

Abstract
When administered near the time of training, protein synthesis inhibitors such as anisomycin impair later memory. A common interpretation of these findings is that memory consolidation requires new protein synthesis initiated by training. However, recent findings support an alternative interpretation that abnormally large increases in neurotransmitter release after injections of anisomycin may be responsible for producing amnesia. In the present study, a local anesthetic was administered prior to anisomycin injections in an attempt to mitigate neurotransmitter actions and thereby attenuate the resulting amnesia. Rats received lidocaine and anisomycin injections into the amygdala 130 and 120 min, respectively, prior to inhibitory avoidance training. Memory tests 48 hr later revealed that lidocaine attenuated anisomycin-induced amnesia. In other rats, in vivo microdialysis was performed at the site of amygdala infusion of lidocaine and anisomycin. As seen previously, anisomycin injections produced large increases in release of norepinephrine in the amygdala. Lidocaine attenuated the anisomycin-induced increase in release of norepinephrine but did not reverse anisomycin inhibition of protein synthesis, as assessed by c-Fos immunohistochemistry. These findings are consistent with past evidence suggesting that anisomycin causes amnesia by initiating abnormal release of neurotransmitters in response to the inhibition of protein synthesis.
Keywords: norepinephrine, memory consolidation, memory modulation, microdialysis, anisomycin, protein synthesis inhibitors, amygdala, lidocaine
Introduction
The predominant model for the molecular basis of memory is that memory formation occurs in two stages: an early transient stage independent of protein synthesis and a long-term memory phase dependent on training-initiated de novo protein synthesis (cf.: Davis & Agranoff, 1966; Davis & Squire, 1984; Schafe & Ledoux, 2000; Kandel, 2001; Dudai, 2002; Izquierdo et al., 2002; Nader, 2003). Support for this model comes from numerous studies in which protein synthesis inhibitors such as anisomycin administered near the time of training have no effect on memory tested during the first few hours after training but impair memory tested at longer time intervals.
However, evidence obtained over the past forty years suggests that drugs that inhibit protein synthesis may lead to secondary dysfunctions in neural activity that might produce amnesia. Several studies have suggested that protein synthesis inhibitors interfere with neurotransmitter synthesis and release. For example, early studies showed that protein synthesis inhibitors reduced the activity of tyrosine hydroxylase, the rate-limiting enzyme involved in the synthesis of norepinephrine and dopamine from tyrosine (Flexner et al., 1973; Flexner & Goodman, 1975; Goodman et al., 1975). Recent studies found that intra-amygdala and intra-hippocampal injections of anisomycin induced abnormally large increases in the release of norepinephrine, dopamine, serotonin, and acetylcholine at the site of the injection (Canal et al., 2007; Qi & Gold, 2009). Related to these findings is evidence that many neurotransmitter-related drugs and hormones reverse amnesias associated with protein synthesis inhibitors and do so without reversing the extent of inhibition of protein synthesis (cf.: Martinez et al., 1981; Davis & Squire, 1984; Gold, 2008).
The present study tested whether a local anesthetic, lidocaine, would attenuate anisomycin-related memory impairments by inhibiting the anisomycin-induced increases in norepinephrine release, though likely not baseline release (Pan et al., 1996). Since the time course for anterograde amnesia observed with lidocaine is shorter than that for anisomycin, we were able to adjust the timing of the injections such that lidocaine had no effect on memory while anisomycin retained its ability to impair memory. Our hypothesis was that lidocaine would reduce the magnitude of anisomycin-induced increases in norepinephrine release and would attenuate anisomycin-induced amnesia.
Methods
Subjects
Male Sprague-Dawley rats (4–5 months old; Harlan Labs, Madison, WI) were individually housed and maintained on a 12:12-h light-dark cycle with free access to food and water. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Urbana-Champaign and were in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals.
Surgery
Rats (total N = 51) were anesthetized with isoflurane and placed in a stereotaxic apparatus with skulls arranged in a horizontal position. For the behavioral experiment, 23-gauge guide cannulae (Plastics One, Roanoke, VA) were aimed bilaterally at the basolateral amygdala (coordinates AP = −2.9 mm from bregma, ML = ±4.8 mm, DV = 5.4 mm below dura) and secured to the skull using dental cement and four small screws. Stylets ending flush with the tips of the cannulae were inserted. For the microdialysis experiment, an injection guide cannula was implanted as above on one side and a guide cannula for a combination microdialysis/injection probe was implanted on the contralateral side. The guide cannula for the SciPro probe was lowered until it was 3 mm above the amygdala (coordinates as above, except DV = −4.4 mm below dura). Rats were allowed to recover from surgery for at least one week prior to behavioral and microdialysis procedures. The rats were handled for 5 days for 3 min/day prior to the days of training and microdialysis sampling.
Inhibitory Avoidance Training and Testing
Rats were trained on a one-trial inhibitory avoidance task using a trough-shaped apparatus (91 cm long, 22.9 cm wide at top, 7.6 cm wide at bottom, and 15.2 cm deep) in which a door separated a well-lit start compartment (31 cm) from a dark shock compartment (60 cm). After placement of rats into the start compartment on the training day, the door was opened and the rats were allowed to cross into the dark compartment where they received a footshock (0.5 mA, 1.5 sec). Forty-eight hr later, rats were again placed into the start compartment and the door was opened. The latency to enter the dark compartment on the test day (maximum = 180 sec) was taken as the measure of memory.
Injections
For behavioral testing, anisomycin (Sigma, St. Louis, MO) was dissolved in 1 N HCl and brought to a final pH of 7.2 with 1 N NaOH. Phosphate-buffered saline (1 mM KH2PO4, 155 mM NaCl, 3 mM Na2HPO4) was added to obtain a final anisomycin concentration of 125 μg/μl; 0.5 μl (62.5 μg) was injected per side into the amygdala of each rat. Lidocaine was dissolved in 0.9% saline for a final concentration of 10 μg/0.5 μl/side. Rats received bilateral injections (0.25 μl/min for 2 min via CMA/100 microinjection pump) of saline or lidocaine 10 min prior to injections of PBS or anisomycin, the second injection coming 2 hr prior to training. The timing of the injections was determined by pilot studies to ensure an absence of anterograde impairments of memory by lidocaine and a presence of memory impairments by anisomycin. We had found that anisomycin impaired 48-hr memory if injected into the amygdala up to 2 hr but not 6 hrs or more before inhibitory avoidance training (Canal et al., 2007). In pilot studies for the present experiment, lidocaine impaired memory if injected within 30 min of training but not if injected at longer pretraining intervals. Four groups were trained and tested in the behavioral experiment: saline + PBS (n=7), lidocaine + PBS (n=4), saline + anisomycin (n=9), lidocaine + anisomycin (n=12). Following injections, cannulae remained in place for an additional 1 min in order to allow diffusion of drugs. Rats were placed back into their home cages until they were trained on the inhibitory avoidance task.
Norepinephrine samples
In the microdialysis experiment, combination microdialysis/injection probes (MAB 9.14, SciPro, Sanborn, NY) were used to inject drugs and to collect microdialysis samples simultaneously. The probes consisted of a 2 mm membrane and an injection port that extended 1 mm below the microdialysis membrane. Combination probes were inserted into the guide cannulae and artificial cerebrospinal brain fluid (128 mM NaCl, 2.5 mM KCl, 1.3 mM CaCl2, 2.1 mM MgCl2, 0.9 mM NaH2PO4, 2.0 mM Na2HPO4, 1.0 mM dextrose; pH= 6.5) was perfused through the brain at a rate of 0.6 μl/min. Each 45-min sample (27 μl) was collected in a tube containing 40 μl of 0.2 N acetic acid for a total volume of 67 μl. Samples collected during the first hour were discarded. Then, three baseline samples were collected before intra-amygdala bilateral injections of anisomycin (n=4), lidocaine (n=3), or both anisomycin and lidocaine (n=4) at concentrations as above. Injection procedures followed those used in behavioral experiments and drugs were injected during the first 4 min of the corresponding microdialysis sample. Five post-injection samples were collected thereafter. Immediately following collection, samples were frozen at −20°C until assayed. Microdialysis samples were analyzed for norepinephrine content using HPLC with electrochemical detection with procedures described previously (Canal et al., 2007). The detection limit for norepinephrine was 0.5 pg.
Histology
The locations of cannula placements were assessed histologically, using 50 μm frozen sections (Leica 1800 cryostat) through the amygdala and cresyl violet staining. Rats with cannulae placements outside the amygdala were excluded from data analyses. While only rats with bilateral cannula placements in the amygdala were included here, the localization of drug effects to the amygdala or to individual nuclei within the amygdala was not specifically determined. Drug diffusion estimates vary with method of assessment, e.g. tissue staining, autoradiography or neurophysiology (Edeline et al., 2002). Lidocaine injections of 1 μl volumes, twice that employed here, exhibit spread of 1.7 mm using autoradiography measures (Martin, 1991), an extent of spread suggesting that the effects in the present experiment were largely confined to amygdala sites of action.
Immunohistochemistry
Separate groups of rats were used to examine c-Fos protein expression in the amygdala, used as a marker of protein synthesis inhibition as employed previously (e.g., Inda et al., 2005; Mamou et al., 2006; Canal et al., 2007, 2008; Canal & Gold, 2007). Three groups received concentrations and times of injections used in inhibitory avoidance testing. These groups included: saline + PBS (N = 4), saline + anisomycin (N = 2), and lidocaine + anisomycin (N = 2). Sixty minutes after footshock training, rats were anesthetized with sodium pentobarbital and perfused with saline followed by freshly prepared 4% paraformaldehyde. The brains were removed and placed in 4% paraformaldehyde overnight. Brains were then transferred to 20% glycerol in 0.1 M phosphate-buffered saline prior to collecting frozen sections (50 μm) approximately 100 μm from the injection site. The sections were washed in 0.05 M PBS, incubated in 1% H2O2 and 0.4% Triton X-100, and blocked with 3% goat serum. The sections were then incubated for 48 hr at 4°C in c-Fos primary antibody (1:7500; c-Fos antibody, rabbit polyclonal; sc-253, Santa Cruz Biotechnology, Santa Cruz, CA), and were subsequently washed in PBS followed by 1-hr incubation in secondary antibody (1:400 goat anti-rabbit; Santa Cruz Biotechnology). Sections were immunostained using the ABC Vectastain Elite kit (Vector Laboratory, Burlingame, CA) and diaminobenzidine.
For quantification, pictures were taken at 10x on a Leica CTR6000 microscope attached to a Leica DFC350FX digital camera and analyzed with Image J (NIH). In Image J, the threshold was set at 2.5x background for all pictures. Using the software, particles were counted within a circle (455 μm diameter) positioned in the basolateral nucleus of the amygdala.
Statistical Analyses
Because of the ceiling inhibitory avoidance scores, parametric statistical tests of the behavioral results were not appropriate. Therefore, non-parametric Mann-Whitney U-tests (two-tailed) (Siegel, 1956) were used to analyze inhibitory avoidance scores. Because norepinephrine means and standard deviations for the groups differed greatly, the neurochemical data were analyzed using log10 transformations of the values. Norepinephrine results were assessed with three-way repeated-measures ANOVAs (Statview software). Scheffe’s post hoc t-tests were used to compare norepinephrine levels across groups. Student’s t-tests were used to compare c-Fos expression levels across groups.
Results
Inhibitory Avoidance Memory
Lidocaine attenuated the magnitude of amnesia produced by anisomycin. Figure 1 shows the latencies to cross into the dark compartment on the memory test day, 48 hr after training and treatments. Rats that received injections of saline + anisomycin into the amygdala exhibited significantly shorter latencies when tested 48 hr after training compared to rats receiving vehicle injections (U7,9=0; p<0.002). Memory scores for rats receiving lidocaine injections into the amygdala did not differ from those of the vehicle controls, with all rats at the maximum (ceiling) latency in both groups. Importantly, lidocaine given 10 min prior to anisomycin injections resulted in test latencies significantly longer than those of the saline + anisomycin group (U12,9=19; p<0.02). The attenuation of the effects of anisomycin by lidocaine was partial; the lidocaine-anisomycin group exhibited memory scores that were still significantly shorter than those of vehicle controls (U12,7=0; p<0.002).
Figure 1.

Effects of intra-amygdala injections of anisomycin (ANI) and lidocaine (LIDO) on inhibitory avoidance memory. Injections of anisomycin into the amygdala 2 hours prior to training impaired memory tested 48 hours after training. Lidocaine injected 10 minutes prior to anisomycin partially attenuated the anisomycin-induced memory impairment. SAL = saline; PBS=phosphate-buffered saline.
Release of norepinephrine
Lidocaine attenuated the anisomycin-induced release of norepinephrine in the amygdala. As shown in Figure 2, the samples collected during and immediately after injections of anisomycin exhibited large increases in release of norepinephrine above baselines. Levels of norepinephrine release returned to baseline values and remained stable in later samples. ANOVAS revealed a significant effect of treatment (F1,5=10.82; p<0.05), time (F8,40=14.30; p<0.0001) and a treatment by time interaction (F8,40=8.65; p<0.0001) between the anisomycin vs. lidocaine + anisomycin groups. Post hoc tests revealed a substantial attenuation of anisomycin-induced increases in norepinephrine release in those rats pretreated with lidocaine (anisomycin 1300% vs. anisomycin-lidocaine 242%; p<0.01). Lidocaine did not itself result in a decrease in norepinephrine release compared to baseline values.
Figure 2.

Effects of intra-amygdala injections on norepinephrine release from the amygdala. Anisomycin (ANI) injections caused a large increase in norepinephrine release. Lidocaine (LIDO) pretreatment substantially and significantly blocked this effect. Lidocaine alone had no effect on norepinephrine release. B= Baseline; P= post-injection.
Immunohistochemistry
c-Fos immunoreactivity in the amygdala, assessed after inhibitory avoidance training, was readily evident in the vehicle-treated rats (Figure 3). Anisomycin appeared to block fully the training-initiated increase in expression of c-Fos. In the lidocaine + anisomycin condition, lidocaine did not attenuate the anisomycin-induced block of c-Fos expression; the inhibition of c-Fos expression remained apparently complete. These qualitative descriptions were confirmed quantitatively. As shown in Figure 4, the particle counts in saline/PBS controls (N = 4) were significantly higher than in either the saline-anisomycin (N = 2) or lidocaine-anisomycin (N=2) groups (161 ± 23 vs. 8 ± 7 and 4 ± 0.5, respectively; t’sdf’s=4 = 4.33 and 4.47, p’s<0.02). Counts in the saline-anisomycin and lidocaine-anisomycin groups did not differ significantly from each other (tdf=2 = 0.64, p>0.5).
Figure 3.

Top: Schematic showing placement of electrodes and sampling area for c-Fos expression (adapted from Paxinos and Watson, 2005, Figure 54). The rectangular dashed line identifies the area in which cannulae placements were accepted for behavioral and immunohistochemistry analyses. The circle (top and bottom of this figure; 455 μm diameter) represents the area analyzed for immunohistochemistry. Bottom: Representative pictures of c-Fos immunohistochemistry. Note that anisomycin reduced c-Fos expression; lidocaine did not attenuate the inhibition of c-Fos immunoreactivity caused by anisomycin. SAL= saline; PBS=phosphate-buffered saline; ANI = anisomycin; LIDO = lidocaine.
Figure 4.
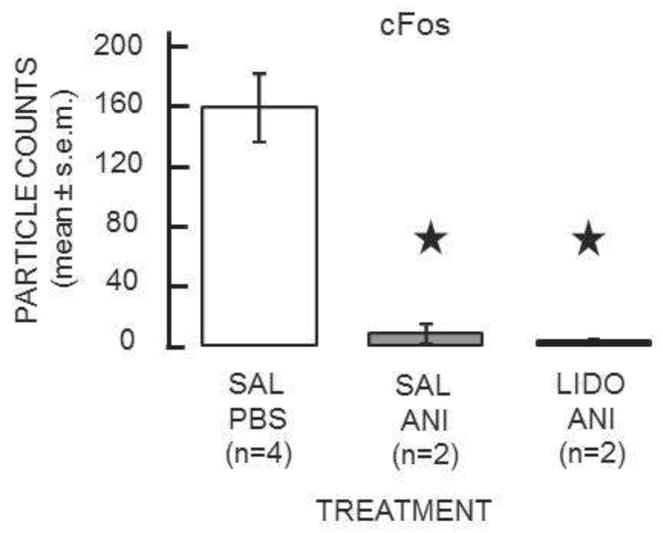
Quantification of c-Fos results. cFos expression was significantly higher in the sal-PBS condition than in either the sal-anisomycin or lidocaine-anisomycin conditions. (Stars indicate p’s < 0.02 vs. sal-PBS.)
Discussion
When lidocaine and anisomycin were administered directly into the amygdala, lidocaine pretreatment attenuated anisomycin-produced amnesia of memory of inhibitory avoidance training. The attenuation of anisomycin-induced amnesia by lidocaine occurred without concomitant attenuation of anisomycin-induced inhibition of local protein synthesis. However, lidocaine effectively blocked the increase in release of norepinephrine at the amygdala site of anisomycin injection.
Lidocaine injections into the amygdala have been used previously to impair memory formation (Vazdarjanova & McGaugh, 1999; Coleman-Mesches et al., 1996; Parent & McGaugh, 1994; Salinas et al., 1993; Keller et al., 2004). In each of these experiments, lidocaine was administered near the time of training. In the present experiment, we took advantage of the finding that anisomycin had anterograde effects on memory lasting at least 2 hr (Canal et al., 2007), while lidocaine had anterograde effects on memory for less than 30 min after injection. This difference in temporal properties permitted us to administer lidocaine soon before anisomycin under conditions in which only anisomycin would be expected to produce amnesia, revealing effects of lidocaine on the efficacy of anisomycin in producing amnesia without direct effects of the local inactivation on memory. The tests of memory in the lidocaine-alone group confirmed the absence of anterograde effects on memory and the tests in the anisomycin-alone group confirmed the presence of later amnesia.
The finding here that injections of anisomycin produce amnesia for memory of inhibitory avoidance is consistent with the results obtained using many tasks (cf.: Martinez et al.,1981; Davis & Squire, 1984; Dudai, 2002; Kandel, 2001; Alberini, 2008; Gold, 2008; Desgranges et al., 2008; De la Cruz, et al., 2008). Previous results show that intra-amygdala injections of anisomycin impair synaptic plasticity within the amygdala (Huang & Kandel, 2007) and impair fear conditioning (Schafe & Ledoux, 2000; Parsons et al., 2006; Maren et al., 2003). Directly related to the present findings is evidence that intra-amygdala injections of anisomycin impair memory for inhibitory avoidance training (Canal et al., 2007; Canal & Gold, 2008; Milekic et al., 2007; Izquierdo et al., 2002). Together with evidence that memory for inhibitory avoidance training is impaired in rats by lesions, electrical stimulation, and by local inactivation and other pharmacological manipulations of the amygdala (Gold et al., 1975, 1978; Izquierdo et al., 1992; Izquierdo & Medina, 1993; Jerusalinsky et al., 1992; Kim & McGaugh, 1992; Lennartz et al., 1996; Liang et al., 1982; Parent & McGaugh, 1994; Walker & Gold, 1994), the findings indicate that, like fear conditioning, memory for inhibitory avoidance training is also dependent on amygdala functions.
The key behavioral result was in the lidocaine + anisomycin group, in which inactivation of the amygdala just prior to the administration of anisomycin blunted the extent of amnesia. There are many treatments that attenuate amnesia produced by protein synthesis inhibitors (cf. Gold, 2006, 2008). Most studies demonstrating attenuation of amnesia produced by protein synthesis inhibitors used stimulant drugs, which themselves can enhance memory, leading to suggestions that pharmacological attenuation of amnesia produced by protein synthesis inhibitors is accomplished by enhancing residual memory that survives the inhibition (Davis & Squire, 1984; Squire, 2006). The present findings are not compatible with this view. Lidocaine itself impairs memory formation under other conditions (e.g., Vazdarjanova & McGaugh, 1999; Coleman-Mesches et al., 1996; Parent & McGaugh, 1994; Salinas et al., 1993; Keller et al., 2004), and to our knowledge does not enhance memory. The results obtained here with lidocaine are therefore consistent with attenuation of amnesia after protein synthesis inhibition by other drugs that similarly do not themselves enhance memory (Canal et al., 2007; Qi & Gold, 2009; Gold & Sternberg, 1978).
The findings of experiments in which protein synthesis inhibitors injected directly into the amygdala impair memory have generally been interpreted as revealing a role for training-initiated protein synthesis important for the formation of new memories (Duvarci et al., 2005; Nader et al., 2000a,b; Milekic et al., 2007; Cammarota et al., 2004; Izquierdo et al., 2002; Schafe & LeDoux, 2000; Chai et al., 2006), an interpretation common to similar tests of other brain areas as well (Rossato et al., 2007; De la Cruz et al., 2008; Desgranges et al., 2008; Izquierdo et al., 1992; Morris et al., 2006). When trained on fear conditioning tasks in particular, the interpretation is often that protein synthesis is necessary at a primary site of neural plasticity essential for memory formation (Duvarci et al., 2005; Maren et al., 2003; Nader, 2003; Nader et al., 2003a,b); Schafe & LeDoux, 2000), although the evidence for the amygdala as the primary site of memory storage for fear conditioning appears to be unconvincing (Weinberger, 2010). Similar interpretations of necessary protein synthesis have also been offered for inhibitory avoidance memory (Milekic et al., 2007; Izquierdo et al., 2002; Izquierdo & Medina, 1995).
While most interpretations of these reports regard the amygdala as an anatomical locus of memory formation, an alternative function for the amygdala is that the amygdala functions to modulate memories that are formed elsewhere in the brain (McGaugh, 2004; McIntyre et al., 2003). The evidence for this position includes findings that manipulations of the amygdala can interfere with memory formation and its modulation at other brain regions such as the hippocampus (McGaugh et al., 2002). According to this view, the amygdala does not serve as a storage site for memories but rather as a site of modulation. Related to the present findings, norepinephrine action within the amygdala is a major component of the mechanisms mediating modulation of memory formation. Modulation of memory by release of norepinephrine within the amygdala is consistent with the present findings showing that large increases in release of norepinephrine may mediate anisomycin-induced memory impairments.
The neurochemical results presented here replicate past findings showing that injections of anisomycin into the amygdala result in abnormally high levels of release of several neurotransmitters including norepinephrine (Canal et al., 2007). Norepinephrine effects on memory appear to follow an inverted-U relationship with memory formation. High levels of release of norepinephrine or high doses of norepinephrine injected in the amygdala near the time of training impair later memory (Gold & van Buskirk, 1978a,b; Canal et al., 2007; Liang et al., 1990; Decker & McGaugh, 1989). In addition, the β-adrenergic receptor antagonist, propranolol, blocks memory impairments produced by anisomycin with co-injections into the amygdala (Canal et al., 2007) and hippocampus (Qi & Gold, 2009). These findings support the view that the large increases in release of norepinephrine in response to anisomycin injections contribute to the amnesia produced by the protein synthesis inhibitor. This possibility is supported further by the present findings that lidocaine, under conditions in which the treatment attenuates anisomycin-induced amnesia, also blocks the abnormal release of norepinephrine in response to injection of the protein synthesis inhibitor.
Release of norepinephrine within the amygdala modulates memory formation apparently based on processing both within and beyond the amygdala. In addition, release of norepinephrine at many brain sites, such as the hippocampus, can also more directly modulate memory formation and synaptic plasticity (e.g.: Bramham et al., 1997; Dommett et al., 2008; Izquierdo et al., 1992; Izquierdo & Medina, 1995; Izumi & Zorumski, 1999; Lashgari et al., 2008; McGaugh et al., 2002; Miyashita & Williams, 2004; Reid & Harley, 2010; Roosevelt et al., 2006; Schimanski et al., 2007). Anisomycin injections directly into the hippocampus, like injections into the amygdala, impair memory for inhibitory avoidance learning (Milekic et al., 2007; Cammorota et al., 2004; Izquierdo et al., 2002; Canal & Gold, 2007; Qi & Gold, 2009). Importantly, amnesia produced by anisomycin injections directly into the hippocampus is also associated with increased release of norepinephrine at the site of injection (Qi & Gold, 2009). In that experiment, amnesia produced by intra-hippocampal injections of anisomycin was attenuated by the β-adrenergic antagonist, propranolol, as also seen with amygdala injections (Canal et al., 2007). These results are analogous to those reported here using lidocaine to attenuation anisomycin-induced amnesia and show that anisomycin-induced impairments of memory may be mediated by non-physiological levels of release of norepinephrine, and apparently other neurotransmitters (Canal et al., 2007; Qi & Gold, 2009), within multiple neural systems.
In contrast to the demonstration that lidocaine reduced the norepinephrine response to anisomycin, lidocaine did not attenuate the extent of inhibition of training-related increases in immunoreactivity of c-Fos, used here as a marker of protein synthesis. Rats that received lidocaine + anisomycin showed little c-Fos expression, with immunohistochemistry apparently very similar to that seen after anisomycin alone. The present results are consistent with those we have seen before (Canal et al., 2007); c-Fos immunoreactivity decreased after anisomycin injections into the amygdala but propranolol, which attenuated the amnesia, did not block the decrease in c-Fos immunoreactivity. Similar uncoupling of attenuating effects of neurotransmitter-related drugs on memory and on protein synthesis inhibition has been shown many times before (e.g., Barondes & Cohen, 1968; Serota et al., 1972; Hall et al., 1976; Quartermain et al., 1977; Flood et al., 1977, 1978; Lundgren & Carr, 1978; Sershen et al., 1982). The finding in the lidocaine + anisomycin group of substantial inhibition of expression of c-Fos while memory remains evident is not consistent with the view (Lamprecht & Dudai, 1996) that the transient expression of c-Fos in the amygdala is required for the formation of memory for amygdala-dependent tasks.
Of note, the large pulse of norepinephrine release evident after anisomycin injection had dissipated by the time training occurred. The return of norepinephrine release to baseline, and sometimes below baseline (Canal et al., 2007), may reflect depletion of releasable stores. Interestingly however, high extracellular levels of norepinephrine at the time of training seem unnecessary for the induction of amnesia, suggesting that norepinephrine initiates a cascade of downstream events that interfere with memory formation. This possibility may be related to gene superinduction seen after anisomycin injection (Edwards & Mahadevan, 1992; Radulovic & Tronson, 2008). There is also evidence that central injections of anisomycin elicit changes in motor cortex maps, synapse size, and number (Kleim et al., 2003) and may induce cell death (Morris et al., 2006). Like the changes in neurotransmitter release, these changes too might contribute to the induction of amnesia by inhibition of protein synthesis, adding to the potential specific cellular mechanisms responsible for amnesia produced by protein synthesis inhibitors. Together, such findings have led to the development of alternatives to the idea that training-initiated protein synthesis is necessary for memory formation (cf.: Routtenberg & Rekart, 2005; Rudy et al., 2006; Holahan & Routtenberg, 2007; Gold, 2006, 2008; Routtenberg, 2008). The evidence supporting these recent interpretations of the effects of protein synthesis inhibitors on memory point to a clear need to distinguish the mechanisms of amnesia from the mechanisms of memory formation; these mechanisms need not be reciprocal.
The present findings add to the evidence that anisomycin may impair memory either as a secondary action of the drug on neurotransmitter release or as a secondary consequence of the inhibition of protein synthesis. If the latter proves to be the case, then amnesias obtained with other protein synthesis inhibitors and possibly with other more specific inhibitors of cell signaling processes may also be secondary to similar neurochemical effects. In this regard, injections of CREB antisense into the amygdala produce amnesia and also perturb local release of norepinephrine and acetylcholine (Canal et al., 2008). Such findings suggest that interference with cell transcription and translation mechanisms might result in disruption of normal neuronal functions in a manner inconsistent with optimal memory and neural plasticity mechanisms. These secondary consequences on memory of interference with transcription and translation may be independent of specific drug, constraining the utility of such experiments for providing specific information regarding the mechanisms underlying memory formation.
Acknowledgments
This work was supported by USPHS NIH research grants from the National Institute on Drug Abuse, DA024129, and the National Institute on Aging, AG007648. We thank Ms. Jamie Richards for her technical assistance with the immunohistochemistry results reported here.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberini CM. The role of protein synthesis during the labile phases of memory: Revisiting the skepticism. Neurobiology of Learning and Memory. 2008;89:234–246. doi: 10.1016/j.nlm.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barondes SH, Cohen HD. Arousal and the conversion of “short-term” to “long-term” memory. Proceedings of the National Academy of Sciences, USA. 1968;61:923–929. doi: 10.1073/pnas.61.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Bacher-Svendsen K, Sarvey JM. LTP in the lateral perforant path is beta-adrenergic receptor-dependent. Neuroreport. 1997;8:719–724. doi: 10.1097/00001756-199702100-00028. [DOI] [PubMed] [Google Scholar]
- Cammorota M, Bevilaqua LR, Medina JH, Izquierdo I. Retrieval does not induce reconsolidation of inhibitory avoidance memory. Learning and Memory. 2004;11:572–578. doi: 10.1101/lm.76804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Amnesia produced by altered release of neurotransmitters after intra-amygdala injections of a protein synthesis inhibitor. Proceedings of the National Academy of Sciences, USA. 2007;104:12500–12505. doi: 10.1073/pnas.0705195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Intra-amygdala injections of CREB antisense impair inhibitory avoidance memory: Role of norepinephrine and acetylcholine. Learning and Memory. 2008;15:677–686. doi: 10.1101/lm.904308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Gold PE. Different temporal profiles of amnesia after intra-hippocampus and intra-amygdala infusions of anisomycin. Behavioral Neuroscience. 2007;121:732–741. doi: 10.1037/0735-7044.121.4.732. [DOI] [PubMed] [Google Scholar]
- Chai SC, Holahan MR, Shyu BC, Wang CC. Differential patterns of extracellular signal-regulated kinase-1 and -2 phosphorylation in rat limbic brain regions after short-term and long-term inhibitory avoidance learning. Neuroscience. 2006;137:1321–1330. doi: 10.1016/j.neuroscience.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Coleman-Mesches K, Salinas JA, McGaugh JL. Unilateral amygdala inactivation after training attenuates memory for reduced reward. Behavioural Brain Research. 1996;77:175–180. doi: 10.1016/0166-4328(95)00231-6. [DOI] [PubMed] [Google Scholar]
- Davis RE, Agranoff BW. Stages of memory formation in goldfish: evidence for an environmental trigger. Proceedings of the National Academy of Sciences, USA. 1966;55:555–559. doi: 10.1073/pnas.55.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychological Bulletin. 1984;96:518–559. [PubMed] [Google Scholar]
- Decker MW, McGaugh JL. Effects of concurrent manipulations of cholinergic and noradrenergic function on learning and retention in mice. Brain Research. 1989;477:29–37. doi: 10.1016/0006-8993(89)91391-7. [DOI] [PubMed] [Google Scholar]
- De la Cruz V, Rodriguez-Ortiz CJ, Falderas I, Bermudez-Rattoni F. Medial temporal lobe structures participate differentially in consolidation of safe and aversive taste memories. European Journal of Neuroscience. 2008;28:1377–1381. doi: 10.1111/j.1460-9568.2008.06432.x. [DOI] [PubMed] [Google Scholar]
- Desgranges B, Levy F, Ferreira G. Anisomycin infusion in amygdala impairs consolidation of odor aversion memory. Brain Research. 2008;1236:166–175. doi: 10.1016/j.brainres.2008.07.123. [DOI] [PubMed] [Google Scholar]
- Dommett EJ, Henderson EL, Westwell MS, Greenfield SA. Methylphenidate amplifies long-term plasticity in the hippocampus via noradrenergic mechanisms. Learning and Memory. 2008;15:580–586. doi: 10.1101/lm.1092608. [DOI] [PubMed] [Google Scholar]
- Dudai Y. Molecular basis of long-term memories: A question of persistence. Current Opinion in Neurobiology. 2002;12:211–216. doi: 10.1016/s0959-4388(02)00305-7. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. Activation of extracellular signal-regulated -- kinase-mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. European Journal of Neuroscience. 2005;21:283–289. doi: 10.1111/j.1460-9568.2004.03824.x. [DOI] [PubMed] [Google Scholar]
- Edeline JM, Hars B, Hennevin E, Cotillon N. Muscimol diffusion after intracerebral microinjections: a reevaluation based on electrophysiological and autoradiographic quantifications. Neurobiology of Learning and Memory. 2002;78:100–124. doi: 10.1006/nlme.2001.4035. [DOI] [PubMed] [Google Scholar]
- Edwards DR, Mahadevan LC. Protein synthesis inhibitors differentially superinduce c-fos and c-jun by three distinct mechanisms: Lack of evidence for labile repressors. EMBO Journal. 1992;11:2415–2424. doi: 10.1002/j.1460-2075.1992.tb05306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner LB, Goodman RH. Studies on memory: inhibitors of protein synthesis also inhibit catecholamine synthesis. Proceedings of the National Academy of Sciences, USA. 1975;72:4660–4663. doi: 10.1073/pnas.72.11.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner LB, Serota RG, Goodman RH. Cycloheximide and acetoxycycloheximide: inhibition of tyrosine hydroxylase activity and amnestic effects. Proceedings of the National Academy of Sciences, USA. 1973;70:354–356. doi: 10.1073/pnas.70.2.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, Bennett EL, Orme AE, Jarvik ME. Protein synthesis dependent gradient of ECS retrograde amnesia. Behavioral Biology. 1977;21:307–328. doi: 10.1016/s0091-6773(77)90101-8. [DOI] [PubMed] [Google Scholar]
- Flood JF, Bennett EL, Orme AE, Rosenzweig MR, Jarvik ME. Memory: modification of anisomycin-induced amnesia by stimulants and depressants. Science. 1978;199:324–326. doi: 10.1126/science.619461. [DOI] [PubMed] [Google Scholar]
- Gold PE. The many faces of amnesia. Learning and Memory. 2006;13:506–514. doi: 10.1101/lm.277406. [DOI] [PubMed] [Google Scholar]
- Gold PE. Protein synthesis inhibition and memory: formation vs. amnesia. Neurobiology of Learning and Memory. 2008;89:201–211. doi: 10.1016/j.nlm.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold PE, Hankins L, Edwards RM, Chester J, McGaugh JL. Memory interference and facilitation with posttrial amygdala stimulation: effect on memory varies with footshock level. Brain Research. 1975;86:509–513. doi: 10.1016/0006-8993(75)90905-1. [DOI] [PubMed] [Google Scholar]
- Gold PE, Rose RP, Hankins LL. Retention impairment produced by unilateral amygdala implantation: reduction by posttrial amygdala stimulation. Behavioral Biology. 1978;22:515–523. doi: 10.1016/s0091-6773(78)92673-1. [DOI] [PubMed] [Google Scholar]
- Gold PE, Sternberg DB. Retrograde amnesia produced by several treatments: evidence for a common neurobiological mechanism. Science. 1978;201:367–369. doi: 10.1126/science.208153. [DOI] [PubMed] [Google Scholar]
- Gold PE, van Buskirk RB. Posttraining brain norepinephrine concentrations: correlation with retention performance of avoidance training and with peripheral epinephrine modulation of memory processing. Behavioral Biology. 1978a;23:509–520. doi: 10.1016/s0091-6773(78)91614-0. [DOI] [PubMed] [Google Scholar]
- Gold PE, van Buskirk RB. Effects of α- and β-adrenergic receptor antagonists on post-trial epinephrine modulation of memory: Relationship to post-training brain norepinephrine concentrations. Behavioral Biology. 1978b;24:168–184. doi: 10.1016/s0091-6773(78)93045-6. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Flexner JB, Flexner LB. The effect of acetoxycycloheximide on rate of accumulation of cerebral catecholamines from circulating tyrosine as related to its effect on memory. Proceedings of the National Academy of Sciences, USA. 1975;72:479–482. doi: 10.1073/pnas.72.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ME, Schlesinger K, Stamm E. Prevention of memory loss following puromycin treatment. Pharmacology Biochemistry & Behavior. 1976;4:353–355. doi: 10.1016/0091-3057(76)90256-2. [DOI] [PubMed] [Google Scholar]
- Holahan MR, Routtenberg A. Post-translational synaptic protein modification as substrate for long-lasting, remote memory: an initial test. Hippocampus. 2007;17:93–97. doi: 10.1002/hipo.20245. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Low-frequency stimulation induces a pathway-specific late phase of LTP in the amygdala that is mediated by PKA and dependent on protein synthesis. Learning and Memory. 2007;14:497–503. doi: 10.1101/lm.593407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, Delgado-García JM, Carrión ÁM. Acquisition, consolidation, reconsolidation, and extinction of eyelid conditioning responses require de novo protein synthesis. Journal of Neuroscience. 2005;25:2070–2080. doi: 10.1523/JNEUROSCI.4163-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo LA, Barros DM, Vianna MR, Coitinho A, deDavid e Silva T, Choi H, Moletta B, Medina JH, Izquierdo I. Molecular pharmacological dissection of short- and long-term memory. Cellular and Molecular Neurobiology. 2002;22:269–287. doi: 10.1023/a:1020715800956. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, da Cunha C, Rosat R, Jerusalinsky D, Ferreira MB, Medina JH. Neurotransmitter receptors involved in post-training memory processing by the amygdala, medial septum, and hippocampus of the rat. Behavioral and Neural Biology. 1992;58:16–26. doi: 10.1016/0163-1047(92)90847-w. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Medina JH. Role of the amygdala, hippocampus and entorhinal cortex in memory consolidation and expression. Brazilian Journal of Medical and Biological Research. 1993;26:573–589. [PubMed] [Google Scholar]
- Izquierdo I, Medina JH. Correlation between the pharmacology of long-term potentiation and the pharmacology of memory. Neurobiology of Learning and Memory. 1995;63:19–32. doi: 10.1006/nlme.1995.1002. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Norepinephrine promotes long-term potentiation in the adult rat hippocampus in vitro. Synapse. 1999;31:196–202. doi: 10.1002/(SICI)1098-2396(19990301)31:3<196::AID-SYN4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Jerusalinsky D, Ferreira MB, Walz R, Da Silva RC, Bianchin M, Ruschel AC, Zanatta MS, Medinia JH, Izquierdo I. Amnesia by post-training infusion of glutamate receptor antagonists into the amygdala, hippocampus, and entorhinal cortex. Behavioral and Neural Biology. 1992;58:76–80. doi: 10.1016/0163-1047(92)90982-a. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Keller M, Perrin G, Meurisse M, Ferreira G, Levy F. Cortical and medial amygdala are both involved in the formation of olfactory offspring memory in sheep. European Journal of Neuroscience. 2004;20:3433–3441. doi: 10.1111/j.1460-9568.2004.03812.x. [DOI] [PubMed] [Google Scholar]
- Kim M, McGaugh JL. Effects of intra-amygdala injections of NMDA receptor antagonists on acquisition and retention of inhibitory avoidance. Brain Research. 1992;585:35–48. doi: 10.1016/0006-8993(92)91188-k. [DOI] [PubMed] [Google Scholar]
- Kleim JA, Bruneau R, Calder K, Pocock D, VandenBerg PM, MacDonald E, Monfils MH, Sutherland RJ, Nader K. Functional organization of adult motor cortex is dependent upon continued protein synthesis. Neuron. 2003;40:167–176. doi: 10.1016/s0896-6273(03)00592-0. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, Dudai Y. Transient expression of c-Fos in rat amygala during training is required for encoding conditioned taste aversion memory. Learning and Memory. 1996;3:31–41. doi: 10.1101/lm.3.1.31. [DOI] [PubMed] [Google Scholar]
- Lashgari R, Khakpour-Taleghani B, Motamedi F, Shahidi S. Effects of reversible inactivation of locus coeruleus on long-term potentiation in perforant path-DG synapses in rats. Neurobiology of Learning and Memory. 2008;90:309–316. doi: 10.1016/j.nlm.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Lennartz RC, Hellems KL, Mook ER, Gold PE. Inhibitory avoidance impairments induced by intra-amygdala propranolol are reversed by glutamate but not glucose. Behavioral Neuroscience. 1996;110:1033–1039. doi: 10.1037//0735-7044.110.5.1033. [DOI] [PubMed] [Google Scholar]
- Liang KC, McGaugh JL, Yao HY. Involvement of amygdala pathways in the influence of post-training intra-amygdala norepinephrine and peripheral epinephrine on memory storage. Brain Research. 1990;508:225–233. doi: 10.1016/0006-8993(90)90400-6. [DOI] [PubMed] [Google Scholar]
- Lundgren P, Carr LA. Effects of anisomycin and CNS stimulants on brain catecholamine synthesis. Pharmacology Biochemistry & Behavior. 1978;9:559–561. doi: 10.1016/0091-3057(78)90059-x. [DOI] [PubMed] [Google Scholar]
- Mamou CB, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nature Neuroscience. 2006;9:1237–1239. doi: 10.1038/nn1778. [DOI] [PubMed] [Google Scholar]
- Maren S, Ferrario CR, Corcoran KA, Desmond TJ, Frey KA. Protein synthesis in the amygdala, but not the auditory thalamus, is required for consolidation of Pavlovian fear conditioning in rats. European Journal of Neuroscience. 2003;18:3080–3088. doi: 10.1111/j.1460-9568.2003.03063.x. [DOI] [PubMed] [Google Scholar]
- Martin JH. Autoradiographic estimation of the extent of reversible inactivation produced by microinjection of lidocaine and muscimol in the rat. Neuroscience Letters. 1991;127:160–164. doi: 10.1016/0304-3940(91)90784-q. [DOI] [PubMed] [Google Scholar]
- Martinez JL, Jr, Jensen RA, McGaugh JL. Attenuation of experimentally-induced amnesia. Progress in Neurobiology. 1981;16:155–186. doi: 10.1016/0301-0082(81)90011-3. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annual Review of Neuroscience. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, McIntyre CK, Power AE. Amygdala modulation of memory consolidation: interaction with other brain systems. Neurobiology of Learning and Memory. 2002;78:539–552. doi: 10.1006/nlme.2002.4082. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Power AE, Roozendaal B, McGaugh JL. Role of the basolateral amygdala in memory consolidation. Annals of the New York Academy of Sciences. 2003;985:273–293. doi: 10.1111/j.1749-6632.2003.tb07088.x. [DOI] [PubMed] [Google Scholar]
- Milekic MH, Pollonini G, Alberini CM. Temporal requirement of C/EBPbeta in the amygdala following reactivation but not acquisition of inhibitory avoidance. Learning and Memory. 2007;14:504–511. doi: 10.1101/lm.598307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T, Williams CL. Peripheral arousal-related hormones modulate norepinephrine release in the hippocampus via influences on brainstem nuclei. Behav Brain Research. 2004;153:87–95. doi: 10.1016/j.bbr.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Morris RG, Inglis J, Ainge JA, Olverman HJ, Tulloch J, Dudai Y, Kelly PA. Memory reconsolidation: Sensitivity of spatial memory to inhibition of protein synthesis in dorsal hippocampus during encoding and retrieval. Neuron. 2006;50:479–489. doi: 10.1016/j.neuron.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces unbound. Trends in Neuroscience. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000a;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. The labile nature of consolidation theory. Nature Reviews in Neuroscience. 2000b;1:216–219. doi: 10.1038/35044580. [DOI] [PubMed] [Google Scholar]
- Pan WHT, Chen NH, Tsai FY, Liao HY. Intrategmental infusion of cocaine decreases dopamine release and enhances norepinephrine release in the medial prefrontal cortex. European Journal of Pharmacology. 1996;317:205–213. doi: 10.1016/s0014-2999(96)00724-8. [DOI] [PubMed] [Google Scholar]
- Parent MB, McGaugh JL. Posttraining infusion of lidocaine into the amygdala basolateral complex impairs retention of inhibitory avoidance training. Brain Research. 1994;661:97–103. doi: 10.1016/0006-8993(94)91186-x. [DOI] [PubMed] [Google Scholar]
- Parsons RG, Gafford GM, Baruch DE, Riedner BA, Helmstetter FJ. Long-term stability of fear memory depends on the synthesis of protein but not mRNA in the amygdala. European Journal of Neuroscience. 2006;23:1853–1859. doi: 10.1111/j.1460-9568.2006.04723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 5. San Diego: Elsevier/Academic Press; 2005. CD-ROM. [Google Scholar]
- Qi Z, Gold PE. Intrahippocampal infusions of anisomycin produce amnesia: contribution of increased release of norepinephrine, dopamine, and acetylcholine. Learning and Memory. 2009;16:308–314. doi: 10.1101/lm.1333409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartermain D, Freedman LS, Botwinick CY, Gutwein BM. Reversal of cycloheximide-induced amnesia by adrenergic receptor stimulation. Pharmacology Biochemistry & Behavior. 1977;7:259–267. doi: 10.1016/0091-3057(77)90143-5. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Tronson NC. Protein synthesis inhibitors, gene superinduction and memory: too little or too much protein? Neurobiology of Learning and Memory. 2008;89:212–218. doi: 10.1016/j.nlm.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AT, Harley CW. An associativity requirement for locus coeruleus-induced long-term potentiation in the dentate gyrus of the urethane-anesthetized rat. Experimental Brain Research. 2010;200:151–159. doi: 10.1007/s00221-009-1955-6. [DOI] [PubMed] [Google Scholar]
- Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Research. 2006;1119:124–132. doi: 10.1016/j.brainres.2006.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Myskiw JC, Medina JH, Izquierdo I, Cammarota M. On the role of hippocampal protein synthesis in the consolidation and reconsolidation of object recognition memory. Learning and Memory. 2007;14:36–46. doi: 10.1101/lm.422607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routtenberg A. The substrate for long-lasting memory: if not protein synthesis, then what? Neurobiology of Learning and Memory. 2008;89:225–233. doi: 10.1016/j.nlm.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routtenberg A, Rekart JL. Post-translational protein modification as the substrate for long-lasting memory. Trends in Neuroscience. 2005;28:12–19. doi: 10.1016/j.tins.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, Moineau J, Bolding K. Anisomycin and the reconsolidation hypothesis. Learning and Memory. 2006;13:1–3. doi: 10.1101/lm.157806. [DOI] [PubMed] [Google Scholar]
- Salinas JA, Packard MG, McGaugh JL. Amygdala modulates memory for changes in reward magnitude: reversible post-training inactivation with lidocaine attenuates the response to a reduction in reward. Behavioral Brain Research. 1993;59:153–159. doi: 10.1016/0166-4328(93)90162-j. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. Journal of Neuroscience. 2000;20(1–5):RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski LA, Ali DW, Baker GB, Nguyen PV. Impaired hippocampal LTP in inbred mouse strains can be rescued by beta-adrenergic receptor activation. European Journal of Neuroscience. 2007;25:1589–1598. doi: 10.1111/j.1460-9568.2007.05376.x. [DOI] [PubMed] [Google Scholar]
- Sershen H, Reith ME, Lajtha A. On the interaction between nicotine and cycloheximide. Brain Research. 1982;251:183–185. doi: 10.1016/0006-8993(82)91290-2. [DOI] [PubMed] [Google Scholar]
- Serota RG, Roberts RB, Flexner L. Acetoxycycloheximide-induced transient amnesia: protective effects of adrenergic stimulants. Proceedings of the National Academy of Sciences, USA. 1972;69:340–342. doi: 10.1073/pnas.69.2.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel S. Nonparametric Statistics for the Behavioral Sciences. New York: McGraw–Hill; 1956. [Google Scholar]
- Squire LR. Lost forever or temporarily misplaced? The long debate about the nature of memory impairment. Learning and Memory. 2006;13:522–529. doi: 10.1101/lm.310306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazdarjanova A, McGaugh JL. Basolateral amygdala is involved in modulating consolidation of memory for classical fear conditioning. Journal of Neuroscience. 1999;19:6615–6622. doi: 10.1523/JNEUROSCI.19-15-06615.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Gold PE. Intra-amygdala kinase inhibitors disrupt retention of a learned avoidance response in rats. Neuroscience Letters. 1994;176:255–258. doi: 10.1016/0304-3940(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Weinberger NM. The medial geniculate, not the amygdala, as the root of auditory fear conditioning. Hearing Research. 2010 doi: 10.1016/j.heares.2010.03.093. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]