methanol with citric acid. However, the rate of conversion was slow, and despite extended reaction times, the reaction did not proceed to completion. Moreover, 43 was not separable from 44 by chromatography.52
Scheme 20.

Yamaguchi macrolactonization
Conversion of 40 to 45 is formally the addition of ketene to the lactone as a nucleophile and also react with the 2° alcohol as an electrophile (Scheme 21). An alkoxyethyne seemed ideally suited for this purpose. Deprotonation of the alkyne terminus would provide an efficient nucleophile, and alkoxyalkynes are known to undergo thermal decomposition to ketenes,53 which are potent electrophiles.54 The lithium anion of ethoxyethyne (46) smoothly added to the carboxyl at C3 to give tetraol 47 (Scheme 22). Slow addition of 47 to refluxing xylenes and Bu3N effected a thermal retro-ene reaction to form ethylene and ketene 48 that then underwent a highly group-selective coupling with the least hindered (yet most remote) of the 4 hydroxyl groups to give the desired macrocycle (45) in excellent yield (90%).
Scheme 21.

Hypothetical ketene-based macrolactonization
Scheme 22.

Macrolactonization by way of in situ retro-ene reaction
This macrolactonization method was first reported by Funk as a mechanistic probe55 but has not been employed previously in the context of total synthesis.56 As alkynyl ethers lack acidic α-hydrogens, they avoid the problem of competing enolate formation that plagues many macrolactonization techniques.57 Because of these features, as well as the fact that macrolactonization is one of the most commonly utilized strategies in complex molecule synthesis, this retro-ene-macrocyclization certainly warrants further consideration in the field of natural product synthesis.
In contrast to 44, methanolysis of 45 proceeded efficiently in 10 hours to give 43 in >99% yield (Scheme 23). Selective silylation of the allylic alcohol, Dess-Martin oxidation,58 and exposure to HF afforded 49. Crystallization from diethyl ether/pentanes allowed for an X-ray crystal structure determination of 49 (Figure 1). This is the only known crystal structure of an (+)-acutiphycin derivative and hopefully the structural information obtained from this compound can be used to further understand the mode of activity of (+)-acutiphycin.59 Finally the TBDPS was removed by treatment with acetic acid-buffered TBAF,2 completing the total synthesis of (+)-acutiphycin (1).
Scheme 23.

Completion of the total synthesis of (+)-acutiphycin
Figure 1.

X-ray Crystal structure of 49
Diethyl ether and disorder at the terminus of the C19–C22 chain omitted for clarity.
Conclusion
Nickel-catalyzed reductive coupling reactions of aldehydes and 1,6-enynes show great potential for use in total synthesis due to the high regioselectivity, good functional group tolerance, and substrate-controlled diastereoselectivity. Due to difficulties associated with the elaboration of the initial retrosynthetic plan, a new highly convergent synthesis of (+)-acutiphycin (1) was developed, with a longest linear sequence of 18 steps from either methyl acetoacetate (4.0%, 84% per step) or isobutyraldehyde (3.1%, 82% per step). Unique features of this work include the first application of an alkynyl ether as a macrolactone precursor in total synthesis, and the first use of an intermolecular, SmI2-mediated Reformatsky reaction as a fragment coupling operation. The modular nature of the route should enable rapid and systematic investigation of the structure-activity relationships of this potent natural product.
Experimental Section
Nickel-catalyzed reductive coupling of 3 with 5/13
Representative procedure for 12 (3 + 5 with no phosphine additive)
In a glovebox, Ni(cod)2 (5.5 mg, 0.02 mmol, 10 mol%) was added to a pre-dried 25 mL round bottom flask, if phosphine was included it was added (10 mol%) at this time (in this example it was not). The flask was then placed under argon on a Schlenk line and neat Et3B was added (0.10 mL, 0.69 mmol, 345 mol%). The solution was cooled to 0 °C and 3 (76.5 mg, 0.2 mmol, 100 mol%) was added followed by the 1,6-enyne (5) (57.5 mg, 0.205 mmol, 102 mol%). The reaction was stirred for 1 hour at 0 °C and then warmed to room temperature and stirred for 3 additional hours. The reaction was diluted with reagent grade EtOAc, opened to the atmosphere and stirred for 30 minutes. Solvent was removed in vacuo and crude material was purified via silica gel purified by silica gel chromatography (50:1 hexanes:diethyl ether ➔ 7:2 hexanes:diethyl ether) to give 112 mg (84%) of 12 as a mixture of diastereomers (c. 80:20 C7 R:S).
A similar procedure (1 mmole scale) was performed for 3 + 13, giving 435 mg (65%) of 20 as a mixture of diastereomers (c. 62:38 C7 S:R).
δ-Lactone (19)
Since the diastereomers could not be separated they were characterized as their δ-lactones. The following is a representative procedure: PPTS (1 mg, 0.004 mmole) was added to a solution of 12 (15 mg, 0.022 mmole) in benzene (1.5 mL), the vessel was sealed and heated to 60 °C for 2 hours, the solvent was removed in vacuo and the crude material purified by silica gel chromatography (10:1 hexanes:diethyl ether) to give 11 mg (79%) of 19 (more polar) and 2.7 mg (19%) of the other diastereomer (less polar). Spectral data for all four lactone products is provided in the supporting information.
19: [α]D +1.2 (c 1.46, 21 °C, CHCl3); IR 2959 (s), 2931 (s), 2858 (s), 1742 (s), 1472 (m), 1428 (m), 1380 (w), 1236 (m), 1112 (s), 1080 (s), 1028 (s), 911 (w), 834 (m), 702 (s); 1H NMR (500 MHz, CDCl3) δ 7.64 (m, 4H), 7.47 (m, 2H), 7.40 (m, 4H), 5.91 (dd, J = 17.6, 10.9 Hz, 1H), 5.51 (nOe 11.7%) (d, J = 9.9 Hz, 1H), 5.16 (nOe 11.7%) (dd, J = 10.6, 3.5 Hz, 1H), 4.97 (dd, J = 17.6, 1.4 Hz, 1H), 4.95 (dd, J = 10.9, 1.4 Hz, 1H), 4.30 (m, 1H), 3.26 (d, J = 2.0 Hz, 1H), 2.64 (m, 1H), 2.60 (dt, Jt = 2.4, Jd = 18.0 Hz, 1H), 2.45 (dd, J = 13.3, 4.5 Hz, 1H), 1.75 (m, 2H), 1.55 (d, J = 1.0 Hz, 3H), 1.09 (s, 9H), 1.01 (s, 3H), 1.00 (s, 3H), 0.95 (s, 9H), 0.91 (d, J = 6.5 Hz, 3H), 0.07 (s, 6H); 13C (125.8 MHz, CDCl3) δ 170.5, 146.5, 136.1, 135.8, 135.8, 133.3, 133.3, 130.3, 130.3, 129.6, 128.1, 128.1, 111.3, 82.6, 81.6, 64.6, 43.4, 39.0, 34.7, 34.4, 27.1, 26.5, 25.7, 23.8, 19.3, 18.9, 15.8, 11.9, –2.6, –3.6; HRMS m/z (ESI, M+H+) calcd 635.3946, found 635.3968.
(+)-δ-Lactone-silyl-enol-ethers (23, 29)
Representative procedure for coupling (23)
In a darkened fume hood a solution of Cp2Zr(H)Cl (694 mg, 2.7 mmol) and 25 (1.10 g, 2.6 mmol) in toluene (20 mL) was heated to 43 °C for 80 minutes then cooled to –65 °C. Dimethylzinc (2 M in toluene, 1.31 mL, 2.62 mmol) was slowly added to the cold solution followed by α,α-diphenyl-N-methyl-D-prolinol (107 mg, 0.40 mmol). The reaction was allowed to gradually warm up to –30 °C over 90 minutes, at which point 3 (768 mg, 2.0 mmol) in 5 mL toluene was added followed by a 3 mL rinse (toluene). The reaction was warmed to –25 °C and stirred for 50 minutes then warmed to –15 °C and stirred overnight. The reaction was warmed to room temperature then heated to 35 °C for 20 minutes, this was done to ensure complete conversion to the lactone. The reaction was cooled to 0 °C and quenched via the careful addition of saturated aqueous ammonium chloride. The aqueous layer was extracted with diethyl ether and the combined organics were washed with 0.1 M NaHSO4 and brine, dried over magnesium sulfate, filtered, concentrated and subject to chromatography: 50:1 hexanes/diethyl ether, 80 mL (removes excess 25 and related) ➔ 9:1 hexanes/diethyl ether, 1 L (23) ➔ 6:1 hexanes/diethyl ether (epi-C(7)-23), to give 810 mg 23 (52%) and 154 mg epi-C(7)-23 (10%) as clear oils (data for 23 only). [α]D +16.0 (c 1.85, 22 °C, CHCl3); IR 2957 (s), 2877 (s), 1747 (s), 1664 (w), 1460 (w), 1381 (w), 1317 (w), 1231 (m), 1111 (s), 1008 (s), 738 (s); 1H NMR (500 MHz, CDCl3) δ 7.65 (m, 4H), 7.46 (m, 2H), 7.40 (m, 4H), 5.63 (nOe 14.5%) (d, J = 9.5 Hz, 1H), 4.51 (q, J = 6.5 Hz, 1H), 4.31 (nOe 14.5%, 9.4%) (dd, J = 6.5, 3.5 Hz, 1H), 4.12 (nOe 9.4%) (m, 1H), 3.77 (s, 1H), 2.70 (ddd, J = 17.0, 6.0, 1.0 Hz, 1H), 2.62 (m, 1H), 2.49 (dd, J = 17.0, 8.0 Hz, 1H), 1.87 (m, 2H), 1.55 (d, J = 1.0 Hz, 3H), 1.50 (d, J = 6.5 Hz, 3H), 1.07 (s, 9H), 1.03–0.96 (m, 21 H), 0.89 (d, J = 7.0 Hz, 3H), 0.82 (s, 3H), 0.73 (q, J = 8.0 Hz, 6H), 0.64 (q, J = 8.0 Hz, 6 H); 13C (125.8 MHz, CDCl3) δ 171.3, 157.4, 135.9, 133.7, 133.4, 133.0, 130.3, 130.2, 128.9, 128.1, 128.0, 99.1, 82.9, 81.2, 77.2, 65.7, 46.1, 40.1, 37.4, 34.2, 27.0, 25.7, 21.2, 20.6, 19.2, 11.6, 7.5, 7.2, 6.4, 5.9; HRMS m/z (ESI, M+Na+) calcd 801.4738, found 801.4721.
(+)-Enone (39)
SmI2 (Strem, 0.1 M THF, 50 mL, 5.0 mmol) was added to a 500 mL teardrop flask that had been thoroughly purged with argon and cooled to –78 °C. A solution of 36 (765 mg, 1.03 mmol) and 24 (282 mg, 1.09 mmol) in THF (52mL) was added to the reaction vessel over 70 minutes. Excess SmI2 was oxidized by bubbling dry air through the solution until the solution turned yellow, the solution was poured into a separation funnel containing aqueous sodium thiosulfate, sodium bicarbonate and diethyl ether, the layers were separated and the aqueous layer extracted with diethyl ether. The combined organic layers were washed with saturated aqueous sodium thiosulfate (2x), dried over magnesium sulfate, filtered, concentrated and purified by chromatography: hexanes (flush out iodine) ➔ 10:1 hexanes/ethyl acetate ➔ 6:1 hexanes/ethyl acetate to give the β-hydroxy ketone as a mixture of 4 diastereomers (852 mg, 90%). To a cold (0 °C) solution of the β-hydroxy ketone (852 mg, 0.924 mmol) in DCM (70 mL) was added Martin sulfurane (Aldrich, 4.3 g, 6.39 mmol) and the reaction was stirred at 0 °C for 2 h, sealed and placed in a freezer (–4 °C) for 50 h. The reaction was quenched with saturated aqueous sodium bicarbonate and the product extracted with diethyl ether. The combined organic layers were washed four times with 1 M NaOH and once with brine, dried over magnesium sulfate, filtered, concentrated and purified by chromatography: hexanes ➔ 15:1 hexanes:diethyl ether (400 mL flush) ➔ 13:1 hexanes:ethyl acetate to give 665 mg (80%) 39 as a single detectable isomer. [α]D +20.5 (c 1.28, 21 °C, CHCl3); IR 2957 (s), 2933 (s), 2876 (s); 1746 (m), 1654 (w), 1463 (w), 1379 (w), 1231 (m), 1112 (s), 1008 (m), 739 (s); 1H NMR (500 MHz, CDCl3) δ 7.65 (m, 4H), 7.46 (m, 2H), 7.40 (ψt, J = 7.5 Hz, 4H), 6.05 (t, J = 9.5 Hz, 1H), 4.29 (dd, J = 7.0, 3.0 Hz, 1H), 4.18 (s, 1H), 4.11 (m, 1H), 3.78 (q, J = 5.5 Hz, 1H), 2.71 (dd, J = 17.5, 6.0 Hz, 1H), 2.49 (dd, J = 17.5, 8.5 Hz, 1H), 2.38 (q, J = 8.0 Hz, 1H), 2.32 (t, J = 6.0 Hz, 2H), 1.91 (m, 1H), 1.83 (m, 1H), 1.78 (s, 3H), 1.56 (s, 3H), 1.42 (bm, 3H), 1.32–1.24 (bm, 6H), 1.17 (s, 3H), 1.07 (s, 9 H), 1.03 (s, 3H), 1.01–0.93 (bm, 20H), 0.88 (t, J = 7.0 Hz, 3H), 0.66 (q, J = 8.0 Hz, 6H), 0.60 (q, J = 8.0 Hz, 6H); 13C (125.8 MHz, CDCl3) δ 211.2, 171.0, 138.0, 135.8, 133.6, 133.4, 132.1, 131.9, 130.8, 130.3, 130.2, 129.7, 128.1, 128.0, 82.3, 80.9, 71.6, 65.7, 54.2, 40.1, 37.3, 37.3, 36.5, 35.3, 32.2, 27.0, 25.5, 25.4, 22.9, 21.1, 20.8, 19.2, 14.8, 14.3, 12.0, 7.4, 7.1, 5.9, 5.2; HRMS m/z (ESI, M+Na+) calcd 927.5781, found 927.5770.
(–)-Alkoxyacetylene-tetraol (47)
n-BuLi (2.5 M in hexanes, 117 µL, 0.29 mmol) was added dropwise to a cold (–10 °C) solution of iPr2NH (40 µL, 0.29 mmol) in THF (7.2 mL) and the solution stirred for 15 minutes then cooled to –78 °C. Ethoxyethyne (63 wt% in hexanes, 45 µL, 0.29 mmol)60 was subsequently added, and the solution stirred for 50 minutes. After dry (azeotroped with anhydrous toluene) 40 (20 mg, 0.029 mmol) in THF (500 µL) was added dropwise down the side of the reaction vessel the reaction was stirred for 10 minutes at –78 °C and then warmed to –42 °C for 45 minutes. The reaction was quenched with pH 7.2 phosphate buffer and diluted with diethyl ether. The aqueous phase was extracted twice with ethyl acetate and the combined organic layers were washed with sodium bicarbonate and brine, and dried over sodium sulfate. The slurry was filtered, concentrated, and purified by chromatography (3:2 hexanes:ethyl acetate ➔ 4:5 hexanes:ethyl acetate) to give 47 as a clear oil (15.9 mg, 72%). Stereochemistry is unassigned, dr >10:1. [α]D –16.1 (c 0.4, 21 °C, CHCl3); IR 3365 (bm), 2929 (s), 2857 (s), 2226 (s), 1717 (w), 1654 (m), 1471 (m), 1428 (m), 1111 (s), 1008 (s), 703 (s); 1H NMR (500 MHz, CDCl3) δ 7.72 (m, 4H), 7.45 (m, 2H), 7.40 (m, 4H), 5.50 (d, J = 10.5 Hz, 1H), 5.46 (t, J = 7.0 Hz, 1H), 4.46 (apparent quint, J = 5.5 Hz, 1H), 4.25 (q, J = 7.0, 2H), 4.16 (d, J = 9.0 Hz, 1H), 3.99 (s, 1H), 3.66 (m, 1H), 3.50 (d, J = 2.0 Hz, 1H), 3.01 (bs, 1H), 2.82 (dd, J = 15.0, 7.5 Hz, 1H), 2.72 (dd, J = 15.0, 6.0 Hz, 1H), 2.65 (m, 1H), 2.48 (d, J = 2.5 Hz, 1H), 2.23 (t, J = 7.0 Hz, 2H), 1.75 (m, 1H), 1.69 (s, 3H), 1.50–1.40 (bm, 8H), 1.35–1.25 (bm, 9H), 1.06 (s, 9H), 0.98 (d, J = 7.0 Hz, 3H), 0.92 (s, 3H), 0.90 (t, J = 7.0 Hz, 3H), 0.72 (s, 3H); 13C (125.8 MHz, CDCl3) δ 185.4, 138.7, 136.2, 136.2, 135.6, 133.5, 133.4, 130.1, 130.1, 127.9, 127.9, 127.0, 125.7, 103.6, 86.9, 84.3, 77.6, 74.0, 71.9, 69.2, 52.2, 45.3, 42.7, 41.3, 37.3, 36.0, 34.1, 32.1, 29.9, 27.1, 25.6, 22.9, 20.6, 19.5, 15.9, 15.2, 14.6, 14.3, 11.9; HRMS m/z (ESI, M+Na+) calcd 771.4627, found 771.4639.
(–)-Macrocycle, hemi-ketal (45)
Dry (azeotroped with anhydrous toluene) 47 (13 mg, 17.4 µmol) in dry xylenes (24 mL) was added dropwise over 5 hours to refluxing (150 °C) xylenes (48 mL) and tri-n-butylamine (48 µL, 0.20 mmol). The reaction was stirred for an additional 20 minutes after the slow addition was complete, then poured into a separation funnel containing ice, and diluted with ethyl acetate. The organic layer was washed with 0.1 M NaHSO4 and brine, dried over magnesium sulfate, filtered, concentrated and purified by silica gel chromatography (20:1 hexanes:ethyl acetate ➔ 4:1 hexanes:ethyl acetate) to give 11.3 mg (90%) of 45 as a single diastereomer. [α]D –8.1 (c 0.19, 21 °C, CHCl3); IR 3452 (bm), 2929 (s), 2858 (s), 1710 (m), 1428 (m), 1378 (m), 1208 (s), 1112 (s), 1058 (s), 998 (s), 702 (s); 1H NMR (500 MHz, CDCl3) δ 7.67 (m, 4H), 7.43 (m, 2H), 7.38 (apparent t, J = 7.5 Hz, 4H), 5.42 (d, J = 10.5 Hz, 1H), 5.17 (d, J = 10.0 Hz, 1H) 5.12 (d, J = 2.0 Hz, 1H), 4.92 (m, 1H), 4.29 (apparent sept, J = 5.0 Hz, 1H), 4.12 (dd, J = 12.0, 2.0 Hz, 1H), 3.92 (s, 1H), 3.56 (s, 1H), 2.88 (m, 1H), 2.55 (d, J = 14.0 Hz, 1H), 2.47 (d, J = 14.0 Hz, 1H), 2.37 (m, 1H), 2.04 (d, J = 14.0 Hz, 1H), 1.99 (dd, J = 12.0, 3.5 Hz, 1H), 1.71 (m, 1H), 1.66 (s, 3H), 1.62 (d, J = 1.0 Hz, 3H), 1.55–1.48 (bm, 2H), 1.40–1.32 (bm, 2H), 1.30–1.23 (bm, 8H), 1.06 (s, 9H), 1.01 (d, J = 7.0 Hz, 3H), 0.99 (s, 3H), 0.87 (t, J = 7.0 Hz, 3H), 0.64 (s, 3H); 13C (125.8 MHz, CDCl3) δ 172.4, 136.3, 135.9, 134.5, 131.9, 131.4, 129.8, 129.8, 127.8, 127.8, 96.6, 81.2, 79.8, 76.5, 74.4, 66.5, 44.8, 44.2, 43.3, 38.6, 35.8, 34.1, 32.8, 31.8, 27.2, 25.1, 22.7, 22.2, 19.4, 19.4, 18.8, 14.2, 13.0, 11.1; HRMS m/z (ESI, M+Na+) calcd 743.4314, found 743.4334.
(+)-Macrocycle, methyl-ketal (43)
45 (11.3 mg, 15.7 µmol), citric acid (3.8 mg, 19.8 µmol) and methanol (30 mL) were combined in a sealed tube and then heated to 75 °C overnight. The crude mixture was concentrated and purified by chromatography (20:1 hexanes:ethyl acetate ➔ 4:1 hexanes:ethyl acetate) to give 11.5 mg (100%) of 43. [α]D +15.2 (c 0.083, 22 °C, CHCl3); IR 3447 (bm), 2926 (s), 2855 (s), 1725 (m), 1462 (m), 1378 (m), 1201 (m), 1113 (s), 1063 (s), 702 (s); 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 7.0 Hz, 4H), 7.44 (m, 2H), 7.38 (m, 4H), 5.63 (t, J = 7.0 Hz, 1H), 5.50 (d, J = 10.5 Hz, 1H), 4.78 (m, 1H), 4.17 (s, 1H), 4.12 (m, 1H), 3.76 (d, J = 11.5 Hz, 1H), 3.41 (d, J = 6.5 Hz, 1H), 2.98 (s, 3H), 2.86 (m, 1H), 2.74 (d, J = 13.5 Hz, 1H), 2.46 (d, J = 13.5 Hz, 1H), 2.44 (m, 1H), 2.20 (m, 1H), 2.01 (dd, J = 7.5, 4.5 Hz, 1H), 1.77 (m, 1H), 1.71 (s, 3H), 1.66 (m, 3H), 1.49 (bm, 2H), 1.37 (q, J = 11.5 Hz, 1H), 1.32–1.20 (bm, 9H), 1.05 (s, 9H), 1.01 (d, J = 7.0 Hz, 3H), 0.99 (s, 3H), 0.89 (s, 3H), 0.88 (t, J = 7.0 Hz, 3H); 13C (125.8 MHz, CDCl3) δ 169.7, 136.4, 136.4, 135.1, 135.0, 132.1, 130.4, 130.3, 128.5, 128.3, 128.3, 126.4, 100.4, 83.4, 80.4, 77.9, 75.3, 75.1, 67.1, 49.6, 44.5, 44.3, 43.7, 39.1, 35.0, 34.4, 32.4, 31.9, 30.4, 27.6, 25.8, 24.6, 23.2, 21.9, 20.7, 19.8, 14.8, 14.7, 13.5, 13.4; HRMS m/z (ESI, M+Na+) calcd 757.4470, found 757.4464.
(+)-Acutiphycin (1)
49 (3.9 mg, 5.4 µmol) was dissolved in THF (2.1 mL) and treated with 980 µL of TBAF/HOAc solution (TBAF 1 M THF, 2.5 mL; acetic acid 0.15 mL). The reaction was stirred at room temperature for 52 hours, diluted with ethyl acetate and washed with sodium bicarbonate (2x) and brine, dried over magnesium sulfate, filtered, concentrated and purified by silica gel chromatography (3:2 diethyl ether:hexanes) to give 1 as a white solid (2.4 mg, 92%). mp 150–151 °C; [α]D +151.6 (c 0.095, 21 °C, CH2Cl2); IR (solution in CDCl3) 3608 (m), 3457 (bw), 2985 (s), 2932 (m), 2902 (s), 1702 (m), 1643 (m), 1562 (m), 1298 (m), 1261 (m), 1216 (s), 1167 (s); 1H NMR (500 MHz, CDCl3) δ 5.39 (d, J =2.4 Hz, 1H), 5.29 (m, 2H), 4.98 (m, 1H), 4.64 (d, J = 3.8 Hz, 1H), 4.33 (dd, J = 12.0, 2.1 Hz, 1H), 4.28 (m, 1H), 4.95 (m, 1H), 2.67 (d, J =14.6 Hz, 1H), 2.62 (d, J = 14.6 Hz, 1H), 2.42 (ddd, J = 15.1, 10.7, 1.9 Hz, 1H), 2.18 (ddd, J = 11.9, 4.6, 1.3 Hz, 1H), 2.10 (apparent t, J = 13.6 Hz, 1H), 1.89 (dt, Jd = 12.2, Jt = 2.2 Hz, 1H), 1.78 (d, J = 1.3 Hz, 3H), 1.67 (s, 3H), 1.61–1.51 (m, 3H), 1.33–1.24 (m, 9H), 1.12 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H), 0.89 (s, 3H), 0.88 (t, J = 6.9 Hz, 3H); 1H NMR (500 MHz, 1:1 C6D6:CDCl3) δ 5.37 (bs, 1H), 5.21 (d, J = 10.4 Hz, 1H0, 5.16 (d, J = 11.2 Hz, 1H), 4.91 (m, 1H), 4.52 (s, 1H), 4.18 (d, J = 11.9 Hz, 1H), 4.01 (tt, J = 11.1, 4.3 Hz, 1H), 3.87 (m, 1H), 2.42 (d, J = 14.4 Hz, 1H), 2.29 (d, J = 14.4 Hz, 1H), 2.20 (ddd, J = 14.9, 10.7, 1.8 Hz, 1H), 1.97 (d, J = 12.8 Hz, 1H), 1.91 (m, 1H), 1.63 (s, 3H), 1.54 (m, 1H), 1.44 (s, 3H), 1.30 (m, 1H), 1.22–1.10 (m, 11H), 1.04 (m, 3H), 1.00 (dt, Jd = 2.2, Jt = 11.5 Hz, 1H), 0.85 (s, 3H), 0.82 (m, 3H); 13C (125.8 MHz, CDCl3) δ 215.7, 172.6, 135.1, 135.0, 131.1, 126.6, 96.8, 79.9, 76.1, 74.4, 64.7, 52.8, 44.8, 43.9, 43.3, 38.2, 35.5, 32.9, 31.8, 25.8, 25.2, 22.7, 19.3, 16.3, 14.2, 13.1, 11.3; 13C (125.8 MHz, DMSO-d6) δ 214.5, 170.9, 136.2, 134.9, 128.0, 123.6, 96.2, 77.3, 74.1, 73.8, 62.7, 53.1, 45.9, 43.5, 41.3, 38.2, 34.4, 31.7, 31.0, 24.4, 23.3, 22.0, 20.7, 16.7, 13.9, 12.9, 11.9; HRMS m/z (ESI, M+Na+) calcd 503.2979, found 503.2987.
Supplementary Material
Figure 2.

nOe evidence for stereochemistry of δ-lactone 19
Figure 3.

nOe evidence for stereochemistry of δ-lactones 23 and 29
Acknowledgment
This work was supported by the National Institute of General Medical Sciences (GM-063755). We thank Dr. Karen Miller-Moslin for thoughtful discussions on 1,6-enyne reductive coupling reactions. We are grateful to Dr. Li Li for obtaining mass spectrometric data for all compounds, and to Dr. Peter Müller for obtaining crystal structures of 27 and 49.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for 1, 3, 5, 6, 9, 10, 13, 18, 19, 21–25, 27, 29, 31, 36, 39–41, 45, 47, and 49 as well as all unknown (with the exception of the diastereomeric mixture of β-hydroxy ketones resulting from the Reformatsky reaction depicted in Scheme 18) intermediates en route to the successful total synthesis of 1. X-ray data (CIF) for 27 and 49. Available free of charge via the Internet at http://pubs.acs.org
References
- 1.Barchi JJ, Jr, Moore RE, Patterson FML. J. Am. Chem. Soc. 1984;106:8193–8197. [Google Scholar]
- 2.(a) Smith AB, III, Chen SS-Y, Nelson FC, Reichert JM, Salvatore BA. J. Am. Chem. Soc. 1995;117:12013–12014. [Google Scholar]; (b) Smith AB, III, Chen SS-Y, Nelson FC, Reichert JM, Salvatore BA. J. Am. Chem. Soc. 1997;119:10935–10946. [Google Scholar]
- 3.(a) Hena MA, Kim C-S, Horiike M, Kiyooka Si. Tetrahedron Lett. 1999;40:1161–1164. [Google Scholar]; (b) Kiyooka S-i, Hena MA. J. Org. Chem. 1999;64:5511–5523. doi: 10.1021/jo990342r. [DOI] [PubMed] [Google Scholar]
- 4.Moslin RM, Jamison TF. J. Am. Chem. Soc. 2006;128:15106–15107. doi: 10.1021/ja0670660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For a review of nickel-catalyzed coupling processes see: Montgomery J. Angew. Chem. Int. Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. Moslin RM, Miller-Moslin KM, Jamison TF. Chem. Commun. Advance article.
- 6.For representative examples nickel-catalyzed reductive coupling reactions of aldehydes and alkynes in total synthesis: Synthesis of (+)-allopumiliotoxin 339A. Tang XQ, Montgomery J. J. Am. Chem. Soc. 1999;121:6098–6099. Synthesis of(−)-terpestacin. Chan J, Jamison TF. J. Am. Chem. Soc. 2003;125:11514–11515. doi: 10.1021/ja0373925. Synthesis of (+)-amphidinolide T1. Colby EA, O’Brien KC, Jamison TF. J. Am. Chem. Soc. 2004;126:998–999. doi: 10.1021/ja039716v.
- 7.Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]
- 8.Miller KM, Jamison TF. J. Am. Chem. Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 9.Colby EA, Jamison TF. J. Org. Chem. 2003;68:156–166. doi: 10.1021/jo0264123. [DOI] [PubMed] [Google Scholar]
- 10.Available in two steps from methyl acetoacetate: Eggen M, Mossman CJ, Buck SB, Nair SK, Bhat L, Ali SM, Reiff EA, Boge TC, Georg GI. J. Org. Chem. 2000;65:7792–7799. doi: 10.1021/jo000767+.
- 11.(a) Lee BH, Biswas A, Miller MJ. J. Org. Chem. 1986;51:106–109. [Google Scholar]; (b) Huckin SN, Weiler L. Can. J. Chem. 1974;52:2157–2164. [Google Scholar]
- 12.Noyori R. Asymmetric Catalysis in Organic Synthesis. New York: John Wiley & Sons; 1994. p. 56. [Google Scholar]
- 13.For a review of chelation controlled additions to aldehydes see: Reetz MT. Angew. Chem., Int. Ed. 1984;23:556–569.
- 14.Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org. Lett. 2005;7:2937–2940. doi: 10.1021/ol050881k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roush WR, Palkowitz AD, Ando K. J. Am. Chem. Soc. 1990;112:6348–6359. [Google Scholar]
- 16.Arakis S, Ito H, Butsugan Y. J. Org. Chem. 1988;53:1831–1833. [Google Scholar]
-
17.Relative stereochemistry was assigned by comparison of coupling constants of the benzylidine derivatives of the major and minor diastereomers.
- 18.Griffith WP, Ley SV, Whitcombe GP, White AD. J. Chem. Soc., Chem. Commun. 1987:1625–1627. [Google Scholar]
- 19.(a) Seyferth D, Hilbert P, Marmor RS. J. Am. Chem. Soc. 1967;89:4811–4812. [Google Scholar]; (b) Gilbert JC, Weerasooriya U. J. Org. Chem. 1979;44:4997–4998. [Google Scholar]
- 20.(a) Tokunaga M, Larrow JF, Kakuchi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 21.The model study featured 1-cyclohexyl-propyne and (±)-3-(tert-butyl-dimethyl-silanyloxy)-3-phenyl-propionaldehyde and gave 77% yield, with 69:31 regioselectivity and 71:29 dr using (+)-NMDPP.
- 22.The highest selectivity observed with an alkyne featuring a 2° and a 1° terminus is 85:15 (Scheme 1; eq 3), and frequently lower selectivity is observed. See ref 9.
- 23.(a) Devasagayaraj A, Stüdemann T, Knochel P. Angew. Chem. Int. Ed. 1995;34:2723–2725. [Google Scholar]; (b) Giovannini R, Stüdemann T, Devasagayaraj A, Dussin G, Knochel P. J. Org. Chem. 1999;64:3544–3553. doi: 10.1021/jo982317b. [DOI] [PubMed] [Google Scholar]
- 24.The orientation of the aldehyde and mode of diastereoinduction has not been fully elucidated.
- 25.It was also determined that regioselectivity was reversed by the addition of PCyp3. For complete details see: Miller KM, Jamison TF. J. Am. Chem. Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. Moslin RM, Miller KM, Jamison TF. Tetrahedron. 2006;62:7598–7610.
- 26.For synthesis of 14 and the mechanistic implications of this study see: Moslin RM, Jamison TF. Org. Lett. 2006;8:455–458. doi: 10.1021/ol052719n. ref 23 (b)
- 27.Marshall JA, Adams ND. J. Org. Chem. 1999;64:5201–5204. doi: 10.1021/jo9823083. [DOI] [PubMed] [Google Scholar]
- 28.15 is available in 3 steps from tiglic acid via deconjugative methylanation: Aurell MJ, Gil S, Mestres R, Parra M, Parra L. Tetrahedron. 1998;54:4357–4366. See Supporting Information.
- 29.Both enantiomers of 3-butyn-2-ol are commercial available from Aldrich and may also be prepared according to: Marshall JA, Schaaf GM. J. Org. Chem. 2001;66:7825–7831. doi: 10.1021/jo015936k.
- 30.nOe analysis was used to confirm stereochemical assignment. See Supporting Information for details.
- 31.For a review detailing the difficulties associated with asymmetric additions to ketones and the difficulties associated with this as compared to aldehydes see: Betancort JM, Garcia C, Walsh P. J. Synlett. 2004:749–760.
- 32.Available in two steps from isobutryaldehyde: Shiojii K, Kawaoka H, Miura A, Okuma K. Synth. Commun. 2001;31:3569–3575.
- 33.Determined by X-ray crystallography. See Supporting Information.
- 34.Compound 24 as well as a TMS, TBS, and acetate-enol ether were all tested with and without a phosphine additive and in no case was the coupling product observed.
- 35.Early work focused on tert-butyl-dimethylsilyl (TBS) protecting groups, although no problems were encountered with this protecting group, triethylsilyl (TES) protecting group was chosen for later strategies to avoid anticipated difficulties in deprotection at the C11 site.
- 36.(a) Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197–5200. [Google Scholar]; (b) Wipf P, Ribe S. J. Org. Chem. 1998;63:6454–6455. [Google Scholar]
- 37.Determined by nOe analysis. See Supporting Information.
- 38.Synthesized in a manner analogous to 6. See Supporting Information for details.
- 39.For a discussion of SmI2-mediated Reformatsky reactions including their remarkable preference to react intramolecularly even in the case of medium-ring lactones see: Tabuchi T, Kawamura K, Inanaga J. Tetrahedron Lett. 1986;27:3889–3890. Inanaga J, Yokoyama Y, Handa Y, Yamaguchi M. Tetrahedron Lett. 1991;32:6371–6374.
- 40.Arhart RJ, Martin JC. J. Am. Chem. Soc. 1972;94:5003–5010. [Google Scholar]
- 41.The M + Na+ was recorded on the HRMS, hence the actual value was 1734.0549; however, for ease of discussion the M+ weights are described.
- 42.33 was not characterized further and its assignment is tentative.
- 43.For a review of intramolecular SmI2 mediated reactions see: Edmonds DJ, Johnston D, Procter D. J. Chem. Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a.
- 44.In this case, Zn/Ag–graphite was employed: Fürsnter A. Synthesis. 1989:571–590.
- 45.Available in five steps from 1-heptene in a manner similar to that described in Scheme 5. See Supporting Information.
- 46.Krief A, Laval AM. Chem. Rev. 1999;99:745–777. doi: 10.1021/cr980326e. [DOI] [PubMed] [Google Scholar]
- 47.Three syn-selective reductions of α,α’-difluoro-β-hydroxyketones using iBu2AlH/ZnCl2/TMEDA appear to be the closest and only precedents Kuroboshi M, Ishihara T. Tetrahedron Lett. 1987;28:6481–6484.
- 48.Chen K-M, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]
- 49.Evans DA, Hoveyda AH. J. Org. Chem. 1990;55:5190–5192. [Google Scholar]
- 50.(a) Rychnovsky SD, Skalitzky D. J. Tetrahedron Lett. 1990;31:945–948. [Google Scholar]; (b) Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;31:7099–7100. [Google Scholar]
- 51.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- 52.The elimination of methanol to give the ene-ester was also the major product in Smiths synthesis. See ref 2.
- 53.For an useful discussion of how different alkoxy substitutients affect the temperature at which ethylene is lost see: Moyano A, Pericàs MA, Serratosa F, Valentí E. J. Org. Chem. 1987;52:5532–5538.
- 54.Vollema G, Arens JF. Recl. Trav. Chim. Pays-Bas. 1963;82:305–321. [Google Scholar]
- 55.Funk RL, Abelman MM, Jellison KM. Synlett. 1989:36–37. [Google Scholar]
- 56.We are aware of only two reports of using this technique to form macrolactones: Magriotis PA, Vourloumis D, Scott ME, Tarli A. Tetrahedron Lett. 1993;34:2071–2074. Liang L, Ramaseshan M, Magee DI. Tetrahedron. 1993;49:2159–2168.
- 57.Parenty A, Moreau X, Campagne J-M. Chem. Rev. 2006;106:911–939. doi: 10.1021/cr0301402. [DOI] [PubMed] [Google Scholar]
- 58.The ketone product of this reaction was previously synthesized by Smith. See ref 2.
- 59.Based on NMR analysis Moore postulated a solution phase structure of (+)-acutiphycin, which closely matches the X-ray structure of 49. See ref 1.
- 60.Although not utilized here, lithiated ethoxyethyne can be generated in situ and added directly to aldehydes and ketones: Raucher S, Bray BL. J. Org. Chem. 1987;52:2332–2333.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.