

Abstract
Several reactions of simple, unactivated alkenes with electrophiles under nickel(0) catalysis are discussed. The coupling of olefins with aldehydes and silyl triflates provides allylic or homoallylic alcohol derivatives, depending on the supporting ligands and, to a lesser extent, the substrates employed. Reaction of alkenes with isocyanates yields N-alkyl acrylamides. In these methods, alkenes act as the functional equivalents of alkenyl- and allylmetal reagents.
INTRODUCTION: FROM ALKYNES TO ALLENES TO ALKENES
Catalytic reactions that effect selective carbon–carbon bond formation allow for the rapid assembly of highly functionalized molecules from simple precursors. Aliphatic terminal alkenes (alpha olefins), industrially produced on megaton scale each year, are highly desirable precursors for such transformations due to their low cost and functional group compatibility [1]. As part of our program directed toward developing C–C bond-forming reactions of simple, ideally commercial, starting materials, we became very interested in transition metal-catalyzed alkene–aldehyde coupling processes. For these imagined methodologies to come to fruition, however, we were required to build on lessons learned from the development of coupling reactions of alkynes and allenes in our laboratory and others.
The nickel-catalyzed coupling of alkynes and aldehydes to form allylic alcohols was first described in 1997 by Montgomery (Scheme 1, eq. 1–2) [2]. In the presence of a phosphine ligand and diethyl zinc, alkynals underwent reductive cyclization. In the absence of ligand, alkylative cyclization occurred, with a carbon substituent installed from the organozinc reagent. At the same time, Montgomery reported a three-component alkylative coupling of alkyne, aldehyde, and organozinc; however, intermolecular reductive coupling was not observed, even in the presence of phosphine.
Scheme 1.

Alkyne–aldehyde reductive coupling.
In 2000, our laboratory reported the first catalytic method for intermolecular reductive coupling of alkynes and aldehydes, utilizing a nickel(0) catalyst, tributylphosphine, and triethylborane as the stoichiometric reducing agent (Scheme 1, eq. 3) [3]. Di- or trisubstituted allylic alcohols can be obtained with high regioselectivity from terminal or internal alkynes, respectively, and aromatic or aliphatic aldehydes.
An enantioselective variant of this reaction using the chiral monophosphine ligand neomenthyldiphenylphosphine (NMDPP, 1) emerged from our laboratory in 2003 (Scheme 2) [4]. In conjunction with ozonolysis, the alkyne–aldehyde reductive coupling is also useful in the preparation of α-hydroxyketones in high enantiopurity (up to 96% ee). Recently, Montgomery has successfully applied a chiral N-heterocyclic carbene (NHC) ligand to the nickel(0)-catalyzed coupling of alkynes, aldehydes, and triethylsilane to give silyl-protected allylic alcohols (up to 85% ee) [5].
Scheme 2.

Enantioselective, nickel(0)-catalyzed reductive coupling of alkynes and aldehydes.
In the course of these investigations of alkynes, it was found that 1,3- and 1,6-enynes reacted with very high degrees of regioselectivity, the result of an alkene directing effect (Scheme 3) [6]. Intriguingly, in the case of 1,6-enynes, not only does the tethered alkene lead to a high degree of regioselectivity, the sense of regioselectivity can be switched by the presence (or absence) of a catalytic amount of a phosphine ligand.
Scheme 3.

Regioselectivity in the reductive coupling of enynes and aldehydes.
In addition to their reactions with aldehydes, alkynes were shown to undergo catalytic reductive coupling reactions with epoxides [7] and imines [8]. These and other studies on the nickel(0)-catalyzed coupling reactions of alkynes have been further detailed in several recent reviews [9].
Like alkynes, allenes contain a carbon with sp-hybridization. However, until 2005, transition metal-mediated multicomponent coupling reactions of unactivated allenes with aldehydes were known to occur only at the sp2-hybridized carbons, affording homoallylic alcohols [10]. We then reported a rare example of intermolecular addition of an electrophile to the central, sp-hybridized, and ostensibly least nucleophilc carbon of an unactivated allene (Scheme 4) [11]. Not only did this reaction afford Z allylic alcohol derivatives with high regioselectivity, it also allowed for the preparation of highly enantioenriched products via efficient chirality transfer from axially chiral allenes. A key finding in this work was the superiority of silanes to triethylborane and other Lewis acidic reducing agents.
Scheme 4.

Enantioselective and regioselective nickel-catalyzed coupling of allenes, aldehydes, and silanes.
Conceptually, we reasoned, the reductive coupling reactions we had developed of alkynes and allenes with aldehydes could be replaced with non-reductive coupling between an alkene and aldehyde. This had potential to eliminate compatibility issues introduced by sometimes-harsh reducing agents. Not only are numerous olefins commercially available, synthetic methods abound for their preparation. However, we had already incorporated alkene units into intermolecular reductive coupling reactions: 1,3- and 1,6-enynes underwent reductive coupling at the alkyne only. Could we find another way to access what seemed to be the lower intrinsic reactivity of alkenes?
NICKEL-CATALYZED COUPLING REACTIONS OF ALKENES
When we began our work with alkenes, several intramolecular alkene–aldehyde coupling processes had been reported, including the titanium-catalyzed reductive cyclization of enals and enones [12]. Ogoshi described a nickel-promoted reductive cyclization of enals, but this process was stoichiometric in nickel [13]. Later on, several examples of catalytic, intramolecular alkene–ketone coupling reactions were reported [14]. Despite this progress, transition-metal catalyzed intermolecular reactions (reductive or otherwise) of this type remained elusive.
Historically, the most direct method for intermolecular coupling of unactivated alkenes and aldehydes to form homoallylic alcohol products is the carbonyl-ene reaction [15]. Typically, the alkenes employed in intermolecular carbonyl-ene reactions are 1,1-disubstituted and trisubstituted olefins. Electron-deficient enophiles such as glyoxylates are generally more efficient than simple aromatic or aliphatic aldehydes. Consequently, we wished to develop a reaction with features not amenable to existing carbonyl-ene methodology: first, the ability to transform less activated alkenes; second, enhanced and complementary electrophile scope; and third, the option to produce allylic alcohols in addition to homoallylic products.
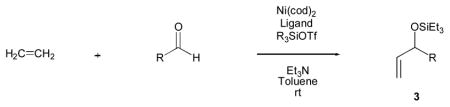
Intermolecular Coupling of Alkenes, Aldehydes, and Silyl Triflates
Pivotal to our work was Ogoshi’s observation that a Lewis acid, such as a silyl triflate, facilitated the formation of an oxanickellacycle through cyclization of an α,ω-enal [13]. We proposed that if the intermolecular coupling of an alkene and aldehyde occurred, the nickel alkyl bond could undergo a β-hydride elimination, followed by the removal of triflic acid from nickel to regenerate the nickel catalyst.
Our investigations began with the simplest alkene, ethylene, and benzaldehyde. After examining a variety of phosphorous-based ligands, we found that we were able to achieve catalysis of intermolecular alkene–aldehyde coupling when Ni(cod)2, tris-(o-methoxyphenyl)-phosphine ((o-anisyl)3P), triethylamine, and triethylsilyl triflate (Et3SiOTf) were used [16–17].
Under 1 atm of ethylene, a variety of aromatic aldehydes undergo efficient coupling to yield the silyl ether of an allylic alcohol (Table 1). Different phosphines, such as dicyclohexylphenylphosphine and triphenylphosphine, give lower yields under the same reaction conditions. Other common silyl triflates can be used in the coupling reaction, providing orthogonal protection of the hydroxyl group when necessary (3f, 3g).
Table 1.
Nickel-catalyzed coupling of ethylene, aldehydes, and silyl triflates.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
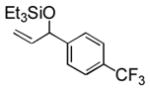
| entry | R (aldehyde) | R3SiOTf | product | isolated yield (%) | entry | R (aldehyde) | R3SiOTf | product | isolated yield (%) |
| 1 | Ph | Et3SiOTf |
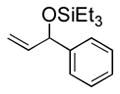 3a |
82 (65) b | 8 |
|
Et3SiOTf |
 3h |
80 |
| 2 | p-tolyl | Et3SiOTf |
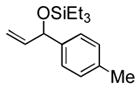 3b |
88 (65) b | 9 | 2-furyl | Et3SiOTf |
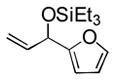 3i |
38 |
| 3 | o-tolyl | Et3SiOTf |
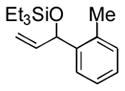 3c |
93 (64) b | 10 f |
|
Et3SiOTf |
 3j |
25 |
| 4 | p-anisyl | Et3SiOTf |
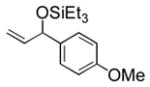 3d |
95 (65) b | 11 f |
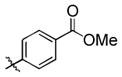
|
Et3SiOTf |
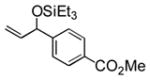 3k |
34 |
| 5 | 2-naphthyl | Et3SiOTf |
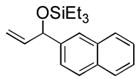 3e |
95 (83) b | 12 | piv | Et3SiOTf |
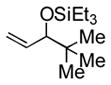 3l |
70 |
| 6 | 2-naphthyl | Me3SiOTf |
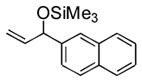 3f |
60 | 13 |
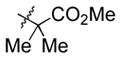
|
Et3SiOTf |
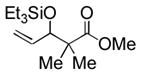 3m |
81 (40) c, d |
| 7 | 2-naphthyl | t-BuMe2SiOTf |
 3g |
67 | 14 | cyclohexyl | Et3SiOTf |
 3n |
25 d (34) d,e |
Standard procedure: Ni(cod)2 (20 mol %) and (o-anisyl)3P (40 mol %) were dissolved in 2.5 mL toluene under argon. Ethylene (balloon, 1 atm) was substituted for argon. Triethylamine (600 mol %), the aldehyde (100 mol %, 0.5 mmol), and silyl triflate (175 mol %) were added. The reaction was stirred for 6–18 h at 23 ºC.
(o-anisyl)3P was replaced by Cy2PhP.
(o-anisyl)3P was replaced by Ph3P.
Yield determined by 1H NMR using DMF as a standard.
Conducted under 2 atm of ethylene.
Stirred at room temperature for 30 h.
Notably, sterically encumbered tertiary aliphatic aldehydes such as pivaldehydes (3l) and 2,2-dimethyl-3-oxo-propionic acid methyl ester (3m) couple with ethylene in good yield. Several heteroaromatic aldehydes (3h, 3i) are also tolerated, even in the presence of electrophilic silyl triflates. A competing (but unsurprising) side reaction occurs in coupling reactions with enolizable aldehydes, which react rapidly with the silyl triflate and triethylamine to form alkenyl silyl ethers. The coupling of ethylene with cyclohexanecarboxaldehyde (3n) is fast enough, however, that a significant amount of coupling product is observed and can be isolated.
Electron-rich aromatic aldehydes are more efficient substrates than their electron-poor counterparts. Aldehydes with electron-donating para-substituents (Me and OMe) reacted in some of the highest yields observed for the coupling reaction. Electron-withdrawing para-substitutents (CF3, CO2Me) suffer from incomplete conversion, even after extended reaction time. In these and only these cases, side products resulting from a pinacol coupling are observed.
The encouraging results obtained with ethylene prompted us to examine the scope of the reaction with respect to other alkenes. Alpha olefins such as 1-octene can afford more than one possible product depending on where the new carbon–carbon bond is formed. Two distinct types of coupling product are typically observed: a 1,1-disubstituted allylic alcohol product (A) and a homoallylic alcohol product (H). By varying the phosphine ligand, one coupling product may be favored over the other (Scheme 5).
Scheme 5.

Nickel-catalyzed coupling of alpha olefins, aldehydes, and silyl triflates leads to two types of product.
The ratio of coupling products appears to be determined by a combined effect of the electron-donating ability and the cone angle of the phosphine ligands. Excellent H:A selectivity (95:5) can be achieved by using a less electron-rich phosphine with small cone angle, such as (EtO)Ph2P [18]. Modest selectivity favoring the A product can be obtained by using an electron-rich phosphine with a large cone angle, such as Cy2PhP. The nature of the alkene may play a role as well, as bulkier alkene substituents appear to increase the H:A ratio.
While the reaction conditions favoring the homoallylic product amount to a carbonyl-ene-type process, we do not believe that a Lewis acid-catalyzed carbonyl-ene reaction mechanism is operating here. In the coupling of β-citronellene and benzaldehyde, the nickel(0)-phosphine system selectively reacts with the monosubstituted olefin, whereas a classical Lewis acid reacts at the more nucleophilic trisubstituted double bond (Scheme 6). Further supporting the idea that the nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates does not involve a carbonyl-ene reaction mechanism is the observation that ethylene, with no allylic hydrogen, also participates in this coupling reaction using the same nickel(0)-phosphine catalyst.
Scheme 6.

Coupling of β-citronellene and benzaldehyde reveals a distinct reactivity pattern in the nickel-catalyzed reaction relative to a traditional Lewis acid-catalyzed carbonyl-ene reaction.
Instead, we propose that the alkene–aldehyde coupling commences with the formation of an oxanickellacycle from a nickel(0) complex (Scheme 7). The regioselectivity of oxanickellacycle assembly determines the product distribution (H:A ratio). A syn β-hydride elimination would afford the coupling product and a nickel–hydride species, analogous to a Heck reaction [19]. Base-promoted reductive elimination of the nickel–hydride intermediate could regenerate the nickel(0) catalyst. A base-mediated β-elimination of the oxanickellacycle via an E2-like mechanism cannot be completely ruled out. However, we believe that the observation of isomerization and dimerization (hydrovinylation) of the starting olefins suggests the presence of a nickel–hydride species, likely formed by a β-hydride elimination.
Scheme 7.

Proposed mechanism for nickel-catalyzed coupling of alkene, aldehyde, and silyl triflate.
While we had attained greater than 95:5 H:A selectivity in the alkene–aldehyde coupling, a general method for high A:H selectivity remained elusive. However, we recently were able to remedy that deficiency by exploiting a synergistic relationship between a strong σ donor and a strong π acceptor [20]. In preliminary investigations of (NHC) ligands in these transformations, we found that IPr (2) was highly selective for the allylic product, even when a branched alkene was used. However, the reaction did not appear to proceed catalytically. Additionally, side products resulting from hydrosilylation of the aldehyde (reduction) and hydrovinylation were formed in significant quantities [21].
We suspected that both the formation of side products and lack of catalysis might be caused by the fact that the electron-rich ligand retards formal reductive elimination of triflic acid to regenerate a nickel(0) species. Stalling the catalytic cycle results in accumulation of a [Ni(NHC)H](OTf) species, which is responsible for the side reactions. We examined a variety of additives reported to accelerate reductive elimination or stabilize nickel catalysts, including electron-poor olefins [22] and electron-withdrawing phosphines [23]. These provided slight catalytic activity, but had significant flaws in terms of competing reaction of added alkene or erosion of the A:H ratio with electron-poor phosphines. However, very electron-deficient phosphite ligands proved to be highly effective additives.
Triphenylphosphite in particular not only rendered the reaction catalytic, but also suppressed formation of the homoallylic product and dramatically reduced hydrosilylation and hydrovinylation (Scheme 8). Both IPr and (PhO)3P were necessary for catalysis: when IPr is absent, no product is observed; when phosphine is absent, no turnover. Unlike other phosphorus-based additives, (PhO)3P did not diminish the A:H selectivity, perhaps because it can not promote the reaction on its own. Remarkably, donor (IPr) and acceptor (phosphite) do not interfere with each other.
Scheme 8.

High selectivity for the allylic product in dual IPr/(PhO)3P system.
This dual NHC/phosphite system resulted in an impressive A:H selectivity of greater than 20:1 in all cases examined. Additionally, more alkene substrates were suitable, including styrenes and relatively encumbered α-branched alkenes. Both the selectivity and the broader substrate scope may be related to the sterically demanding and highly σ-donating nature of IPr. The ligand orients the alkene substituents away from the Ni center, and also is expected to accelerate the oxidative coupling. We propose that the phosphite additive promotes reductive elimination by reducing the electron density at a coordinatively unsaturated Ni center, rescuing the catalytic cycle by a shunt mechanism (Scheme 9).
Scheme 9.

Proposed mechanism for coupling of alkenes and aldehydes catalyzed by [Ni(IPr){P(OPh)3}].
Unlike the related transition metal-catalyzed reductive coupling reactions developed by our group and others, the nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates in its various forms is not an overall reductive process. In theory, the coupling of alkene and aldehyde does not necessitate a third component to form the allylic or homoallylic alcohol derivatives. However, both alkenes and aldehydes are generally unreactive toward each other, meaning that activation of one or both of the components is necessary. The Lewis acidic nature of silyl triflate apparently provides sufficient activation of the electrophile for the nickel catalyst to promote the coupling reaction.
The nickel-catalyzed reaction we have described offers an alternative to both alkenylmetal reagents (when Cy2PhP or IPr/(PhO)3P are used) or allylmetal reagents (when EtOPh2P is used). The substitution of alkenes for these organometallic species should offer considerable advantages in ease of preparation and functional group compatibility.
Intermolecular Coupling of Alkenes and Isocyanates
Following our initial work with aldehydes, we became interested in expanding the scope of electrophiles with which simple alkenes could be coupled. Our attention was drawn to pioneering studies by Hoberg, who reported the stoichiometric and catalytic coupling reactions of phenyl isocyanate and ethylene on nickel(0) with trialkyl phosphine ligands, yielding N-phenylacrylamide [24–25]. A variety of other olefins, both activated and unactivated, have been reported to react with phenyl isocyanate with nickel(0) and phosphine ligands; the major product is the trans-disubstituted α,β-unsaturated amide [26]. Only in two cases is a 1,1-disubstituted acrylamide observed (as a minor product, in 3% and 13% yield).
Hoberg proposed that the reaction proceeds via an azanickelacyclopentanone intermediate, which then undergoes β-hydrogen elimination. Presumably, in order to obtain 1,2-disubstitutedα,β-unsaturated amides, the alkene substituent would be on the carbon adjacent to nickel in the relevant nickellacycle. We felt that, as in the coupling of alkenes with aldehydes, a bulky NHC ligand might result in the formation of the alternative nickellacycle, in which the olefin substituent is pointed away from the metal center.
Indeed, in the presence of catalytic nickel(0) and the NHC ligand IPr, carbon–carbon bond formation was observed to form selectively at the 2-position of the olefin, yielding N-alkylated acrylamides [27]. The NHC ligand gives products with the opposite sense of regioselectivity compared to those obtained when phosphine ligands are used (Scheme 10). We found that excellent conversion could be obtained upon heating. The scope of the reaction appears to be limited to bulky, electron-rich alkyl isocyanates; a common side reaction is oligomerization of the isocyanate, which may be catalyzed by free NHC ligands in solution [28].
Scheme 10.

Proposed regioselectivity of nickellacycle formation in the catalytic coupling reaction of alkenes and isocyanates.
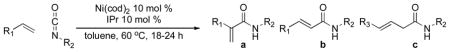
While unbranched α-olefins have a fairly modest preference for carboxamidation at the 2-position of the olefin, alkenes with branching at the allylic or homoallylic position proceed with very good selectivity (Table 2). A third, homoallylic product is formed from allylbenzene in which the double bond has moved into conjugation with the aromatic ring, but this is not observed in other cases. Again, the reaction appears to be selective for monosubstituted olefins. Low conversion is observed in the cases of 1,1-disubstituted, cis-, and trans-olefins. Esters and silyl-protected alcohols, and in some cases even ketones, are tolerated under the reaction conditions.
Table 2.
Scope and selectivity in nickel-catalyzed coupling of alkenes and isocyanates.
 | |||||
|---|---|---|---|---|---|
| entrya | R1 | R2 | R3 | product(s) | yield a;b;c (%)b |
| 1 | n-hexyl | Cy | - | 6a, 6b | 79; 14; 0 |
| 2 | n-hexyl | t-Bu | - | 7a, 7b | 74; 17; 0 |
| 3 |
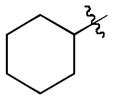
|
t-Bu | - | 8a | 91; 0; 0 |
| 4c |
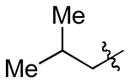
|
Cy | - | 9a, 9b | 74; 5; 0 |
| 5c | PhCH2 | Cy | Ph | 10a, 10b, 10c | 65; 8; 22 |
| 6c | PhCH2 | t-Bu | Ph | 11a, 11b, 11c | 83; 1d; 5d |
| 7c |
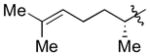
|
Cy | - | 12a | 86; 0; 0; |
| 8 |
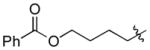
|
Cy | - | 13a, 13b | 74; 13; 0 |
| 9 |
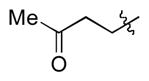
|
t-Bu | - | 14a, 14b | 70; 2; 0 |
| 10 |
|
t-Bu | - | 15a, 15b | 65; 9; 0 |
Standard conditions: Reactions were run with 0.5 mmol of 1-octene, 1.0 mmol of isocyanate, 0.05 mmol of Ni(cod)2, and 0.05 mmol of IPr in 0.5 mL toluene under Ar(g) in a sealed tube at 60 ºC for 18–24 h.
Isolated yields.
Reaction was run using 2 mL of toluene.
Isolated as a mixture of 10b and 10c, with relative ratios determined by 1H NMR.
The alkene–isocyanate coupling is unique among the systems we have examined thus far in that it does not require a third component to proceed. However, as in our work with aldehydes, what are ostensibly the least activated alkenes react. The NHC ligand seems to play an important role in favoring reaction at the more substituted position of the alkene. In this reaction, we again can utilize a simple alkene as the functional equivalent of a 2-alkenylmetal reagent.
CONCLUSIONS
We have described several new reactions of simple alkenes catalyzed by nickel(0). An NHC ligand allows us for the first time to selectively produce 1,1-disubstituted acrylamides in the coupling of alkenes and isocyanates. In the intermolecular reaction of alkenes, aldehydes, and silyl triflates, we have the ability to produce either allylic or homoallylic alcohol derivatives with excellent selectivity by judicious choice of supporting ligands. We have studied the attributes of ligand and substrate that tend to favor one product over the other, and we have documented a notable case of synergy between two distinct ligand types. In these reactions, simple alkenes are conceptual, and indeed practical, surrogates for alkenyl- and allylmetal reagents.
Acknowledgments
Financial support for this research was provided by the National Institute of General Medical Sciences (GM-063775 and GM-072566), NSF (CAREER CHE-0134704), Boehringer Ingelheim, Merck Research Laboratories, and MIT. C.-Y. H. thanks The Croucher Foundation for a postdoctoral fellowship. S.-S. N. thanks Bristol-Myers Squibb for a graduate fellowship.
References
- 1.Lappin GR, Sauer JD. Alpha Olefins Applications Handbook. Marcel Dekker; New York: 1989. [Google Scholar]
- 2.Oblinger E, Montgomery J. J Am Chem Soc. 1997;119:9065. [Google Scholar]
- 3.Huang WS, Chan J, Jamison TF. Org Lett. 2000;2:4221. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]
- 4.Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]; Van Dyke AR, Miller KM, Jamison TF. Org Synth. 2007;84:111. [Google Scholar]
- 5.Chaulagain MR, Sormunen GJ, Montgomery J. J Am Chem Soc. 2007;129:9568. doi: 10.1021/ja072992f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130. doi: 10.1021/ja0491735. [DOI] [PubMed] [Google Scholar]; Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 7.Molinaro C, Jamison TF. J Am Chem Soc. 2003;125:8076. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]
- 8.Patel SJ, Jamison TF. Angew Chem, Int Ed. 2003;42:1364. doi: 10.1002/anie.200390349. [DOI] [PubMed] [Google Scholar]; Patel SJ, Jamison TF. Angew Chem, Int Ed. 2004;43:3941. doi: 10.1002/anie.200460044. [DOI] [PubMed] [Google Scholar]
- 9.Montgomery J. Angew Chem, Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634. [DOI] [PubMed] [Google Scholar]; Montgomery J, Sormunen GJ. Top Curr Chem. 2007;279:1. [Google Scholar]; Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun Advance Article. doi: 10.1039/b707737h 2007. [DOI] [Google Scholar]
- 10.Palladium: Anwar U, Grigg R, Rasparini M, Sridharan V. Chem Commun. 2000:645.Ha YH, Kang SK. Org Lett. 2002;4:1143. doi: 10.1021/ol025557t.Hopkins D, Malinakova HC. Org Lett. 2004;6:2221. doi: 10.1021/ol0492795.Nickel Montgomery J, Song M. Org Lett. 2002;4:4009. doi: 10.1021/ol026670m.Kang SK, Yoon SK. Chem Commun. 2002:2634. doi: 10.1039/b207620a.Wu MS, Rayabarapu DK, Cheng CH. J Am Chem Soc. 2003;125:12426. doi: 10.1021/ja037718+.
- 11.Ng SS, Jamison TF. J Am Chem Soc. 2005;127:7320. doi: 10.1021/ja0521831. [DOI] [PubMed] [Google Scholar]; Ng SS, Jamison TF. Tetrahedron. 2006;61:11405. [Google Scholar]
- 12.Kablaoui NM, Buchwald SL. J Am Chem Soc. 1995;117:6785. [Google Scholar]; Crowe WE, Rachita MJ. J Am Chem Soc. 1995;117:6787. [Google Scholar]
- 13.Ogoshi S, Oka MA, Kurosawa H. J Am Chem Soc. 2004;126:11802. doi: 10.1021/ja0460716. [DOI] [PubMed] [Google Scholar]
- 14.Ogoshi S, Ueta M, Arai T, Kurosawa H. J Am Chem Soc. 2005;127:12810. doi: 10.1021/ja0542486. [DOI] [PubMed] [Google Scholar]
- 15.Alder K, Pascher F, Schmitz A. Ber Dtsch Chem Ges. 1943;76:27. [Google Scholar]; Snider B. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; Oxford: p. 527.p. 1991. [Google Scholar]
- 16.Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194. doi: 10.1021/ja055363j. [DOI] [PubMed] [Google Scholar]
- 17.Ng SS, Ho CY, Jamison TF. J Am Chem Soc. 2006;128:11513. doi: 10.1021/ja062866w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho CY, Ng SS, Jamison TF. J Am Chem Soc. 2006;128:5362. doi: 10.1021/ja061471+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Negishi E-I. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley-Interscience; New York: 2002. [Google Scholar]; Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; Hills ID, Fu GC. J Am Chem Soc. 2004;126:13178. doi: 10.1021/ja0471424. [DOI] [PubMed] [Google Scholar]
- 20.Ho CY, Jamison TF. Angew Chem, Int Ed. 2007;46:782. doi: 10.1002/anie.200603907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajanbabu TV. Chem Rev. 2003;103:2845. doi: 10.1021/cr020040g. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T, Yamamoto A, Ikeda S. J Am Chem Soc. 1971;93:3350. [Google Scholar]
- 23.Komiya S, Abe Y, Yamamoto A, Yamamoto T. Organometallics. 1983;2:1466. [Google Scholar]; Sawaki R, Sato Y, Mori M. Org Lett. 2004;6:1131. doi: 10.1021/ol049908a. [DOI] [PubMed] [Google Scholar]
- 24.Hoberg H, Sümmermann K, Milchereit A. Angew Chem, Int Ed Engl. 1985;24:325. [Google Scholar]
- 25.Hoberg H, Hernandez E. J Chem Soc, Chem Commun. 1986:544. [Google Scholar]
- 26.Hoberg H. J Organomet Chem. 1988;358:507. references cited therein. [Google Scholar]
- 27.Schleicher KD, Jamison TF. Org Lett. 2007;9:875. doi: 10.1021/ol063111x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duong HA, Cross MJ, Louie J. Org Lett. 2004;6:4679. doi: 10.1021/ol048211m. [DOI] [PubMed] [Google Scholar]