

Abstract
Described in this work are total syntheses of amphidinolides T1 and T4 using two nickel-catalyzed reductive coupling reactions of alkynes, with an epoxide in one case (intermolecular) and with an aldehyde in another (intramolecular). The latter was used to effect a macrocyclization, form a C-C bond and install a stereogenic center with >10:1 selectivity in both natural product syntheses. Alternative approaches in which intermolecular alkyne-aldehyde reductive coupling reactions would serve to join key fragments were investigated and are also discussed; it was found that macrocyclization (i.e. intramolecular alkyne-aldehyde coupling) was superior in several respects (diastereoselectivity, yield, and length of syntheses). Alkyne-epoxide reductive couplings were instrumental in the construction of key fragments corresponding to approximately half of the molecule of both natural products. In one case (T4 series), the alkyne-epoxide coupling exhibited very high site selectivity in a coupling of a diyne. A model for the stereoselectivity observed in the macrocyclizations is also proposed.
Introduction
The amphidinolide family of marine macrolide natural products has attracted a great deal of interest due to the potent cellular effects and structural diversity displayed by its members.1 Several amphidinolides exhibit extremely potent cytotoxicity against murine lymphoma L1210 cells as well as human epidermoid carcinoma KB cells. To date, numerous subsets of amphidinolides have been identified (A–Y), each of which features a highly oxygenated macrolactone of varying ring sizes. Since the first reports of the amphidinolide family, considerable effort has been focused on synthesizing these macrolides, resulting in several innovative and efficient total syntheses.2
In particular, the amphidinolide T class (Figure 1) has garnered significant attention since its discovery in 2000.3 Members of this subclass, amphidinolides T1–5 (1–5), contain a 19-membered macrocycle, a trisubstituted tetrahydrofuran moiety, α-hydroxy ketone, exocyclic methylene group, and a homoallylic ester linkage. T3-T5 are the most closely related molecules, all containing a ketone at C13, hydroxyl group at C12, and methyl group at C14, and they differ only in their configuration at C12 and C14. Amphidinolide T2 (2) displays the same functionality at C12-C14 as 3–5, but contains an additional hydroxymethyl substituent at C18 where the other four members have an n-propyl group. Amphidinolide T1 (1) differs from 3–5 in the oxidation states at C12 and C13, possessing the reversed hydroxy ketone moeity.
Figure 1.

Amphidinolide T family of natural products.
Total syntheses of 1 and 3–5 have been reported to date. Amphidinolide T4 was synthesized in 2002 by Fürstner and coworkers by taking advantage of an efficient ring-closing metathesis to form the macrocycle.2k Related strategies were applied by the same group to the syntheses of T1, T3, and T5 reported in 2003.2l Amphidinolide T1 was first synthesized by Ghosh and Liu in 2003, utilizing a macrolactonization reaction to form the 19-membered ring.2j
Our interest in the amphidinolide T natural products stemmed from the presence of the α-hydroxy ketone and homoallylic ester moieties, both of which are patterns of functional groups that we have prepared using nickel-catalyzed, alkyne-electrophile reductive coupling reactions developed in our laboratory.4,5,6 Our synthetic strategy represents a novel approach to the T natural products, based on an alternate ring-closing method (Scheme 1). In contrast to all previously reported syntheses of amphidinolide natural products (i.e. T1 or otherwise), this approach features the installation of a stereogenic center (>10:1 diastereoselectivity) during the macrocyclization event. Herein, we disclose the implementation of this strategy in a full account of our synthesis of amphidinolide T17 and a previously unreported synthesis of T4.
Scheme 1.

Macrocyclization Bond Disconnections for Reported Syntheses of 1.
Retrosynthetic Analysis
The foundation of our strategy for the synthesis of both amphidinolide T frameworks (the hydroxy ketone array found in 1 and its reversed positioning in 2–5) is the use of nickel-catalyzed reductive coupling reactions of alkynes, the aforementioned intramolecular alkyne-aldehyde coupling reaction, as well as an intermolecular reductive coupling of an alkyne and epoxide. As shown in Scheme 2, the product of the former, an allylic alcohol, would serve as a latent α-hydroxy ketone moiety.
Scheme 2.

Intramolecular Nickel-Catalyzed α,ω-Alkynal Reductive Coupling Strategies.
Our plan for installing the hydroxy ketone of 1 and the “reversed” hydroxy ketone of 4 hinged upon analogous nickel-catalyzed reductive cyclizations of alkynals 8 and 9 to form the requisite macrocyclic allylic alcohols 6 and 7. As noted above, this process would not only close the 19-membered ring, but also concurrently install a stereogenic center. Control over the configuration of this center would thus be critical to the syntheses.
Although the positions of the alkyne and aldehyde portion of the corresponding alkynal cyclization substrates 8 and 9 are inverted relative to one another, earlier synthetic intermediates nevertheless share many structural and stereochemical features (Scheme 3). The synthesis of amphidinolide T1 would necessitate alkynyl acid 10 and homoallylic alcohol 11, while amphidinolide T4 requires alkenyl acid 12 and hydroxyenyne 13.
Scheme 3.

Intermolecular Alkyne-Epoxide Coupling Strategies.
Both homoallylic alcohols (11 and 13) were predicted to be available from intermolecular nickel-catalyzed reductive coupling reactions of alkynes and epoxides.5 Alcohol 11, for example, would be the product of the reaction between alkyne 14 and (R)-n-propyloxirane (16). While aryl alkynes couple with epoxides with excellent regioselectivity and good yield, in preliminary investigations of alkynylsilanes as coupling partners, the homoallylic alcohol product is not formed under the same conditions, returning only recovered alkyne and epoxide.8 We planned to exploit this difference in reactivity of alkynes in a group-selective coupling of diyne 15 with epoxide 16 to form a hydroxyenyne, which would be elaborated to 13.
Results and Discussion
Synthesis of Amphidinolide T1: Construction of C13-C21 Fragment via Intermolecular Alkyne-Epoxide Reductive Coupling
We commenced our studies by targeting α,ω-alkynal 8 in order to investigate catalytic, stereoselective reductive macrocyclization en route to amphidinolide T1 (1). Chiral alkyne 14 was synthesized in a three-step sequence (Scheme 4), beginning with alkylation of an Evans oxazolidinone with 3-bromo-1-phenyl-1-propyne.9 Cleavage of the auxiliary with lithium aluminum hydride followed by TBS protection furnished 14 in 70% yield and 98% ee. The requisite enantiomerically enriched epoxide 16 was easily prepared by way of Jacobsen’s hydrolytic kinetic resolution (HKR).10 Nickel-catalyzed union of these fragments proceeded smoothly, delivering homoallylic alcohol 11 in very good yield in >98:2 dr and with >98:2 regioselectivity with respect to both addition across the alkyne and opening of the epoxide. Optimal conditions were found when excess epoxide was employed (700 mol% relative to the alkyne) and the reaction was conducted without additional solvent. This transformation constitutes an efficient fragment coupling reaction providing rapid access to the C13-C21 portion of amphidinolide T1 in just four steps.
Scheme 4.

Synthesis of Amphidinolide T1: Assembly of C1-C12 Fragment
We next turned our attention to the synthesis of alkynyl acid 10. The key issues to address would be the installation of a highly substituted tetrahydrofuran ring as well as a remote carboxylic acid moiety with a stereogenic center at the α-position. We predicted that the first of these problems could be solved by a Lewis acid-mediated addition of a propargyl equivalent to a five-membered cyclic oxocarbenium ion. This type of reaction has been well-studied by Reiβig, who demonstrated that substituted five-membered lactols undergo smooth addition reactions with a variety of nucleophiles when treated with a Lewis acid, presumably through an oxocarbenium ion intermediate.11 Woerpel and coworkers have also conducted extensive studies regarding the stereochemical course of similar transformations (Scheme 5) in which 2-acetoxy-4,5-cis-disubstituted tetrahydrofuran derivatives undergo allylation with a high degree of stereocontrol to deliver products possessing a 2,4,5-trans-cis relationship across the tetrahydrofuran ring.12
Scheme 5.

Woerpel’s Study of Nucleophilic Addition to Oxocarbenium Ions (ref. 12).
Encouraged by this stereochemical precedent, we needed to choose an appropriate propargyl nucleophile equivalent13 to effect the desired addition reaction. At the onset of our investigations, we focused on Danheiser’s reports14 of allenylsilanes as effective propargyl nucleophiles in related Lewis acid-mediated additions to acyclic acetals, aldehydes and ketones.
Synthesis of the target oxocarbenium ion precursors began with an enantioselective addition of a chiral (Z)-crotyl borane developed by Brown15 to aldehyde 19,16 ultimately setting the C7-C8 syn relationship (Scheme 6). Hydroboration-oxidation of the crotylation product furnished a diol which was oxidatively cyclized, delivering lactone 20 in good yield over the three step sequence.17 Reduction of the lactone furnished lactol 21a, while direct acetate protection of this reduction product provided acetoxy acetal 21b.
Scheme 6.

With lactol 21a, acetal 21b, and allenylsilane 2218 in hand, an investigation of propargylation conditions was conducted. Gratifyingly, we found that the desired propargyl addition was indeed effected with concomitant removal of the TBS protective group (Scheme 7) when lactol 21a was treated with boron trifluoride diethyletherate and allenylsilane 22, providing the desired alcohol 23 in 41% yield and in very high diastereoselectivity (>95:5).
Scheme 7.

Allenylsilane Addition to Lactol 21a.
Several parameters were investigated in an endeavor to improve the yield of the reaction including concentration, temperature, different Lewis acids, addition rate of Lewis acid, and the use of acetal 21b, but no such efforts were fruitful in this regard (see Supporting Information for details). Accordingly, we concentrated on the nucleophilic component, aiming to increase the nucleophilicity19 of the allene toward the oxocarbenium ion by using allenylstannane 2420 in place of allenylsilane 22. To our satisfaction, the addition proceeded very smoothly with concomitant complete protiodesilylation, delivering terminal alkyne 25 in excellent yield (Scheme 8). Subsequent Sonogashira coupling with iodobenzene furnished target alcohol 23 in near quantitative yield, constituting a vast improvement over the one-step propargylation with allenylsilane 22 that we had reported previously.7
Scheme 8.

Allenylstannane Addition to Lactol 21a.
With the installation of the substituted tetrahydrofuran ring achieved by the allenylstannane addition, introduction of the remaining stereogenic center of carboxylic acid 10 was the next challenge to address. This was accomplished using an auxiliary-controlled, diastereoselective alkylation reaction. The pseudoephedrine-based method developed by Myers displays very high diastereoselectivity, allows for use of the iodide as the limiting reagent, and is well-suited for the alkylation of unactivated primary alkyl electrophiles.21 Alcohol 23 was converted to alkyl iodide 26 which was an effective electrophile for the desired alkylation reaction, proceeding in near quantitative yield and in excellent diastereoselectivity (Scheme 9). The alkylated amide underwent base-promoted hydrolysis to furnish alkynyl acid 10 in excellent yield and in >95:5 dr.
Scheme 9.

Synthesis of Alkynyl Acid 10.
Synthesis of Amphidinolide T1: Fragment Coupling and Reductive Macrocyclization
Enantiomerically pure homoallylic alcohol 11 and enantiomerically pure alkynyl acid 10 were joined in a DCC-mediated ester formation, affording 27 in 72% yield (Scheme 10). While this C–O bond formation was certainly satisfactory, we investigated an alternative, stereoselective fragment coupling via alkylation of an enolate derived from the propionate ester of alcohol 11. This approach would indeed save one step relative to the route described above, but it would nevertheless have to surpass the very high efficiency and complete stereocontrol provided by the Myers’ asymmetric alkylation method. Treatment of ester 28 with LDA and iodide 26 did indeed afford 27 (converging with the original route), but this process was nearly nonselective (1.6:1 dr, favoring the undesired diastereomer). Clearly, therefore, this strategy modification did not result in any improvement in the preparation of ester 27.
Scheme 10.

Contrasting Fragment Coupling Strategies.
After two functional group manipulations (TBS removal with TBAF and oxidation with the Dess-Martin periodinane reagent), we were poised to investigate conditions to form the 19-membered ring via an intramolecular alkyne-aldehyde reductive coupling reaction (Scheme 11). We began our studies with an achiral catalyst consisting of Ni(cod)2 and tributylphosphine, which had been very successful in other alkyne-aldehyde intramolecular reductive coupling reactions in our group.22 A dilute solution of alkynal 8 (0.05 M in toluene), 10 mol% nickel, 20 mol% phosphine and triethylborane was stirred at ambient temperature, but no reaction was observed (only 8 was isolated). A screening of phosphine ligands was conducted, but the desired cyclization was not observed in any case. We next explored the variables of concentration and temperature and were pleased to find that tributylphosphine/Ni(cod)2 promoted cyclization when the reaction mixture was heated to 60 °C, albeit in low yield (25–30%). Nevertheless, the stereoselectivity of the cyclization was outstanding; macrocyclic allylic alcohol 6 was isolated in greater than 10:1 diastereoselectivity possessing the (S)-carbinol configuration, corresponding to that found in the natural product, amphidinolide T1 (1).23 Further optimization efforts revealed that a higher catalyst loading improved the yield; the best conditions proved to be a 20 mol% Ni(cod)2/40 mol% tributylphosphine combination, giving 6 in a 44% yield again in >10:1 dr.24
Scheme 11.

Nickel-Catalyzed Reductive Macrocyclization.
Two features of the macrocyclization are noteworthy. First, we observed that the sense of induction matches the Felkin-Anh model25 of nucleophilic addition to chiral aldehydes, and we therefore hypothesized that the configuration at C14 of the alkynal is largely responsible for the sense of stereoinduction in the intramolecular coupling (macrocyclization). Moreover, the complete diastereocontrol observed in the reductive macrocyclization was quite surprising as intermolecular couplings of aldehyde 29 and alkyne 30 were virtually nonselective (Scheme 12), affording allylic alcohol 31 in 1.5:1 dr. Second, the intramolecular alkyne-aldehyde reductive coupling reaction used in Scheme 11 shortens the overall synthesis by several steps relative to an intermolecular coupling strategy that would require protection of both alkynyl acid 10 and alcohol 11 (and subsequent deprotection (prior to macrolactonization)).
Scheme 12.

Intermolecular Reductive Coupling Strategy.
Synthesis of Amphidinolide T1: Endgame
Macrocyclization of alkynal 8 overcame a significant hurdle in the synthesis of 1; all of the stereogenic centers of 1 had been set, and all of the carbon atoms within the ring framework of 1 were present in 6. The remaining challenge was differentiation of the two benzylidene groups at C12 and C16. Specifically, oxidative cleavage of the C12 benzylidene to a ketone and conversion of the C16 benzylidene to an exo-methylidene group were required.
We decided to exploit the difference in steric congestion around the carbon atoms at issue and accordingly protected the allylic hydroxyl group at C13 with a TBS group (Scheme 13). Subsequent global ozonolysis produced diketone 32 possessing very different environments around the two carbonyl groups. We predicted that the resultant C16 carbonyl (flanked by two CH2 groups) would be more susceptible to nucleophilic attack than the C12 carbonyl (flanked by a bulky silyl ether and a tetrahydrofuran β to C12), and treatment with a methylenating reagent would lead to the functional group pattern present in 1.
Scheme 13.

Synthesis of 1 via Selective Methylenation.
As we set our sights upon selective methylenation, we took into account that both carbonyl groups possessed β-oxygen substituents that might act as leaving groups under strongly basic conditions. We first investigated the Oshima reagent (Zn, CH2Br2, TiCl4),26 as well as the Lombardo-modified reagent “low-temperature aged”,27 but in both cases only diketone was recovered. Standard Takai conditions (Zn, CH2I2, TiCl4)28 were also unsuccessful, but replacement of TiCl4 by ZrCl4 and the addition of lead (II) chloride29 resulted in clean methylenation at the C16 carbonyl furnishing the desired monoketone in 65–75% yield.30
Finally, treatment of this product with HF-pyridine promoted clean conversion (>90% yield) to amphidinolide T1 (Scheme 13), whose spectroscopic, spectrometric, and physical properties matched those reported for natural product 1.
Synthesis of Amphidinolide T4 Framework
Completion of the total synthesis of 1 indicated that our strategy of reductive macrocyclization was indeed a viable route to amphidinolide T1. Nevertheless, a critical issue in using this strategy for the synthesis of other members (T2–5) was the reversed hydroxy ketone pattern at C12 and C13 (see Figure 1). In our next synthetic undertaking, we set out to address this issue by studying the reductive cyclization of alkynal 9, and whether it would exert a high degree of substrate diastereocontrol in a cyclization using an achiral catalyst system as it contains β-branching with respect to the aldehyde instead of α-branching (Scheme 4).
Synthesis of Amphidinolide T4 Framework: Construction of C13-C21 Fragment via Site-Selective, Intermolecular Diyne-Epoxide Reductive Coupling
Synthesis of the requisite alkynal 9 began with an investigation of the viability of a site-selective alkyne-epoxide reductive coupling of diyne 15. Alcohol 33 (previously synthesized in the T1 synthesis, Scheme 4) was oxidized to the aldehyde, converted to the dibromoolefin and finally exposed to methyllithium and chlorotrimethylsilane to arrive at diyne 15 (Scheme 14). We found that 15 did indeed undergo coupling with (R)-propyloxirane to give the desired hydroxyenyne 34 with >95:5 regioselectivity and >95:5 diastereoselectivity. While the crude product mixture was contaminated with products of polymerization of the alkyne, there was no evidence of epoxide coupling at the alkynylsilane. Coupling product 34 was converted to the target homoallylic alcohol 13 by treatment with TBAF and subsequent Sonogashira coupling with iodobenzene. The site-selective epoxide coupling streamlined the synthesis of the C13-C21 fragment by directly introducing the necessary alkyne at C13 rather than starting from T1 coupling product 11 and elaborating to the alkyne. The former route saves several steps, thus compensating for the moderate yield of the diyne coupling reaction.
Scheme 14.

Site-Selective Diyne-Epoxide Reductive Coupling.
Synthesis of Amphidinolide T4 Framework: Construction of C1-C12 Alkenyl Acid, Fragment Coupling and Alkynal Synthesis
Alkenyl acid 12 was targeted next to advance the investigation of the T3/T4 synthesis. This acid was synthesized in a route similar to alkynyl acid 10 (Scheme 15). Lewis acid-mediated allylation of lactol 21a by allyltrimethylsilane proceeded in excellent yield to deliver the primary alcohol, which underwent smooth iodination to afford iodide 35. The iodide was incorporated into an asymmetric Myers’ alkylation-hydrolysis sequence similar to that used in the T1 synthesis affording acid 12 in 88% yield over two steps and in >95:5 dr.
Scheme 15.

Synthesis of Alkenyl Acid 12.
Fragment coupling was carried out by way of a DCC-mediated esterification which proceeded in 72% yield (Scheme 16). As we sought to study ring-closing reductive cyclizations, we examined methods of selective, oxidative cleavage of the terminal alkene that would afford alkynal 9a. This target compound (with the benzylidene group at C16 intact) was the logical starting point for the “reversed” reductive coupling strategy as the T1 alkynal also possessed a benzylidene at this position and underwent cyclization successfully. We found that 9a could indeed be prepared by a dihydroxylation-periodate cleavage sequence (A, Scheme 16), but over-oxidation produced 9b, and moreover, the overall mass recovery of the process was low (< 20% yield on 10 mg scale, 3:2 ratio of 9a to 9b). Given these difficulties, we reconsidered our tactics and realized that a ketone might possibly be tolerated at C16 as ketones are much less reactive than aldehydes under our nickel-catalyzed reductive coupling conditions and, in most cases, inert.31 This alternative oxidation was accomplished via ozonolysis (B, Scheme 16) which cleaved both alkenes while leaving the alkyne intact to deliver 9b in very good yield.32
Scheme 16.

Routes to Alkynals 9a–b.
Synthesis of Amphidinolide T4 Framework: Reductive Macrocyclization and Elaboration to T4
Both 9a and 9b were subjected to the nickel-catalyzed reductive coupling conditions found to promote macrocyclization in our earlier synthesis of 1. Upon addition to a warm solution of Ni(cod)2, tributylphosphine and triethylborane, 9a and 9b underwent reductive cyclization to afford the desired macrocyclic allylic alcohols (Scheme 17). In both cases, the diastereoselectivity was very high (>10:1 dr). TBS protection and ozonolysis of the two alcohols led to diketone 36 which was selectively methylenated and deprotected with HF-pyridine. The 1H NMR data of the final product matched that of amphidinolide T4 (4) and differed significantly from its C12 diastereomer T3 (3), revealing the stereochemical outcome of the reductive cyclization. A comparison of the diagnostic region of the 1H NMR spectra of natural 3, natural 4, and synthetic 4 is shown in Figure 2.
Scheme 17.

Reductive Macrocyclizations of 9a–b and Synthesis of 4.
Figure 2.

Diagnostic Region of the 1H NMR spectra of 3 and 4.
Synthesis of Amphidinolide T4 Framework: Comparison of Inter- and Intramolecular Reductive Coupling Strategies
As we had done in the T1 series (Scheme 12), we sought to compare the corresponding intermolecular alkyne-aldehyde reductive coupling for the 3–4 framework. Interestingly, nickel-catalyzed coupling of ketoalkyne 37 and tetrahydrofuranyl aldehyde 38 resulted in a mixture of products that have been tentatively assigned as various elimination products and the intramolecular hemiketal of the desired allylic alcohol. In attempt to identify reaction products with certainty, the coupling was repeated with immediate silyl protection of the crude product mixture. As shown in Scheme 18, the isolated product was dihydropyran 39 as a 2:1 mixture of diastereomers, confirming our hypothesis that the coupling product was undergoing hemiketal formation and dehydration upon treatment with TBSCl. Not only did this result once again point to a strong conformational bias in the cyclization process that does not exist in the intermolecular process, but also that the macrocycle prevents formation of undesired byproducts such as dihydropyrans related to 39.
Scheme 18.

Intermolecular Coupling Study.
Investigation of Substrate Control in Reductive Macrocyclizations and Development of a Stereoselectivity Model
With the successful nickel-catalyzed macrocyclization of 9b using an achiral catalyst system, we endeavored to employ a chiral catalyst to overcome the inherent bias for the (S)-carbinol configuration of T4 (4) which, if successful, would provide an entry into the T3 stereochemical pattern. We first examined the ligand (S)-neomenthyldiphenylphosphine (NMDPP)33 in combination with Ni(cod)2 and triethylborane, but were disappointed to find that the catalyst system was not stable toward heating and cyclization did not occur. We next turned to P-chiral ferrocenyl ligands developed in our laboratory.4b We were pleased to find that 9b did indeed undergo cyclization when heated with a catalytic amount of Ni(cod)2 and (S)-ferrocenylmethylphenylphosphine and stoichiometric triethylborane, producing allylic alcohol 7b in 30% yield and >10:1 dr. Interestingly, when the opposite enantiomer of the ligand was utilized, 7b was also afforded in >10:1 dr (and 25% yield) with the same configuration at C12 (T4 configuration, Scheme 19).
Scheme 19.

Reductive Cyclizations of 9b with P-Chiral Ferrocenyl Ligands.
This result demonstrated that these stereoselective macrocyclizations are under complete substrate control, and are consistent with our growing hypothesis that both alkynals 8 and 9b are highly conformationally constrained during the reductive macrocyclization process. As noted above, we initially suspected that the configuration of the C14 methyl group in both cases (adjacent to the aldehyde group in 8 and adjacent to the alkyne in 9) played a significant role in inducing this bias.
To probe this hypothesis, i.e. the relevance of the Felkin-Anh model of stereoselectivity to these macrocyclizations, we synthesized alkynals 40 and 41 with the opposite configuration at C14 (Figure 3). Syntheses of both epi-C14 alkynals commenced with ent-33 (prepared using the R enantiomer of the Evans auxiliary) and followed the respective T1 and T4 syntheses exactly. The inverted methyl configuration did not affect either of the alkyne-epoxide or selective diyne-epoxide reductive coupling reactions and subsequent elaboration afforded the alkynals (details included in the Supporting Information).
Figure 3.

Epi-C14 Alkynals.
With the alkynals in hand, we were poised to examine reductive macrocyclization conditions. We found that alkynal 40 underwent cyclization when heated in the presence of 20 mol% Ni(cod)2, 40 mol% tributylphosphine and excess triethylborane in 35% yield with high diastereoselectivity (8:1 dr). Analysis of macrocyclic alcohol 42 using the Mosher method34 revealed that the major diastereomer possessed the S configuration at the carbinol center (Scheme 20). Interestingly, under the same conditions, alkynal 41 did not undergo cyclization (the only recovered compound was unreacted 41).
Scheme 20.

Inverted C14 Methyl Effect on Reductive Macrocyclizations.
The results of this set of coupling reactions (summarized in Table 1) required significant revision of our model of stereoinduction in the catalytic macrocyclizations. Contrary to our original hypothesis and as evidenced by entries 1 and 2, the Felkin-Anh model clearly does not apply in the case of the T1 series. That is, the configuration of the stereogenic center adjacent to the aldehyde undergoing reductive coupling with the alkyne has no effect whatsoever on the sense of induction. A minimum of 8:1 diastereoselectivity favoring the (S) configuration was observed in both cases.
Table 1.
Macrocyclization Experiments.
| entry | macrocyclization experiment | ligand | dr | configuration of major diastereomer |
|---|---|---|---|---|
| 1 | T1 (8, Scheme 11) | PBu3 | >10:1 | (S) C13 |
| 2 | 14-epi-T1 (40, Scheme 20) | PBu3 | 8:1 | (S) C13 |
| 3 | T4 (9a, Scheme 17) | PBu3 | >10:1 | (S) C12 |
| 4 | T4 (9b, Scheme 17) | PBu3 | >10:1 | (S) C12 |
| 5 | T4 (9b, Scheme 19) | 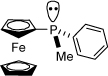 |
>10:1 | (S) C12 |
| 6 | T4 (9b, Scheme 19) | 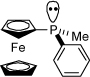 |
>10:1 | (S) C12 |
| 7 | 14-epi-T4 (41, Scheme 20) | PBu3 | n.a. | (macrocyclization not observed) |
The results of the T4 series of macrocyclizations (entries 3–6) are especially striking since the same sense and degree of stereoselectivity are observed for two different alkynals (9a (benzylidine at C16) and 9b (ketone at C16)) and, in the case of 9b, for three very different ligands (achiral, (R), or (S)). That the analogous “epi-methyl” (entry 7) experiment did not afford the desired product unfortunately prevents direct analysis of the effect of this stereogenic center upon the sense and degree of induction in the reductive macrocyclizations.
While we hesitate to infer too much information from a direct comparison of the T1 and T4 series of macrocyclizations, in which the positions of the alkyne and aldehyde have been reversed, we nevertheless propose a simple model of stereoinduction that is applicable to both of these cases (Figure 4). The elements of the analysis that led to this rationale are as follows:
Figure 4.

Conformational Analysis of Possible Diastereomers in Catalytic, Stereoselective Macrocyclizations Leading to Amphidinolides T1 (1) and T4 (4).
We have invoked an oxanickellacyclopentene4c,6e,22,35 as an approximation of the transition state of the reductive coupling.
The primary consideration in our conformational analysis was strain (torsional and steric) developing in the vicinity of the site of reactivity.36
Both possible product diastereomers were subjected to the same analysis, and a rough comparison was made between the two.
Figure 4a represents our proposed model that minimizes the unfavorable interactions in the transition state of the catalytic macrocyclization used to prepare amphidinolide T1. For clarity, three key 1,3-interactions are highlighted although many more are incorporated into the proposed model. The conformation shown possesses the fewest number of gauche+–gauche− (“syn-pentane”),37 A1,3 strain38 and related 1,3-interactions for this diastereomer. For example, orienting the bonds highlighted in red in the same direction and doing the same for the blue pair and the three black (bold) bonds generates no syn-pentane-like interactions in any of these three cases. (Not emphasized in this drawing is the fact that several other 1,3-interactions are also avoided.) On the contrary, all other possible conformations induce at least one syn-pentane-like interaction.
The net effect of this arrangement is that the carbon-carbon bonds connected by the loop representing the rest of the macrocycle are pointed in the same direction (see asterisked bonds). The remaining 12 atoms in the macrocycle are thus easily accommodated without inducing severe steric and/or torsional interactions.
When the same analysis was applied to the diastereomer not observed in this cyclization, the structure in Figure 4b was generated. As can be seen, the bonds that are connected by the loop are now pointed in different directions, approximately 120° relative to one another. Moreover, the loop in this case represents only 9 additional atoms, and model building suggests that requiring this shorter chain to span the required distance is not nearly as straightforward as was the case in the other diastereomer (Figure 4a).
A particularly satisfying feature of this model is that it can in fact be used to explain the observed stereochemical outcomes of the macrocyclizations that were used in the synthesis of amphidinolide T4. In other words, despite the fact that the “ends are reversed” so to speak, minimization of a very similar set of developing steric interactions accounts for the sense of induction in both the T1 and T4 series. As shown in Figure 4c and 4d, the net effect of this analysis for the T4 series is also strikingly similar to that in the T1 series. In Figures 4a (T1) and 4c (T4), 12 atoms are available to join the two ends (which are pointed in the same direction), whereas Figures 4b and 4d suggest that a much greater demand would be required of the 9 atoms in the chain.
A comparison of the results of the T1 series of macrocyclizations and of the 14-epi-T1 series is also worthy of comment (Figures 4a–b, 5, and 6). The goal of these exercises was to account for the initially counterintuitive observation that the stereogenic center adjacent to the site of the aldehyde had no effect on the sense of induction, i.e. that the Felkin-Anh model did not apply in this case. One must be careful, however, in drawing too much from this comparison, especially since the diastereoselectivity in the 14-epi-T1 series was not “complete”, as it was in the T1 and T4 series. To a first approximation, 8:1 dr at 60 °C represents a difference in energy of the two transition states of about 1.4 kcal•mol−1, a rather small value when compared to all the possible interactions in these complex systems.
Figure 5.

Conformational Analysis of the Products of Macrocyclization of an Alkynal Diasteromeric (at C14) to That Used in Synthesis of 1.
Figure 6.

Unified Model of Stereoinduction for Catalytic Macrocyclizations.
Nevertheless, as shown in Figure 5, low energy conformations for the diastereomers observed in the macrocyclizations in the “epi-methyl-T1” series were generated using the criteria in the other cases, and they are reminiscent of those for the corresponding diastereomers in the T1 series (Figures 4a and 4b). The essence of this comparison is summarized in Figure 6.
To summarize, our conclusion from these analyses is that the strongest effect upon the sense of induction is minimization of 1,3-interactions near the site of reactivity in the transition state. Moreover, branching (i.e. 2 substituents that are not hydrogen) at the carbon adjacent to the aldehyde (C14 in these cases) can be accommodated without energetic penalty since C12 is sp2 hybridized. That is, because of the absence of a substituent at C12, there are no 1,3-interactions with the substituents at C14, and thus the effect on stereoselectivity of branching adjacent to the aldehyde is dramatically attenuated. In other words, as long as the remainder of the nascent macrocycle can be accommodated without inducing severe unfavorable interactions, the configuration adjacent to the reacting aldehyde is of no consequence in determining the stereochemical outcome. Rather, it is the minimization of other interactions around the site of reaction that has the greatest effect. This hypothesis is not new in its own right of course,39 but it does suggest a unifying strategy that might be exploited in related stereoselective macrocyclizations.
Conclusion
In summary, we have found that intramolecular alkyne-aldehyde couplings are efficient methods for closing the 19-membered ring present in the amphidinolide T framework. The simultaneous installation of the carbinol stereogenic center found in 1 and 4 occurred with complete diastereocontrol. This reductive macrocyclization approach proved to be far superior to the corresponding intermolecular reductive coupling strategy not only with respect to stereoselectivity, but also in the overall number of synthetic steps necessary to access the natural products.
Supplementary Material
Acknowledgment
We are grateful to Anna K. Hirsch (Cambridge University-MIT Exchange Program) for experimental assistance, and Dr. Li Li for obtaining mass spectrometric data for all compounds. E.A.C. thanks the American Society for Engineering Education (NDSEG Fellowship) and K.C.O. thanks the Thomas A. Spencer Endowed UROP Fund (MIT). We also thank the National Institute of General Medical Sciences (GM-063755), NSF (CAREER CHE-0134704), Amgen, Boehringer Ingelheim, GlaxoSmithKline, Johnson & Johnson, Merck Research Laboratories, and Pfizer for generous financial support. The NSF (CHE-9809061 and DBI-9729592) and NIH (1S10RR13886-01) provide partial support for the MIT Department of Chemistry Instrumentation Facility.
Footnotes
Supporting Information Available: Experimental procedures and data; 1H NMR spectra for 1, 4, 6–15, 20–21, 23, 25–28, 32–36, 40–42 and naturally occurring 1 and 4. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews of the amphidinolides, see Kobayashi J, Tsuda M. Nat. Prod. Rep. 2004;21:77–93. doi: 10.1039/b310427n. Chakraborty TK, Das S. Curr. Med. Chem.-Anti-Cancer Agents. 2001;1:131–149. doi: 10.2174/1568011013354660. Kobayashi J, Ishibashi M. In: Comprehensive Natural Products Chemistry. Mori K, editor. Vol. 8. New York: Elsevier; 1999. pp. 619–649.
- 2.Proposed structure of A: Lam HW, Pattendon G. Angew. Chem. Int. Ed. 2002;41:508–511. doi: 10.1002/1521-3773(20020201)41:3<508::aid-anie508>3.0.co;2-7. Maleczka RE, Jr, Terrell LR, Geng F, Ward JS., III Org. Lett. 2002;4:2841–2844. doi: 10.1021/ol0262284. Trost BM, Chisholm JD, Wrobleski ST, Jung M. J. Am. Chem. Soc. 2002;124:12420–12421. doi: 10.1021/ja027883+. Structural revision and synthesis of Amphidinolide A: Trost BM, Harrington PE. J. Am. Chem. Soc. 2004;126:5028–5029. doi: 10.1021/ja049292k. Amphidinolide J: Williams DR, Kissel WS. J. Am. Chem. Soc. 1998;120:11198–11199. Amphidinolide K: Williams DR, Meyer KG. J. Am. Chem. Soc. 2001;123:765–766. doi: 10.1021/ja005644l. Amphidinolide P: Williams DR, Myers BJ, Mi L. Org. Lett. 2000;2:945–948. doi: 10.1021/ol0000197. Trost BM, Papillon JPN. J. Am. Chem. Soc. 2004;126:13618–13619. doi: 10.1021/ja045449x. Amphidinolide R: Kissel WS. “The Asymmetric Total Syntheses of Amphidinolides J and R,” Ph.D. Thesis. Indiana University; 1998. Amphidinolide T1: Ghosh AK, Liu C. J. Am. Chem. Soc. 2002;125:2374–2375. doi: 10.1021/ja021385j. Amphidinolide T1, T3, T4, T5: Fürstner A, Aïssa C, Riveiros R, Ragot J. Angew. Chem. Int. Ed. 2002;41:4763–4766. doi: 10.1002/anie.200290042. Aïssa C, Riveiros R, Ragot J, Fürstner A. J. Am. Chem. Soc. 2003;125:15512–15520. doi: 10.1021/ja038216z. Amphidinolide W: Ghosh AK, Gong G. J. Am. Chem. Soc. 2004;126:3704–3705. doi: 10.1021/ja049754u. Amphidinolide X: Lepage O, Kattnig E, Fürstner A. J. Am. Chem. Soc. 2004;126:15970–15971. doi: 10.1021/ja044130+.
- 3.For the isolation, structure determination, and biological studies of Amphidinolides T1-5, see: Tsuda M, Endo T, Kobayashi J. J. Org. Chem. 2000;65:1349–1352. doi: 10.1021/jo991393r. Kobayashi J, Kubota T, Endo T, Tsuda M. J. Org. Chem. 2001;66:134–142. doi: 10.1021/jo005607c. Kubota T, Endo T, Tsuda M, Shiro M, Kobayashi J. Tetrahedron. 2001;57:6175–6179.
- 4.(a) Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]; (b) Colby EA, Jamison TF. J. Org. Chem. 2003;68:156–166. doi: 10.1021/jo0264123. [DOI] [PubMed] [Google Scholar]; (c) Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]; (d) Chan J, Jamison TF. J. Am. Chem. Soc. 2003;125:11514–11515. doi: 10.1021/ja0373925. [DOI] [PubMed] [Google Scholar]
- 5.Molinaro C, Jamison TF. J. Am. Chem. Soc. 2003;125:8076–8077. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]
- 6.Several other nickel-catalyzed carbon-carbon bond-forming reactions of alkynes have been developed by Montgomery: Montgomery J. Angew. Chem. Int. Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n. Lozanov M, Montgomery J. J. Am. Chem. Soc. 2002;124:2106–2107. doi: 10.1021/ja0175845. Tang X-Q, Montgomery J. J. Am. Chem. Soc. 1999;121:6098–6099. Oblinger E, Montgomery J. J. Am. Chem. Soc. 1997;119:9065–9066.
- 7.Colby EA, O’Brien KC, Jamison TF. J. Am. Chem. Soc. 2004;126:998–999. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]
- 8.Molinaro C, Jamison TF. Unpublished results [Google Scholar]
- 9.(a) Evans DA, Ennis MD, Mathre DJ. J. Am. Chem. Soc. 1982;104:1737–1739. [Google Scholar]; (b) Savignac M, Durand J-O, Genêt J-P. Tetrahedron: Asymmetry. 1994;5:717–722. [Google Scholar]
- 10.(a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 11.Schmitt A, Reiβig H-U. Synlett. 1990:40–42. [Google Scholar]
- 12.Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J. Am. Chem. Soc. 1999;121:12208–12209. [Google Scholar]
- 13.Lewis acid-mediated addition of propargyl nucleophiles to 2-acetoxy tetrahydrofurans is precedented. For the use of propargylmagnesium bromide, see: Franck X, Hocquemiller R, Figadère B. Chem. Commun. 2002:160–161. doi: 10.1039/b109930m. For the use of a propargyl aluminum species, see: Dankwardt SM, Dankwardt JW, Schlessinger RH. Tetrahedron Lett. 1998;39:4975–4978.
- 14.Danheiser RL, Carini DJ. J. Org. Chem. 1980;45:3925–3927. [Google Scholar]
- 15.Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:5919–5923. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 16.Marshall JA, Shearer BG, Crooks SL. J. Org. Chem. 1987;52:1236–1245. [Google Scholar]
- 17.This sequence of functional group manipulations was performed as reported in reference 12.
- 18.Prepared according to: Westmijze H, Vermeer P. Synthesis. 1979:390–392.
- 19.Allylstannanes are reported to be much more nucleophilic compared to corresponding allylsilanes: Denmark SE, Weber EJ. J. Am. Chem. Soc. 1984;106:7970–7971. Yamamoto Y, Nishii S, Yamada J. J. Am. Chem. Soc. 1986;108:7116–7117. Sato T, Otera J, Nozaki H. J. Org. Chem. 1990;55:6116–6121. Allenylstannanes have also been shown to add to thioacetals under Lewis acidic conditions: Sato T, Okura S, Otera J, Nozaki H. Tetrahedron Lett. 1987;28:6299–6302. Takeda T, Ohshima H, Inoue M, Togo A, Fujiwara T. Chem. Lett. 1987:1345–1348.
- 20.Reported to propargylate enones in a 1, 4 addition mode under Lewis acidic conditions: Haruta J, Nishi K, Matsuda S, Akai S, Tamura Y, Kita Y. J. Org. Chem. 1990;55:4853–4859.
- 21.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am Chem. Soc. 1997;119:6496–6511. [Google Scholar]
- 22.Chan J, Jamison TF. J. Am. Chem. Soc. 2004;126:10682–10691. doi: 10.1021/ja0470968. [DOI] [PubMed] [Google Scholar]
- 23.Determined by Mosher ester analysis and ultimate conversion to 1
- 24.Remaining material appeared to be a number of decomposition products of the alkynal that could not be identified.
- 25.(a) Chérest M, Felkin H, Prudent N. Tetrahedron Lett. 1968;18:2199–2204. [Google Scholar]; (b) Chérest M, Felkin H. Tetrahedron Lett. 1968;18:2204–2208. [Google Scholar]; (c) Anh NT, Eisenstein O. Nouv. J. Chimie. 1977;1:61–70. [Google Scholar]; (d) Anh NT. Topics in Current Chemistry. 1980;88:145–162. [Google Scholar]
- 26.Takai K, Hotta Y, Oshima K, Nozaki H. Tetrahedron Lett. 1978;19:2417–2420. [Google Scholar]
- 27.Lombardo L. Org. Syn. 1987;65:81. [Google Scholar]
- 28.Hibino J, Okazoe T, Takai K, Nozaki H. Tetrahedron Lett. 1985;26:5579–5580. [Google Scholar]
- 29.Lead effects: Takai K, Kakiuchi T, Kataoka Y, Utimoto K. J. Org. Chem. 1994;59:2668–2670. Zirconium alkylidene reagents: Hartner FW, Jr, Schwartz J, Clift SM. J. Am. Chem. Soc. 1983;105:640–641. Tucker CE, Knochel P. J. Am. Chem. Soc. 1991;113:9888–9890.
- 30.Work in our laboratory has shown that this system is effective for the methylenation of sterically hindered ketones. Jeso V, Jamison TF. Unpublished results
- 31.Many alkyne-aldehyde couplings may be conducted in acetone without a decrease in yield. Nevertheless, a notable exception exists, that being an intramolecular, Ni-catalyzed alkyne-ketone coupling observed during studies directed toward the synthesis of terpestacin, reference 22.
- 32.Ozonolysis was followed by exposure to dimethylsulfide affording 9b and another product, determined to be the ozonide at terminal position. Treatment of the ozonide with triphenylphosphine promoted clean conversion to 9b giving an 80% combined yield of 9b. Direct reductive work-up with triphenylphosphine resulted in much lower yields. See Supporting Information for more detail.
- 33.During intermolecular reductive coupling studies of aldehydes and aryl alkynes in our laboratory (reference 4c), NMDPP emerged as a uniquely powerful promoter of asymmetric Ni-catalyzed coupling reactions.
- 34.Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512–519. [Google Scholar]
- 35.Miller KM, Jamison TF. J. Am. Chem. Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 36.This approach is likely an oversimplification, as it ignores the effects of other stereogenic centers, e.g. those in the tether linking the alkyne and the aldehyde. Related experiments with other sets of diastereomers would directly test these influences and possibly result in further refinement (or revision) of the model proposed herein.
- 37.Hoffman RW. Chem. Rev. 1989;89:1841–1860. [Google Scholar]
- 38.Wiberg KB, Murcko MA. J. Am. Chem. Soc. 1988;110:8029–8038. [Google Scholar]
- 39.For an in-depth study of situations in which developing 1,3-interactions (syn-pentane-like) better account for the observed sense of stereoinduction in additions to chiral aldehydes than the Felkin-Anh model, see: Roush WR. J. Org. Chem. 1991;56:4151–4157.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.