

Abstract
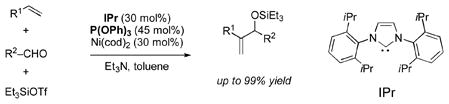
Both a strong electron donor (IPr) and a strong electron acceptor (P(OPh)3) are necessary for a highly selective, nickel-catalyzed coupling reaction between alkenes, aldehydes, and silyltriflates. Without the phosphite, catalysis is not observed and several side reactions are observed. The phosphite appears to suppress the formation of these byproducts and rescue the catalytic cycle by accelerating reductive elimination from an (IPr–Ni–H)(OTf) complex.
Keywords: C–C coupling reactions, N-heterocyclic carbene, nickel, phosphite, reductive elimination
Recently we described two new nickel-catalyzed, three-component coupling reactions involving alpha olefins, aldehydes, and silyltriflates.[1] The appropriate choice of ligand favors the production of either allylic or homoallylic alcohol derivatives (A or H, respectively), both of which are synthetically useful intermediates.[2] In these previous studies we had achieved as high as >95:5 H:A selectivity using EtOPPh2 or Ph3P, but a general method for the reverse, high A:H selectivity, has remained elusive.
 |
(1) |
Herein we describe a solution to this heretofore deficiency and document a synergistic relationship between substoichiometric amounts of a strong σ-donor (IPr, eq 1) and a strong π-acceptor ((PhO)3P). We believe that this phenomenon is the result of attenuation of the strong electron-donating ability of IPr by the electron-withdrawing (PhO)3P at a specific point in the catalytic cycle that consequently accelerates reductive elimination. This new strategy may have many broader applications in certain metal-catalyzed reactions, including the Heck reaction.[3]
In preliminary investigations of imidazolinyl (N-heterocyclic carbene, NHC) ligands in these transformations, we found that IPr was exceptionally selective for the allylic product (eq 2 and Table 1, entry 1), even with a rather encumbered alkene (vinylcyclohexane).[4] With our previous phosphine-based system, α–branched alkenes afforded only traces of the A product. However, catalysis was not observed in these or any other cases. An additional complication was that significant quantities of two side products were also observed, hydrosilylation of the aldehyde (reduction) and hydrovinylation.[5]
Table 1.
Evaluation of additives in Ni–IPr-mediated coupling reactions
| ||||||
|---|---|---|---|---|---|---|
| entry[a] | additive | conversion [%][b] | yield A [%][b,c] | yield H [%][b] | ||
| 1 | none | 55 | 18 (33) | 0 | ||
| 2 | mCF3-styrene | 79 | 39 (49) | 0 | ||
| 3 | CyPPh2 | 61 | 19 (31) | 0 | ||
| 4 | PPh3 | 100 | 34 (34) | 44 | ||
| 5 | EtOPPh3 | 100 | 34 (34) | 37 | ||
| 6 | (EtO)2PPh | 84 | 30 (35) | 29 | ||
| 7 | P(OBu)3 | 68 | 32 (47) | 9 | ||
| 8 | P(OPh)3 | 59 | 45 (76) | 0 | ||
| 9 | P(OPh)3 (no IPr) | 7 | 0 (0) | 0 | ||
See Supporting Information for details.
Determined by 1H NMR (amount of remaining aldehyde relative to an external standard (CH3NO2)).
Values in parentheses are yields based on conversion of aldehyde.
We surmised that both problems (product distribution and absence of catalysis) might have a common cause (Scheme 1). Formal reductive elimination of triflic acid (H–OTf) from 3 to regenerate a Ni0 species (e.g., 1) is likely retarded by the electron-rich IPr ligand.[3b–c,6] Stalling of the catalytic cycle at this point would result in an accumulation of an (NHC–Ni–H)(OTf) species (3) that is responsible for the aforementioned side reactions. Our first attempts to render this process catalytic involved the use of stronger organic and/or inorganic bases in order to facilitate reductive elimination. Unfortunately, however, this strategy summarily failed, perhaps because IPr is a very strong σ-donor.[7]
Scheme 1.

Proposed Mechanism
We thus required an alternative means to rescue the catalytic cycle and considered the possibility of shunting 3 into a different manifold in order to make reductive elimination more facile (Scheme 1). Since the pioneering investigations of Yamamoto had demonstrated that electron-poor alkenes dramatically accelerate reductive elimination of R1–R2 from R1–Ni–R2 species by decreasing the electron density at the metal center,[8] we examined the effects of these olefins in the reaction.
With mCF3-styrene and 30 mol% catalyst loading, a 39% yield of the desired coupling product was observed (Table 1, entry 2), but coupling products derived from the added styrene were also observed in the reaction.[9a–c] Nevertheless, catalysis had been demonstrated for the first time, and in order to avoid undesired coupling reactions between the additive and the aldehyde, we expanded our search to include electron-withdrawing phosphorus-containing compounds.[10]
We were mindful of the fact that this approach could very well have a fatal flaw related to ligand compatibility. In our previous work, we had found that electron-poor phosphinites, among others, disfavored the formation of allylic alcohol derivatives and, as mentioned above, were in fact superior to all other additives for high H:A selectivity.[11] Thus the electron-deficient additive could erode or overturn the high A:H selectivity provided by the IPr ligand.
Table 1 summarizes the results of these studies. Moderately electron-donating phosphines such as CyPPh2 were ineffective (Table 1, entry 3). Substoichiometric amounts of certain electron-poor phosphines (relative to CyPPh2) provided marginal catalytic activity (entries 4–6). Unfortunately, a significant amount of the undesired homoallylic alcohol derivative (H) was observed, presumably to the competing, homoallylic-favoring reaction, the potential pitfall mentioned above.
However, highly electron-deficient phosphite ligands were found to be most efficient additives (entries 7–8). Not only did triphenylphosphite ((PhO)3P) render the system catalytic, but it also completely suppressed formation of the homoallylic byproduct (1H NMR, limit of detection approx. 2%). Furthermore, hydrosilylation of the aldehyde was not observed, and the amount of alkene hydrovinylation was dramatically reduced.
It should be emphasized that both IPr and (PhO)3P are necessary for catalysis; the absence of one results in either no product (IPr absent) or no turnover (phosphite absent). Thus, unlike other electron-poor phosphorus additives, (PhO)3P does not erode the allylic selectivity perhaps because it does not promote the reaction on its own (entry 9).
The scope of this synergistic NHC–phosphite effect and new Ni–IPr-catalyzed transformation are summarized in Table 2. Particularly noteworthy is that A:H selectivity of ≥ 20:1 was observed in all cases examined. Also, the alkene scope is much broader than that in our previous Ni–phosphine-catalyzed processes.
Table 2.
IPr–Ni–P(OPh)3-catalyzed alkene-aldehyde coupling reactions
| entry[a] | alkene (R1) | product | conversion[%][b] | yield[%][b,c] |
|---|---|---|---|---|
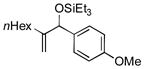
| 1 | nHex |
|
95[d] | 96 |
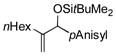
| 2 | nHex |
|
92[d,e] | 82 |
| 3 | nHex |
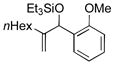
|
80 | 75 |
| 4 | nHex |
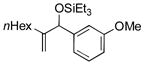
|
86 | 98 |
| 5 | Ph |
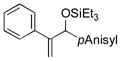
|
69 | 84 |
| 6[d] | PhCH2 |
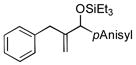
|
75 | 99 |
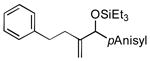
| 7 | PhCH2CH2 |
|
88 | 99 |
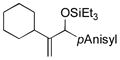
| 8 | Cy |
|
100 | 96 |
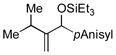
| 9[f,g] | iPr |
|
100 | 93 |
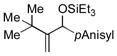
| 10[g,h] | tBu |
|
32 | 41 |
| 11 | iBu |
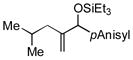
|
100 | 99 |
| 12 |
|
|
100 | 94 |
| 13 | nHex |
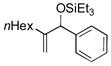
|
95 | 96 |
| 14 | nHex |
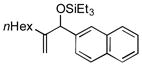
|
74 | 99 |
| 15[h] | nHex |
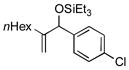
|
88 | 73 |
| 16 | Cy |
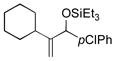
|
66 | 32 |
| 17[h,i] | nHex |
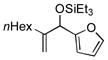
|
100 | 36 |
See Experimental Section and Supporting Information for details.
Determined by integration (1H NMR) relative to an external standard (CH3NO2).
Based on conversion.
150 mol% of alkene used.
t-BuMe2SiOTf used in place of Et3SiOTf.
1 mL of alkene used.
Reaction carried out in a sealed tube.
40 mol% of catalyst used.
Reaction carried out at room temperature.
Para-, meta- and ortho- substituted anisaldehydes are good-to-excellent substrates (entries 1–4). Bulky silyl triflates such as TBDMSOTf can also be used as activating reagents (entry 2). Styrenes, which readily underwent self-hydrovinylation in our earlier studies, become competent educts in the presence of the phosphite (entry 5).
Allylbenzene and homoallylbenzene both display excellent selectivity (entries 6 and 7), despite a reduced reaction rate. Sterically demanding α–branched substrates undergo highly regioselective coupling (entries 8–10). Similarly, coupling reactions of β-branched alkenes also enjoy a marked increase in the A:H selectivity with the phosphite additive (entry 11). A trisubstituted double bond is inert in the reaction, enabling its use as a masked functional group for subsequent modification (entry 12).
Benzaldehyde and 2-napthaldehyde also show good reactivity and high A:H ratios (entries 13–14). A chlorine atom attached to the aldehyde is tolerated (entries 15–16), but the reaction rate is much slower, and the side reactions of hydrosilylation and hydrovinylation become significant. Furfural behaves similarly (entry 17).
We believe the high allylic alcohol selectivity and broader substrate scope in this new reaction system are related to the sterically demanding and highly electron-donating nature of the IPr ligand (Scheme 1). It not only orients the substituents of the alkene and aldehyde away from Ni, but also accelerates oxidative coupling (1 to 2).
Conversely, the price paid for this latter effect, perhaps, is that in the absence of the phosphite additive, reductive elimination is much more difficult for the same reason that oxidative addition is more facile. Thus, we propose that the role of the phosphite promotes this step in the catalytic cycle by reducing the electron density at a coordinatively unsaturated Ni. A complex such as 4 (Scheme 1) would be expected to undergo reductive elimination to 5 more readily than would 3 to 1.
While many electron-deficient olefins have been used as additives for purposes similar to those of Yamamoto mentioned above, this work represents a rare example of using an additive other than an alkene in this manner in a catalytic reaction.[9,12] Remarkably, IPr and the phosphite are compatible with one another in this two-ligand system; neither seems to interfere with the role of the other. For this reason in particular, this phosphite–NHC combination may have several other applications and broader implications in NHC–metal-catalyzed transformations. For example, other reactions that rely upon the reductive elimination of H–X from H–M–X, particularly the Heck reaction, or of R1–M–R2 in general, such as catalytic cross coupling reactions, may also enjoy an accelerating effect by the use of a phosphite.[13] While an electron-deficient alkene may not be appropriate (i.e., it may react) for the Heck reaction, an organophosphite may, in contrast, be compatible. We are currently exploring such possibilities.
Experimental Section
General Procedure (See Supporting Information for examples): In a glove box, Ni(cod)2 (30 mol%), 1,3-bis(2,6-di-isopropylphenyl)imidazol-2-ylidene (IPr) (30 mol%), and a stir bar were added to an oven-dried 10 mL test tube. The tube was sealed with a rubber septum, removed from the glove box, and connected to an Ar line. The catalyst mixture was dissolved in degassed toluene (3 mL) under Ar and stirred 1 h at room temperature. The alkene (500 mol% or indicated amount), triethylamine (600 mol%), aldehyde (0.25 mmol, 100 mol%), triphenylphosphite (45 mol%) were added sequentially. Triethylsilyltriflate (175 mol%) was added dropwise, and the mixture was stirred 48 h at 35 °C. After cooling to room temperature, the mixture was diluted with ether (5 mL) and was allowed to stir 30 min open to air. The resulting mixture was filtered through a short plug of silica gel and rinsed with 20% EtOAc/hexane (50 mL). The solvent was removed under reduced pressure, and purification by flash chromatography (SiO2, 1% EtOAc in hexane) afforded the product.
Supplementary Material
Footnotes
Support for this work was provided by the National Institute of General Medical Sciences (GM-063755). C.–Y. H. thanks The Croucher Foundation for a postdoctoral fellowship. We are grateful to Dr. Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194. doi: 10.1021/ja055363j. [DOI] [PubMed] [Google Scholar]; b) Ho CY, Ng SS, Jamison TF. J Am Chem Soc. 2006;128:5362. doi: 10.1021/ja061471+. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ng SS, Ho CY, Jamison TF. J Am Chem Soc. 2006;128:11513. doi: 10.1021/ja062866w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.See ref. 1 and a) allylic alcohols: Yeston JS. The Value of a Nickel. Science. 2005;309:2139.reviews of the preparation of homoallylic alcohols by carbonyl ene reactions: Snider BB. Acc Chem Res. 1980;13:426.Mikami K, Shimizu M. Chem Rev. 1992;92:1021.Dias LC. Curr Org Chem. 2000;4:305.
- 3.Review of the Heck reaction and Pd-H chemistry: Negishi Ei. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley-Interscience; New York: 2002. Ni(0) species are not effectively regenerated by mechanism common to Pd-catalyzed Heck reaction through deprotonation of H-Ni-X by base: Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048.For theoretical comparison of Pd- and Ni-catalyzed Heck reactions: Lin BL, Liu L, Fu Y, Luo SW, Chen Q, Guo QX. Organometallics. 2004;23:2114.
- 4.Recent reviews of NHC ligands in transition metal catalysis: Weskamp T, Bohm VPW, Herrmann WA. J Organomet Chem. 2000;600:12.Herrmann WA. Angew Chem. 2002;114:1342.Angew Chem Int Ed. 2002;41:1290.Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247.Viciu MS, Nolan SP. Top Organomet Chem. 2005;14:241.Crabtree RH. J Organomet Chem. 2005;690:5451.
- 5.Blom R, editor. Organometallic Catalysts and Olefin Polymerization. Springer; New York: 2001. Nickel-catalyzed hydrovinylation: RajanBabu TV. Chem Rev. 2003;103:2845. doi: 10.1021/cr020040g.
- 6.NHC-M-H complexes of unusually high stability: Clement ND, Cavell KJ, Jones C, Elsevier CJ. Angew Chem. 2004;116:1297. doi: 10.1002/anie.200353409.Angew Chem Int Ed. 2004;43:1277.Viciano M, Mas-Marza E, Poyatos M, Sanaffl M, Crabtree RH, Peris E. Angew Chem. 2005;117:448. doi: 10.1002/anie.200461918.Angew Chem Int Ed. 2005;44:444.
- 7.Bases including K2CO3, Cs2CO3, diisopropylethylamine, pyridine, 2,6-lutidine, DBU, N-methylpyrrolidine, and N-methylmorpholine were examined.
- 8.Yamamoto T, Yamamoto A, Ikeda S. J Am Chem Soc. 1971;93:3350. [Google Scholar]
- 9.Uses of electron-deficient styrenes as additives in catalysis: Giovannini R, Studemann T, Dussin G, Knochel P. Angew Chem. 1998;110:2512. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2387::AID-ANIE2387>3.0.CO;2-M.Angew Chem, Int Ed Engl. 1998;37:2387.Giovannini R, Studemann T, Devasagayaraj A, Dussin G, Knochel P. J Org Chem. 1999;64:3544. doi: 10.1021/jo982317b.Bercot EA, Rovis T. J Am Chem Soc. 2002;124:174. doi: 10.1021/ja017086w.methyl acrylate: Lau J, Sustmann R. Tetrahedron Lett. 1985;26:4907.fumaronitrile: Sustmann R, Lau J, Zipp M. Tetrahedron Lett. 1986;27:5207.dimethyl fumarate: van Asselt R, Elsevier CJ. Tetrahedron. 1994;50:323.
- 10.Yamamoto showed that certain organophosphorus compounds accelerated reductive elimination from a nickel complex (but not in a catalytic reaction) and attributed the effect to the size of the phosphorus additive, rather than its electronic nature: Komiya S, Abe Y, Yamamoto A, Yamamoto T. Organometallics. 1983;2:1466.Use of PPh3 as an additive to stabilize an Ni-NHC catalyst: Sawaki R, Sato Y, Mori M. Org Lett. 2004;6:1131. doi: 10.1021/ol049908a.
- 11.Steric and electronic properties of organophosphines: Rahman M, Liu HY, Eriks K, Prock A, Giering WP. Organometallics. 1989;8:1.Tolman CA. Chem Rev. 1977;77:313.Otto S. J Chem Crystallogr. 2001;31:185.Riihimaki H, Kangas T, Suomalainen P, Reinius HK, Jaaskelainen S, Haukka M, Krause AOI, Pakkanen TA, Pursiainen JT. J Mol Catal A: Chem. 2003;200:81.Steinmetz WE. Quant Struct-Act Relat. 1996;15:1.
- 12.The effects of phosphorous ligands upon reductive elimination from NHC–M–alkyl complexes to give alkyl-imidazolium salts have been studied. In contrast, our observation that the NHC was not consumed suggests that one of the other ligands (e.g., H or OTf) significantly affects the properties and behavior of the metal complex. McGuinness DS, Saendig N, Yates BF, Cavell KJ. J Am Chem Soc. 2001;123:4029. doi: 10.1021/ja003861g.Clement ND, Cavell KJ. Angew Chem. 2004;116:3933. doi: 10.1002/anie.200454166.Angew Chem Int Ed. 2004;43:3845.
- 13.Recently reported Ni-catalyzed Heck reactions using P(OPh)3 or NHC as ligand (150 °C reaction temperature) Iyer S, Ramash C, Ramani A. Tetrahedron Lett. 1997;38:8533.Inamoto K, Kuroda J-i, Danjo T, Sakamoto T. Synlett. 2005:1624.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.